A Closer Look at Estrogen Receptor Mutations in Breast Cancer and Their Implications for Estrogen and Antiestrogen Responses



Abstract
:1. Introduction
1.1. Breast Cancer Types
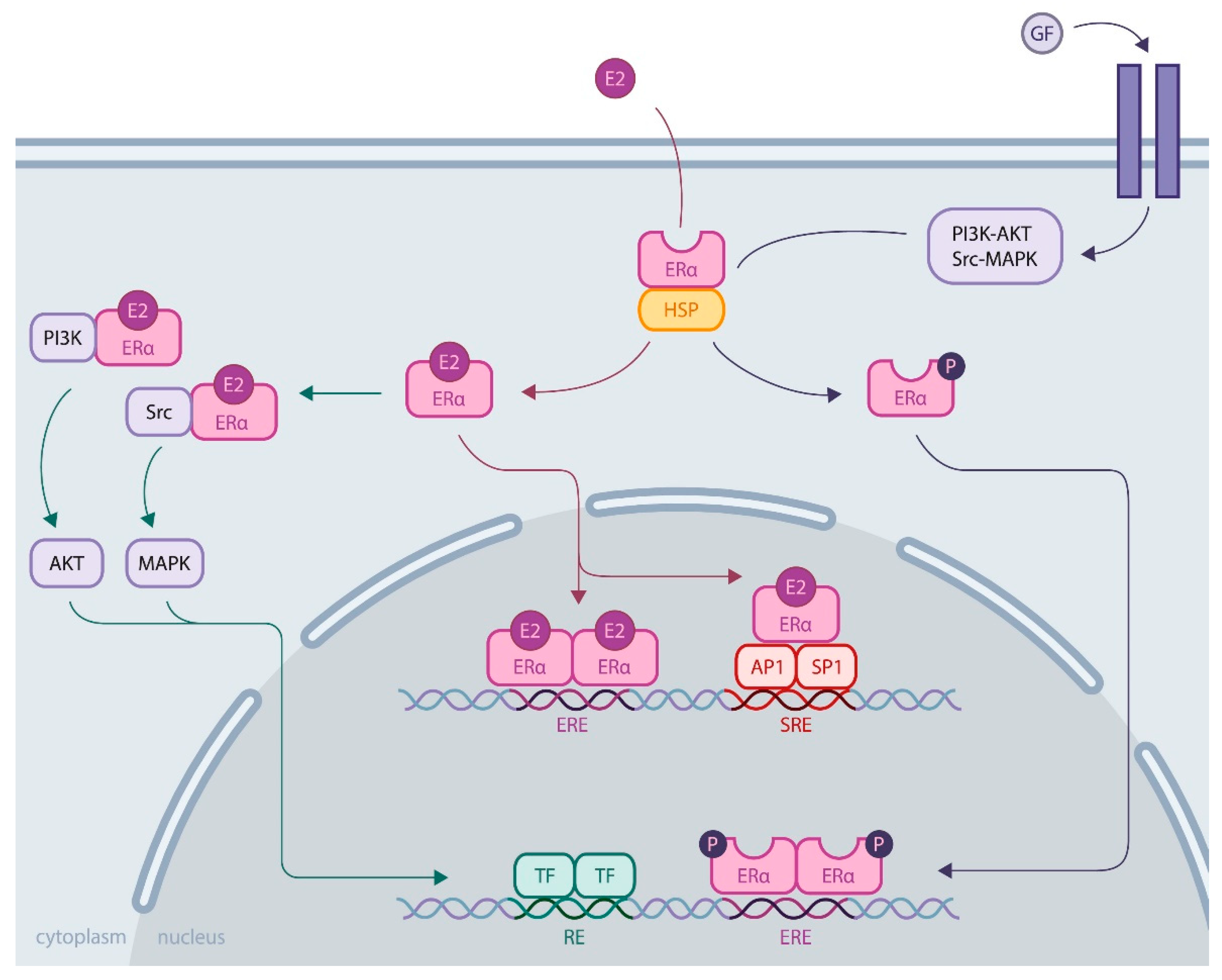
1.2. ERα Activity
1.3. Role of ERα in Breast Cancer
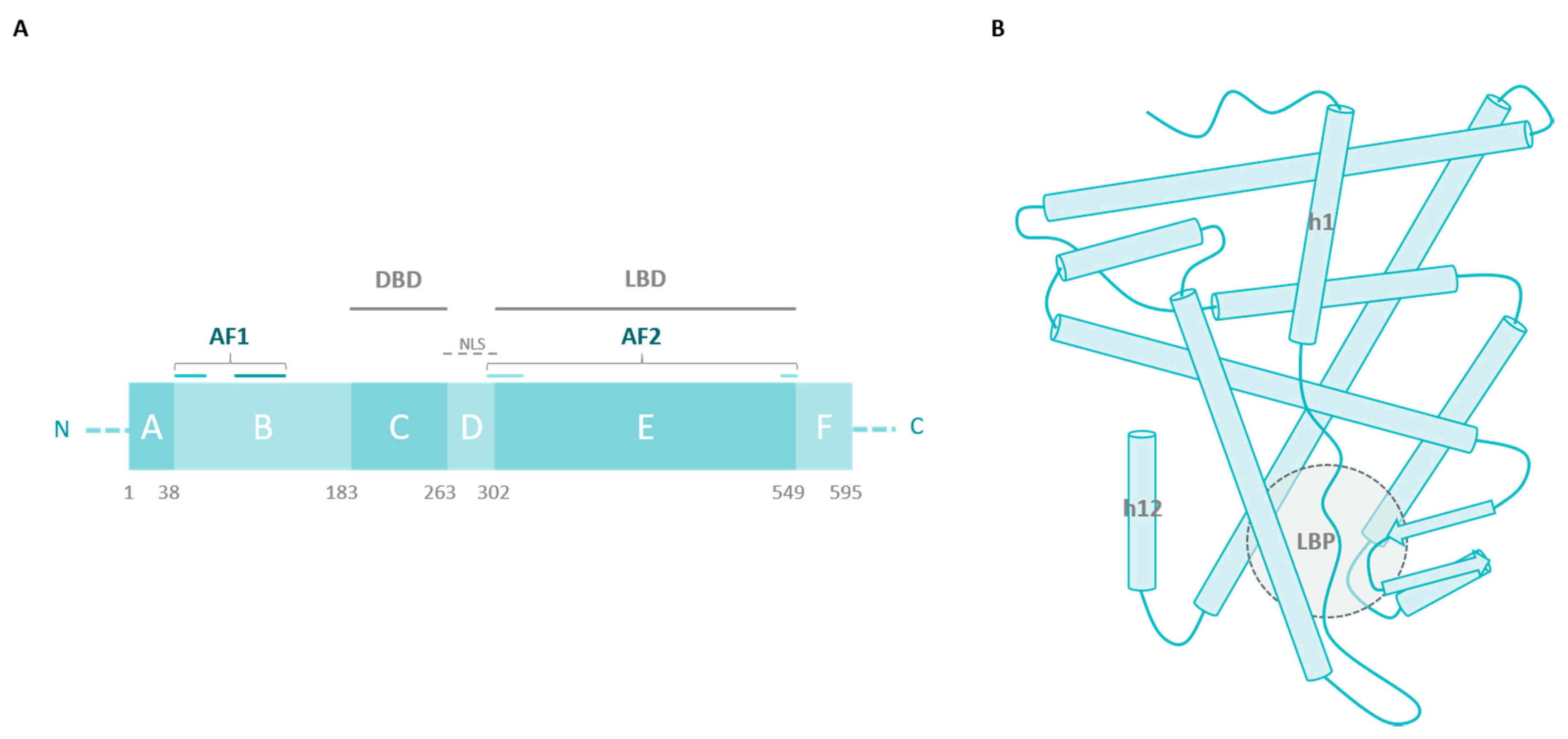
2. ERα Missense Mutations
2.1. The Ligand-Binding Domain
2.2. Outside the LBD
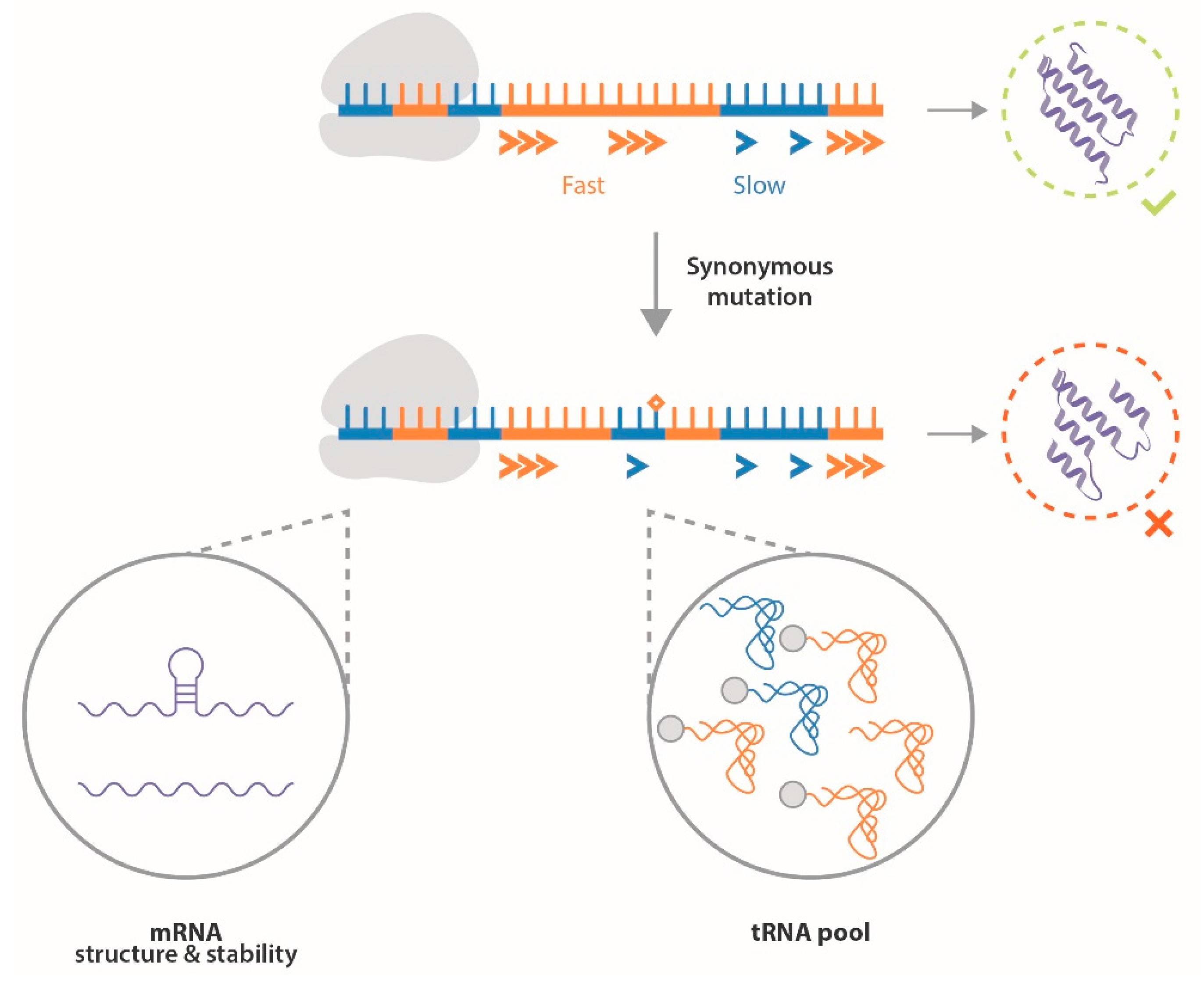
3. ERα Synonymous Mutations
- Small sample size: Most studies included only a few hundred patients and controls, resulting in low statistical power for determining associations;
- Control source: Some studies compared data from breast cancer patients to controls originating from the entire population, whereas other studies used data from the hospital as a control, which could induce bias in the observed associations [59];
- Analytic methods: The heterogeneity of methods employed to analyze the association of ESR1 silent mutations with breast cancer development plays a role in the inconsistency of conclusions as well.
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AF1 | transcriptional activation function 1 |
| DBD | DNA-binding domain |
| E2 | estrogen |
| ERα | estrogen receptor alpha |
| ERBS | estrogen receptor binding site |
| ERE | estrogen responsive element |
| GF | growth factor |
| HER2 | human epidermal growth factor receptor 2 |
| HSP | heat shock protein |
| LBD | ligand-binding domain |
| LBP | ligand-binding pocket |
| PDX | patient derived xenograft |
| PFAR | protein folding activity of ribosomes |
| PR | progesterone receptor |
| PROTAC | proteolysis-targeting chimera |
| PTM | posttranslational modification |
| RFLP | restriction fragment length polymorphism |
| SERCA | selective ER covalent antagonist |
| SERD | selective estrogen receptor downregulator |
| SERM | selective estrogen receptor modulator |
| SRE | serum responsive element |
| TF | transcription factor |
References
- Lakhani, S.; Ellis, I.; Schnitt, S.; Tan, P.; van de Vijver, M. WHO Classification of Tumours of the Breast, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2012; ISBN 978-92-832-2433-4. [Google Scholar]
- Vuong, D.; Simpson, P.T.; Green, B.; Cummings, M.C.; Lakhani, S.R. Molecular Classification of Breast Cancer. Virchows Arch. 2014, 465, 1–14. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Rougemont, J.; Charafe-Jauffret, E.; Cervera, N.; Tarpin, C.; Nguyen, C.; Xerri, L.; Houlgatte, R.; Jacquemier, J.; et al. Gene Expression Profiling Identifies Molecular Subtypes of Inflammatory Breast Cancer. Cancer Res. 2005, 65, 2170–2178. [Google Scholar] [CrossRef] [Green Version]
- Gajulapalli, V.N.R.; Malisetty, V.L.; Chitta, S.K.; Manavathi, B. Oestrogen Receptor Negativity in Breast Cancer: A Cause or Consequence? Biosci. Rep. 2016, 36, e00432. [Google Scholar] [CrossRef]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine Resistance in Breast Cancer—An Overview and Update. Mol. Cell. Endocrinol. 2015, 418, 220–234. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.L.B.; Czerniecki, B.J. Clinical Development of Immunotherapies for HER2 + Breast Cancer: A Review of HER2-Directed Monoclonal Antibodies and Beyond. NPJ Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Diana, A.; Carlino, F.; Franzese, E.; Oikonomidou, O.; Criscitiello, C.; De Vita, F.; Ciardiello, F.; Orditura, M. Early Triple Negative Breast Cancer: Conventional Treatment and Emerging Therapeutic Landscapes. Cancers 2020, 12, 819. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.V.; Jordan, V.C. The Estrogen Receptor: A Model for Molecular Medicine. Clin. Cancer Res. 2003, 9, 1980–1989. [Google Scholar]
