
Estrogen Receptor Signaling Pathways Involved in Invasion and Colony Formation of Androgen-Independent Prostate Cancer Cells PC-3


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blot Analysis for Detection of Total and Phosphorylated SRC and Non-Phosphorylated β-Catenin
2.3. Protein Assays
2.4. Cell Invasion Analysis
2.5. Colony Formation Analysis (Soft Agar)
2.6. Immunofluorescence Analysis for the Detection of Non-Phosphorylated β-Catenin
2.7. Statistical Analysis
3. Results
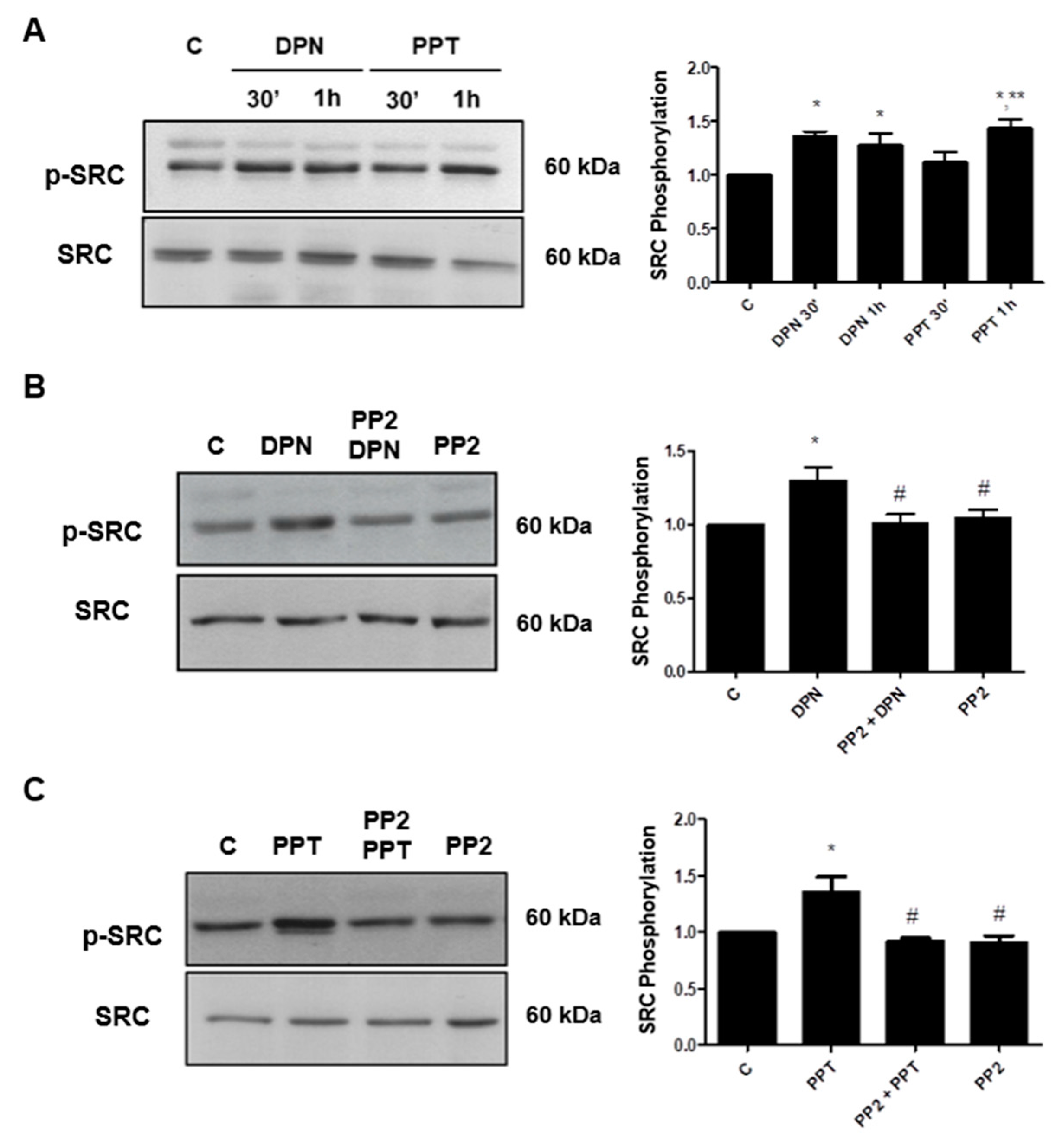
3.1. Activation of ERα and ERβ Increases the Phosphorylation of SRC in PC-3 Cells
3.2. SRC is Involved on Invasion and Colony Formation of PC-3 Cells Induced by ER Activation
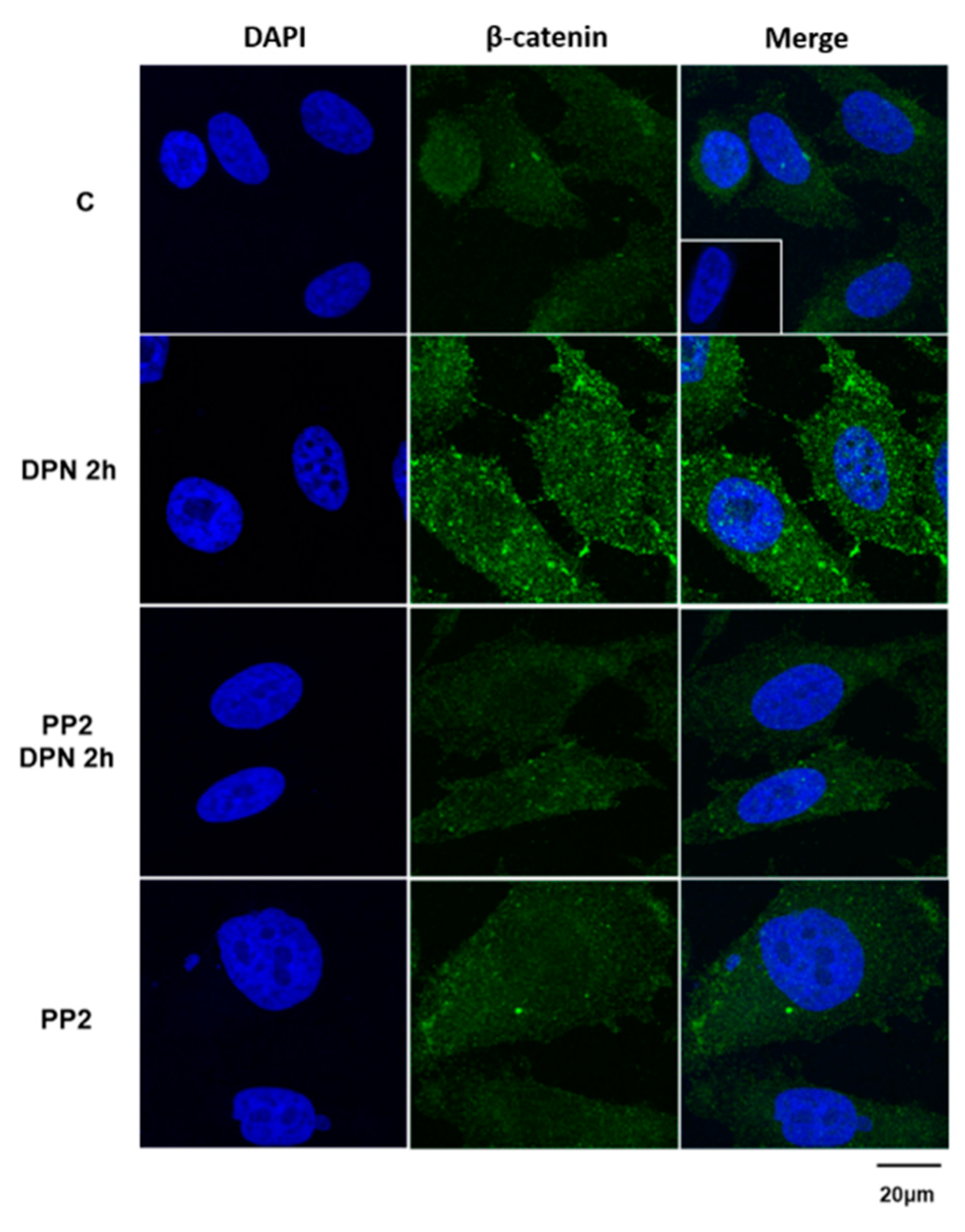
3.3. The Increase in the Expression of the Non-Phosphorylated β-Catenin Induced by Activation of ERβ is Mediated by SRC
3.4. PI3K/AKT are also Required for Invasion and Colony Formation of PC-3 Cells Simulated by ER
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogens and their receptors in prostate cancer: Therapeutic implications. Front. Oncol. 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbs, R.W.; Malhotra, N.R.; Greenwald, D.T.; Wang, A.Y.; Prins, G.S.; Abern, M.R. Prostate, Eetrogens and prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.D.; Bauman, T.M.; Nicholson, T.M.; Vellky, J.E.; Ricke, E.A.; Tang, W.; Xu, W.; Huang, W.; Ricke, W.A. Tissue-specific quantification and localization of androgen and estrogen receptors in prostate cancer. Hum. Pathol. 2019, 89, 99–108. [Google Scholar] [CrossRef]
- Vellky, J.E.; Ricke, W.A. Development and prevalence of castration-resistant prostate cancer subtypes. Neoplasia 2020, 22, 566–575. [Google Scholar] [CrossRef]
- Pisolato, R.; Lombardi, A.P.G.; Vicente, C.M.; Lucas, T.F.G.; Lazari, M.F.M.; Porto, C.S. Expression and regulation of the estrogen receptors in PC-3 human prostate cancer cells. Steroids 2016, 107, 74–86. [Google Scholar] [CrossRef]
- Souza, D.S.; Lombardi, A.P.G.; Vicente, C.M.; Lucas, T.F.G.; Erustes, A.G.; Pereira, G.J.S. Estrogen receptors localization and signaling pathways in DU-145 human prostate cancer cells. Mol. Cell. Endocrinol. 2019, 483, 11–23. [Google Scholar] [CrossRef]
- Lombardi, A.P.; Pisolato, R.; Vicente, C.M.; Lazari, M.F.; Lucas, T.F.; Porto, C.S. Estrogen receptor beta (ERβ) mediates expression of b-catenin and proliferation in prostate cancer cell line PC-3. Mol. Cell. Endocrinol. 2016, 430, 12–24. [Google Scholar] [CrossRef]