- Maximov, P.; Lee, T.; Jordan, V. The Discovery and Development of Selective Estrogen Receptor Modulators (SERMs) for Clinical Practice. CCP 2013, 8, 135–155. [Google Scholar] [CrossRef] [Green Version]
- Jordan, V.C.; Brodie, A.M.H. Development and Evolution of Therapies Targeted to the Estrogen Receptor for the Treatment and Prevention of Breast Cancer. Steroids 2007, 72, 7–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnal, J.-F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef] [PubMed]
- Haldosén, L.-A.; Zhao, C.; Dahlman-Wright, K. Estrogen Receptor Beta in Breast Cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.-Å. Differential Expression of Estrogen Receptor α, Β1, and Β2 in Lobular and Ductal Breast Cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, P.; Nadig, N.; Nambiar, S. Non-Canonical Estrogen Signaling in Endocrine Resistance. Front. Endocrinol. 2019, 10, 708. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.N.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-Wide Analysis of Estrogen Receptor α DNA Binding and Tethering Mechanisms Identifies Runx1 as a Novel Tethering Factor in Receptor-Mediated Transcriptional Activation. MCB 2010, 30, 3943–3955. [Google Scholar] [CrossRef] [Green Version]
- Métivier, R.; Penot, G.; Hübner, M.R.; Reid, G.; Brand, H.; Koš, M.; Gannon, F. Estrogen Receptor-α Directs Ordered, Cyclical, and Combinatorial Recruitment of Cofactors on a Natural Target Promoter. Cell 2003, 115, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]
- Murphy, L.; Cherlet, T.; Lewis, A.; Banu, Y.; Watson, P. New Insights into Estrogen Receptor Function in Human Breast Cancer. Ann. Med. 2003, 35, 614–631. [Google Scholar] [CrossRef]
- Manavathi, B.; Dey, O.; Gajulapalli, V.N.R.; Bhatia, R.S.; Bugide, S.; Kumar, R. Derailed Estrogen Signaling and Breast Cancer: An Authentic Couple. Endocr. Rev. 2013, 34, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Kerdivel, G.; Flouriot, G.; Pakdel, F. Modulation of Estrogen Receptor Alpha Activity and Expression During Breast Cancer Progression. In Vitamins & Hormones; Elsevier: Cambridge, MA, USA, 2013; Volume 93, pp. 135–160. ISBN 978-0-12-416673-8. [Google Scholar]
- Barone, I.; Brusco, L.; Fuqua, S.A.W. Estrogen Receptor Mutations and Changes in Downstream Gene Expression and Signaling. Clin. Cancer Res. 2010, 16, 2702–2708. [Google Scholar] [CrossRef] [Green Version]
- Musgrove, E.A.; Sutherland, R.L. Biological Determinants of Endocrine Resistance in Breast Cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Reinert, T.; Chmielewska, I.; Ellis, M.J. Mechanisms of Aromatase Inhibitor Resistance. Nat. Rev. Cancer 2015, 15, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-X.; Borg, Å.; Wolf, D.M.; Oesterreich, S.; Fuqua, S.A.W. An Estrogen Receptor Mutant with Strong Hormone-Independent Activity from a Metastatic Breast Cancer. Cancer Res. 1997, 57, 1244–1249. [Google Scholar] [PubMed]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of Constitutively Active Estrogen Receptor-α Mutations in Pretreated Advanced Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [Green Version]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 Mutation in Circulating Tumor DNA Demonstrates Evolution during Therapy for Metastatic Breast Cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Hamadeh, I.S.; Patel, J.N.; Rusin, S.; Tan, A.R. Personalizing Aromatase Inhibitor Therapy in Patients with Breast Cancer. Cancer Treat. Rev. 2018, 70, 47–55. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 Mutations—A Mechanism for Acquired Endocrine Resistance in Breast Cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Spoerke, J.M.; Gendreau, S.; Walter, K.; Qiu, J.; Wilson, T.R.; Savage, H.; Aimi, J.; Derynck, M.K.; Chen, M.; Chan, I.T.; et al. Heterogeneity and Clinical Significance of ESR1 Mutations in ER-Positive Metastatic Breast Cancer Patients Receiving Fulvestrant. Nat. Commun. 2016, 7, 11579. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural Underpinnings of Oestrogen Receptor Mutations in Endocrine Therapy Resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen Receptor Alpha Somatic Mutations Y537S and D538G Confer Breast Cancer Endocrine Resistance by Stabilizing the Activating Function-2 Binding Conformation. eLife 2016, 5, e12792. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 Mutations in Hormone-Resistant Metastatic Breast Cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 Ligand-Binding Domain Mutations in Hormone-Resistant Breast Cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.-A.; Ribas, R.; Simigdala, N.; Schuster, E.; Pancholi, S.; Tenev, T.; Gellert, P.; Buluwela, L.; Harrod, A.; Thornhill, A.; et al. Discovery of Naturally Occurring ESR1 Mutations in Breast Cancer Cell Lines Modelling Endocrine Resistance. Nat. Commun. 2017, 8, 1865. [Google Scholar] [CrossRef] [PubMed]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G Mutation in Estrogen Receptor-α: A Novel Mechanism for Acquired Endocrine Resistance in Breast Cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-Therapy-Resistant ESR1 Variants Revealed by Genomic Characterization of Breast-Cancer-Derived Xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [Green Version]
- Pakdel, F.; Reese, J.C.; Katzenellenbogen, B.S. Identification of Charged Residues in an N-Terminal Portion of the Hormone-Binding Domain of the Human Estrogen Receptor Important in Transcriptional Activity of the Receptor. Mol. Endocrinol. 1993, 7, 1408–1417. [Google Scholar] [CrossRef]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Chigira, T.; Nagatoishi, S.; Tsumoto, K. Differential Binding of Prohibitin-2 to Estrogen Receptor α and to Drug-Resistant ERα Mutants. Biochem. Biophys. Res. Commun. 2015, 463, 726–731. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310. [Google Scholar] [CrossRef] [Green Version]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018, 33, 173–186.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuqua, S.A.W.; Gu, G.; Rechoum, Y. Estrogen Receptor (ER) α Mutations in Breast Cancer: Hidden in Plain Sight. Breast Cancer Res. Treat. 2014, 144, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, K.; Parrish, E.; Edmiston, S.N.; Tolbert, D.; Tse, C.