- Silva, R.S.; Lombardi, A.P.G.; de Souza, D.S.; Vicente, C.M.; Porto, C.S. Activation of estrogen receptor beta (ERβ) regulates the expression of N-cadherin, E-cadherin and b-catenin in androgen-independent prostate cancer cells. Int. J. Biochem. Cell Biol. 2018, 96, 40–50. [Google Scholar] [CrossRef]
- Lombardi, A.P.G.; Vicente, C.M.; Porto, C.S. Estrogen receptors promote migration, invasion and colony formation of the Androgen-independent prostate cancer cells PC-3 through b-Catenin pathway. Front. Endocrinol. 2020, 11, 184. [Google Scholar] [CrossRef]
- Acconcia, F.; Marino, M. The effects of 17β-estradiol in cancer are mediated by estrogen receptor signaling at the plasma membrane. Front. Physiol. 2011, 2, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, L.; Kim, J.E.; Mao, C.; Shapiro, D.J. Src couples estrogen receptor to the anticipatory unfolded protein response and regulates cancer cell fate under stress. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118765. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [Green Version]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Mattei, V.; Botta, L.; Angelucci, A. Src family kinases as therapeutic targets in advanced solid tumors: What we have learned so far. Cancers 2020, 12, 1448. [Google Scholar] [CrossRef]
- Goldenberg-Furmanov, M.; Stein, I.; Pikarsky, E.; Rubin, H.; Kasem, S.; Wygoda, M.; Weinstein, I.; Reuveni, H.; Ben-Sasson, S.A. Lyn is a target gene for prostate cancer: Sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004, 64, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.; Kim, D.; Cheng, J.Q.; Zhang, S.; Lee, J.H.; Buettner, R.; Mirosevich, J.; Lee, F.Y.; Jove, R. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005, 65, 9185–9189. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Siddiqui, I.A.; Hafeez, B.B.; Baniahmad, A.; Mukhtar, H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene 2008, 27, 3596–3604. [Google Scholar] [CrossRef] [Green Version]
- Tatarov, O.; Mitchell, T.J.; Seywright, M.; Leung, H.Y.; Brunton, V.G.; Edwards, J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin. Cancer Res. 2009, 15, 3540–3549. [Google Scholar] [CrossRef] [Green Version]