-K.; Geradts, J.; Livasy, C.A.; Singh, H.; Newman, B.; Millikan, R.C. The Estrogen Receptor-α A908G (K303R) Mutation Occurs at a Low Frequency in Invasive Breast Tumors: Results from a Population-Based Study. Breast Cancer Res. 2005, 7, R871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, S.; Rasouli, M.; Nouri, M.; Kalbasi, S. Association of Estrogen Receptor-α A908G (K303R) Mutation with Breast Cancer Risk. Int. J. Clin. Exp. Med. 2013, 6, 39–49. [Google Scholar] [PubMed]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef] [Green Version]
- Michalides, R.; Griekspoor, A.; Balkenende, A.; Verwoerd, D.; Janssen, L.; Jalink, K.; Floore, A.; Velds, A.; van’t Veer, L.; Neefjes, J. Tamoxifen Resistance by a Conformational Arrest of the Estrogen Receptor α after PKA Activation in Breast Cancer. Cancer Cell 2004, 5, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Iacopetta, D.; Covington, K.R.; Cui, Y.; Tsimelzon, A.; Beyer, A.; Andò, S.; Fuqua, S.A.W. Phosphorylation of the Mutant K303R Estrogen Receptor α at Serine 305 Affects Aromatase Inhibitor Sensitivity. Oncogene 2010, 29, 2404–2414. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Cui, Y.; Barone, I.; Ando, S.; Mancini, M.A.; Berno, V.; Fuqua, S.A.W. Growth Factor-Induced Resistance to Tamoxifen Is Associated with a Mutation of Estrogen Receptor α and Its Phosphorylation at Serine 305. Breast Cancer Res. Treat. 2010, 119, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Johnston, S.R.; Kilburn, L.S.; Ellis, P.; Dodwell, D.; Cameron, D.; Hayward, L.; Im, Y.-H.; Braybrooke, J.P.; Brunt, A.M.; Cheung, K.-L.; et al. Fulvestrant plus Anastrozole or Placebo versus Exemestane Alone after Progression on Non-Steroidal Aromatase Inhibitors in Postmenopausal Patients with Hormone-Receptor-Positive Locally Advanced or Metastatic Breast Cancer (SoFEA): A Composite, Multicentre, Phase 3 Randomised Trial. Lancet Oncol. 2013, 14, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus Palbociclib versus Fulvestrant plus Placebo for Treatment of Hormone-Receptor-Positive, HER2-Negative Metastatic Breast Cancer That Progressed on Previous Endocrine Therapy (PALOMA-3): Final Analysis of the Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Fanning, S.W.; Greene, G.L. Next-Generation ERα Inhibitors for Endocrine-Resistant ER+ Breast Cancer. Endocrinology 2019, 160, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Bihani, T.; Patel, H.K.; Arlt, H.; Tao, N.; Jiang, H.; Brown, J.L.; Purandare, D.M.; Hattersley, G.; Garner, F. Elacestrant (RAD1901), a Selective Estrogen Receptor Degrader (SERD), has Antitumor Activity in Multiple ER + Breast Cancer Patient-Derived Xenograft Models. Clin. Cancer Res. 2017, 23, 4793–4804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardia, A.; Aftimos, P.; Bihani, T.; Anderson-Villaluz, A.T.; Jung, J.; Conlan, M.G.; Kaklamani, V.G. EMERALD: Phase III Trial of Elacestrant (RAD1901) vs Endocrine Therapy for Previously Treated ER + Advanced Breast Cancer. Future Oncol. 2019, 15, 3209–3218. [Google Scholar] [CrossRef] [PubMed]
- Herynk, M.H.; Fuqua, S.A.W. Estrogen Receptor Mutations in Human Disease. Endocr. Rev. 2004, 25, 869–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, T.; Heimdal, K.; Skrede, M.; Tveit, K.; Berg, K.; Børresen, A.-L. Oestrogen Receptor (ESR) Polymorphisms and Breast Cancer Susceptibility. Hum. Genet. 1994, 94, 665–670. [Google Scholar] [CrossRef]
- Roodi, N.; Bailey, L.R.; Kao, W.-Y.; Verrier, C.S.; Yee, C.J.; Dupont, W.D.; Parl, F.F. Estrogen Receptor Gene Analysis in Estrogen Receptor-Positive and Receptor-Negative Primary Breast Cancer. J. Nat. Cancer Inst. 1995, 87, 446–451. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Yuan, X.; Zhang, Z.; Zhang, P.; Chao, H.; Jiang, L.; Jiang, J. Association Between ESR1 PvuII, XbaI, and P325P Polymorphisms and Breast Cancer Susceptibility: A Meta-Analysis. Med. Sci. Monit. 2015, 21, 2986–2996. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Chen, D.; Hu, L.-P.; Zhou, L.-L.; Xu, H.-Y.; Bai, Y.-H.; Lin, X.-Y. Estrogen Receptor Alpha Gene Polymorphisms and Breast Cancer Risk: A Case-Control Study with Meta-Analysis Combined. Asian Pac. J. Cancer Prev. 2013, 14, 6743–6749. [Google Scholar] [CrossRef] [Green Version]
- Quan, L.; Hong, C.-C.; Zirpoli, G.; Roberts, M.R.; Khoury, T.; Sucheston-Campbell, L.E.; Bovbjerg, D.H.; Jandorf, L.; Pawlish, K.; Ciupak, G.; et al. Variants of Estrogen-Related Genes and Breast Cancer Risk in European and African American Women. Endocr. Relat. Cancer 2014, 21, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shu, X.; Shu, X.; Bolla, M.K.; Kweon, S.-S.; Cai, Q.; Michailidou, K.; Wang, Q.; Dennis, J.; Park, B.; et al. Re-Evaluating Genetic Variants Identified in Candidate Gene Studies of Breast Cancer Risk Using Data from Nearly 280,000 Women of Asian and European Ancestry. EBioMedicine 2019, 48, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, C.; Li, Y.; Zhao, Z.; Yang, S. Association between ERα Gene Pvu II Polymorphism and Breast Cancer Susceptibility: A Meta-Analysis. Medicine 2018, 97, e0317. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon Optimality, Bias and Usage in Translation and MRNA Decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Hia, F.; Yang, S.F.; Shichino, Y.; Yoshinaga, M.; Murakawa, Y.; Vandenbon, A.; Fukao, A.; Fujiwara, T.; Landthaler, M.; Natsume, T.; et al. Codon Bias Confers Stability to Human MRNAs. EMBO Rep. 2019, 20, e48220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Medina, S.G.; Kushawah, G.; DeVore, M.L.; Castellano, L.A.; Hand, J.M.; Wright, M.; Bazzini, A.A. Translation Affects MRNA Stability in a Codon-Dependent Manner in Human Cells. eLife 2019, 8, e45396. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A. The Yin and Yang of Codon Usage. Hum. Mol. Genet. 2016, 25, R77–R85. [Google Scholar] [CrossRef]
- Marín, M.; Fernández-Calero, T.; Ehrlich, R. Protein Folding and TRNA Biology. Biophys. Rev. 2017, 9, 573–588. [Google Scholar] [CrossRef]
- Dittmar, K.A.; Goodenbour, J.M.; Pan, T. Tissue Specific Differences in Human Transfer RNA Expression. PLoS Genet. 2005, 2, e221. [Google Scholar] [CrossRef]
- Gingold, H.; Tehler, D.; Christoffersen, N.R.; Nielsen, M.M.; Asmar, F.; Kooistra, S.M.; Christophersen, N.S.; Christensen, L.L.; Borre, M.; Sørensen, K.D.; et al. A Dual Program for Translation Regulation in Cellular Proliferation and Differentiation. Cell 2014, 158, 1281–1292. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.; Fidalgo, A.; Varanda, A.S.; Oliveira, C.; Santos, M.A.S. TRNA Deregulation and Its Consequences in Cancer. Trends Mol. Med. 2019, 25, 853–865. [Google Scholar] [CrossRef]