- Piedra, J.; Miravet, S.; Castaño, J.; Pálmer, H.G.; Heisterkamp, N.; García de Herreros, A.; Duñach, M. p120 Catenin-associated Fer and Fyn tyrosine kinases regulate beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin Interaction. Mol. Cell. Biol. 2003, 23, 2287–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coluccia, A.M.; Benati, D.; Dekhil, H.; De Filippo, A.; Lan, C.; Gambacorti-Passerini, C. SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp60(c-Src)-dependent tyrosine phosphorylation of beta-catenin and its nuclear signaling. Cancer Res. 2006, 66, 2279–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, R.L.; Gottardi, C.J. Phospho-regulation of Beta-catenin adhesion and signaling functions. Physiology 2007, 22, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell. Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J. Biol. Chem. 2005, 280, 32081–32089. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Zhang, Z.; Jiang, B.H.; Shi, X. Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem. Biophys. Res. Commun. 2003, 310, 1124–1132. [Google Scholar] [CrossRef]
- Kim, R.J.; Bae, E.; Hong, Y.K.; Hong, J.Y.; Kim, N.K.; Ahn, H.J.; Oh, J.J.; Park, D.S. PTEN loss-mediated Akt activation increases the properties of cancer stem-like cell populations in prostate cancer. Oncology 2014, 87, 270–279. [Google Scholar] [CrossRef]
- Vo, B.T.; Morton, D.; Komaragiri, S.; Millena, A.C.; Leath, C.; Khan, S.A. TGF-β effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology 2013, 154, 1768–1779. [Google Scholar] [CrossRef]
- Stauffer, S.R.; Coletta, C.J.; Tedesco, R.; Nishiguchi, G.; Carlson, K.; Sun, J.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Pyrazole ligands: Structure-affinity/activity relationships and estrogen receptor-alpha-selective agonists. J. Med. Chem. 2000, 43, 4934–4947. [Google Scholar] [CrossRef] [PubMed]
- Meyers, M.J.; Sun, J.; Carlson, K.E.; Marriner, G.A.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor-beta potency-selective ligands: Structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J. Med. Chem. 2001, 44, 4230–4251. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.F.; Siu, E.R.; Esteves, C.A.; Monteiro, H.P.; Oliveira, C.A.; Porto, C.S.; Lazari, M.F. 17beta-estradiol induces the translocation of the estrogen receptors ESR1 and ESR2 to the cell membrane, MAPK3/1 phosphorylation and proliferation of cultured immature rat Sertoli cells. Biol. Reprod. 2008, 78, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente, C.M.; Lima, M.A.; Nader, H.B.; Toma, L. SULF2 overexpression positively regulates tumorigenicity of human prostate cancer cells. J. Exp. Clin. Cancer Res. 2015, 14, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizazi, K. The role of Src in prostate cancer. Ann. Oncol. 2007, 18, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.C.; Song, L.; Haura, E.B. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M. Src signaling in cancer invasion. J. Cell. Physiol. 2010, 223, 14–26. [Google Scholar] [CrossRef]