- Goodarzi, H.; Nguyen, H.C.B.; Zhang, S.; Dill, B.D.; Molina, H.; Tavazoie, S.F. Modulated Expression of Specific TRNAs Drives Gene Expression and Cancer Progression. Cell 2016, 165, 1416–1427. [Google Scholar] [CrossRef] [Green Version]
- Pavon-Eternod, M.; Gomes, S.; Geslain, R.; Dai, Q.; Rosner, M.R.; Pan, T. TRNA Over-Expression in Breast Cancer and Functional Consequences. Nucleic Acids Res. 2009, 37, 7268–7280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purvis, I.J.; Bettany, A.J.E.; Santiago, T.C.; Coggins, J.R.; Duncan, K.; Eason, R.; Brown, A.J.P. The Efficiency of Folding of Some Proteins is Increased by Controlled Rates of Translation In Vivo. J. Mol. Biol. 1987, 193, 413–417. [Google Scholar] [CrossRef]
- Thommen, M.; Holtkamp, W.; Rodnina, M.V. Co-Translational Protein Folding: Progress and Methods. Curr. Opin. Struct. Biol. 2017, 42, 83–89. [Google Scholar] [CrossRef]
- Truitt, M.L.; Ruggero, D. New Frontiers in Translational Control of the Cancer Genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penzo, M.; Galbiati, A.; Treré, D.; Montanaro, L. The Importance of Being (Slightly) Modified: The Role of RRNA Editing on Gene Expression Control and Its Connections with Cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 2016, 1866, 330–338. [Google Scholar] [CrossRef]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of MRNAs Genome-Wide. Mol. Cell 2017, 67, 71–83.e7. [Google Scholar] [CrossRef] [Green Version]
- Buhr, F.; Jha, S.; Thommen, M.; Mittelstaet, J.; Kutz, F.; Schwalbe, H.; Rodnina, M.V.; Komar, A.A. Synonymous Codons Direct Cotranslational Folding toward Different Protein Conformations. Mol. Cell 2016, 61, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-H.; Dang, Y.; Zhou, Z.; Wu, C.; Zhao, F.; Sachs, M.S.; Liu, Y. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-Translational Protein Folding. Mol. Cell 2015, 59, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Sauna, Z.E.; Kimchi-Sarfaty, C. Understanding the Contribution of Synonymous Mutations to Human Disease. Nat. Rev. Genet. 2011, 12, 683–691. [Google Scholar] [CrossRef]
- Rauscher, R.; Ignatova, Z. Timing during Translation Matters: Synonymous Mutations in Human Pathologies Influence Protein Folding and Function. Biochem. Soc. Trans. 2018, 46, 937–944. [Google Scholar] [CrossRef]
- Fu, J.; Dang, Y.; Counter, C.; Liu, Y. Codon Usage Regulates Human KRAS Expression at Both Transcriptional and Translational Levels. J. Biol. Chem. 2018, 293, 17929–17940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savas, S. Useful Genetic Variation Databases for Oncologists Investigating the Genetic Basis of Variable Treatment Response and Survival in Cancer. Acta Oncol. 2010, 49, 1217–1226. [Google Scholar] [CrossRef]
- Horjales, S.; Cota, G.; Señorale-Pose, M.; Rovira, C.; Román, E.; Artagaveytia, N.; Ehrlich, R.; Marín, M. Translational Machinery and Protein Folding: Evidence of Conformational Variants of the Estrogen Receptor Alpha. Arch. Biochem. Biophys. 2007, 467, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Calero, T.; Astrada, S.; Alberti, Á.; Horjales, S.; Arnal, J.F.; Rovira, C.; Bollati-Fogolín, M.; Flouriot, G.; Marin, M. The Transcriptional Activities and Cellular Localization of the Human Estrogen Receptor Alpha Are Affected by the Synonymous Ala87 Mutation. J. Steroid Biochem. Mol. Biol. 2014, 143, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.L.; Henry, N.L.; Kidwell, K.M.; Thomas, D.; Goddard, A.; Azzouz, F.; Speth, K.; Li, L.; Banerjee, M.; Thibert, J.N.; et al. ESR1 and PGR Polymorphisms are Associated with Estrogen and Progesterone Receptor Expression in Breast Tumors. Physiol. Genom. 2016, 48, 688–698. [Google Scholar] [CrossRef]
- AlFakeeh, A.; Brezden-Masley, C. Overcoming Endocrine Resistance in Hormone Receptor–Positive Breast Cancer. Curr. Oncol. 2018, 25, 18. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Munuganti, R.; Lallous, N.; Dalal, K.; Yoon, J.; Sharma, A.; Yamazaki, T.; Cherkasov, A.; Rennie, P. Benzothiophenone Derivatives Targeting Mutant Forms of Estrogen Receptor-α in Hormone-Resistant Breast Cancers. Int. J. Mol. Sci. 2018, 19, 579. [Google Scholar] [CrossRef] [Green Version]
- Furman, C.; Hao, M.-H.; Prajapati, S.; Reynolds, D.; Rimkunas, V.; Zheng, G.Z.; Zhu, P.; Korpal, M. Estrogen Receptor Covalent Antagonists: The Best is Yet to Come. Cancer Res. 2019, 79, 1740–1745. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Xiang, H.; Luo, G. Targeting Estrogen Receptor α for Degradation with PROTACs: A Promising Approach to Overcome Endocrine Resistance. Eur. J. Med. Chem. 2020, 206, 112689. [Google Scholar] [CrossRef]
- Revankar, C.M.; Bologa, C.G.; Pepermans, R.A.; Sharma, G.; Petrie, W.K.; Alcon, S.N.; Field, A.S.; Ramesh, C.; Parker, M.A.; Savchuk, N.P.; et al. A Selective Ligand for Estrogen Receptor Proteins Discriminates Rapid and Genomic Signaling. Cell Chem. Biol. 2019, 26, 1692–1702.e5. [Google Scholar] [CrossRef]
- Huang, W.; Peng, Y.; Kiselar, J.; Zhao, X.; Albaqami, A.; Mendez, D.; Chen, Y.; Chakravarthy, S.; Gupta, S.; Ralston, C.; et al. Multidomain Architecture of Estrogen Receptor Reveals Interfacial Cross-Talk between Its DNA-Binding and Ligand-Binding Domains. Nat. Commun. 2018, 9, 3520. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Pareja, F.G. New Advances in Molecular Breast Cancer Pathology. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; De Angelis, C.; Brown, M.; Schiff, R. The Evolving Role of the Estrogen Receptor Mutations in Endocrine Therapy-Resistant Breast Cancer. Curr. Oncol. Rep. 2017, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating Tumour DNA Analysis to Direct Therapy in Advanced Breast Cancer (PlasmaMATCH): A Multicentre, Multicohort, Phase 2a, Platform Trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- Gilles, A.; Frechin, L.; Natchiar, K.; Biondani, G.; Loeffelholz, O.; von Holvec, S.; Malaval, J.-L.; Winum, J.-Y.; Klaholz, B.P.; Peyron, J.-F. Targeting the Human 80S Ribosome in Cancer: From Structure to Function and Drug Design for Innovative Adjuvant Therapeutic Strategies. Cells 2020, 9, 629. [Google Scholar] [CrossRef] [Green Version]
- Reis, S.D.; Pang, Y.; Vishnu, N.; Voisset, C.; Galons, H.; Blondel, M.; Sanyal, S. Mode of Action of the Antiprion Drugs 6AP and GA on Ribosome Assisted Protein Folding. Biochimie 2011, 93, 1047–1054. [Google Scholar] [CrossRef]