- Migliaccio, A.; Castoria, G.; Di Domenico, M.; de Falco, A.; Bilancio, A.; Lombardi, M.; Barone, M.V.; Ametrano, D.; Zannini, M.S.; Abbondanza, C.; et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000, 19, 5406–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef]
- Chen, F.; Wu, J.; Teng, J.; Li, W.; Zheng, J.; Bai, J. HCRP-1 regulates cell migration, invasion and angiogenesis via Src/ FAK signaling in human prostate cancer. Int. J. Biol. Sci. 2020, 16, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnato, G.; Leopizzi, M.; Urciuoli, E.; Peruzzi, B. Nuclear functions of the tyrosine kinase Src. Int. J. Mol. Sci 2020, 21, 2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; de Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulard, C.; Rambaud, J.; Le Romancer, M.; Corbo, L. Proximity ligation assay to detect and localize the interactions of ERα with PI3-K and Src in breast cancer cells and tumor samples. Methods Mol. Biol. 2014, 1204, 135–143. [Google Scholar] [CrossRef]
- Miyabayashi, T.; Teo, J.L.; Yamamoto, M.; McMillan, M.; Nguyen, C.; Kahn, M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2007, 104, 5668–5673. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardi, A.P.G.; Cavalheiro, R.P.; Porto, C.S.; Vicente, C.M. Estrogen Receptor Signaling Pathways Involved in Invasion and Colony Formation of Androgen-Independent Prostate Cancer Cells PC-3. Int. J. Mol. Sci. 2021, 22, 1153. https://doi.org/10.3390/ijms22031153
Lombardi APG, Cavalheiro RP, Porto CS, Vicente CM. Estrogen Receptor Signaling Pathways Involved in Invasion and Colony Formation of Androgen-Independent Prostate Cancer Cells PC-3. International Journal of Molecular Sciences. 2021; 22(3):1153. https://doi.org/10.3390/ijms22031153
Chicago/Turabian StyleLombardi, Ana Paola G., Renan P. Cavalheiro, Catarina S. Porto, and Carolina M. Vicente. 2021. "Estrogen Receptor Signaling Pathways Involved in Invasion and Colony Formation of Androgen-Independent Prostate Cancer Cells PC-3" International Journal of Molecular Sciences 22, no. 3: 1153. https://doi.org/10.3390/ijms22031153
APA StyleLombardi, A. P. G., Cavalheiro, R. P., Porto, C. S., & Vicente, C. M. (2021). Estrogen Receptor Signaling Pathways Involved in Invasion and Colony Formation of Androgen-Independent Prostate Cancer Cells PC-3. International Journal of Molecular Sciences, 22(3), 1153. https://doi.org/10.3390/ijms22031153