- Hunt, R.C.; Simhadri, V.L.; Iandoli, M.; Sauna, Z.E.; Kimchi-Sarfaty, C. Exposing Synonymous Mutations. Trends Genet. 2014, 30, 308–321. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Breast Cancer Type | Proportion | Biological Profile | Therapy of Choice |
|---|---|---|---|
| Luminal | Endocrine therapy | ||
| A | 60% | ERα+ PR+/− HER2− | |
| B | 10% | ERα+ PR+/− HER2+ | |
| HER2-enriched [6] | 20% | ERα+/− PR+/− HER2+ | Anti-HER2 therapy |
| Triple negative [7] | ERα− PR− HER2− | Chemotherapy | |
| Basal-like | 7% | + basal markers | |
| Non-basal-like | 3% | − basal markers |
| ERα Substitution | Y537S/N/C | D538G | L536R/Q/P/H | E380Q | S463P | K303R |
|---|---|---|---|---|---|---|
| Structural data obtained | Stabilization of the agonist conformation | Stabilization of the agonist conformation | ||||
| Ligand independent activity | ↑ target genes transcription ↑ coactivator recruitment ↑ proliferation | ↑ target genes transcription ↑ coactivator recruitment ↑ proliferation ↑ migratory properties | ↑ target genes transcription ↑ coactivator recruitment | ↑ target genes transcription ↑ proliferation | ↑ target genes transcription ↑ proliferation | ↑ ERα stability ↑ coactivator recruitment ↑ interactions with growth factor receptors |
| Estrogen and antiestrogen responses | AI resistance ↓ SERD sensitivity SERM resistance | AI resistance SERD sensitivity SERM resistance | AI resistance SERD sensitivity | AI resistance SERD sensitivity SERM sensitivity ↑ E2 sensitivity | SERD sensitivity SERM sensitivity | AI resistance SERD sensitivity SERM = agonist activity ↑ E2 sensitivity |
| References | [26,30,31,32,33,34,35,36,38,40,42,43] | [26,30,31,32,33,34,35,37,40,42,43] | [32,34,40] | [30,31,32,38,39,40,41] | [32,35,40] | [44,45,46,47,48,49,50] |
| RFLP | rsID | Domain | Codon | Major Allele | Minor Allele | Amino Acid |
|---|---|---|---|---|---|---|
| PvuII | rs2234693 | 397 (Intron 1) | T | C | ||
| XbaI | rs9340799 | 351 (Intron 1) | A | G | ||
| rs2077647 | A/B | 10 (Exon 1) | TCT | TCC | Ser | |
| BstUI | rs746432 | A/B | 87 (Exon 1) | GCG | GCC | Ala |
| C | 243 (Exon 3) | CGC | CGT | Arg | ||
| rs1801132 | E | 325 (Exon 4) | CCG | CCC | Pro | |
| rs2228480 | 594 (Exon 8) | ACG | ACA | Thr |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clusan, L.; Le Goff, P.; Flouriot, G.; Pakdel, F. A Closer Look at Estrogen Receptor Mutations in Breast Cancer and Their Implications for Estrogen and Antiestrogen Responses. Int. J. Mol. Sci. 2021, 22, 756. https://doi.org/10.3390/ijms22020756
Clusan L, Le Goff P, Flouriot G, Pakdel F. A Closer Look at Estrogen Receptor Mutations in Breast Cancer and Their Implications for Estrogen and Antiestrogen Responses. International Journal of Molecular Sciences. 2021; 22(2):756. https://doi.org/10.3390/ijms22020756
Chicago/Turabian StyleClusan, Léa, Pascale Le Goff, Gilles Flouriot, and Farzad Pakdel. 2021. "A Closer Look at Estrogen Receptor Mutations in Breast Cancer and Their Implications for Estrogen and Antiestrogen Responses" International Journal of Molecular Sciences 22, no. 2: 756. https://doi.org/10.3390/ijms22020756
APA StyleClusan, L., Le Goff, P., Flouriot, G., & Pakdel, F. (2021). A Closer Look at Estrogen Receptor Mutations in Breast Cancer and Their Implications for Estrogen and Antiestrogen Responses. International Journal of Molecular Sciences, 22(2), 756. https://doi.org/10.3390/ijms22020756