
The SARS-Coronavirus Infection Cycle: A Survey of Viral Membrane Proteins, Their Functional Interactions and Pathogenesis


Abstract
:1. Introduction
2. Viral Proteins and Their Roles in the Infection Cycle
3. The CoV Replication Cycle (from Entry to Exit)
4. Coronavirus Genome Structure, Replication and Expression
5. CoV Replication Organelle Formation: A Dance of Membrane Rearrangements
6. Structures of nsp3 and nsp4
7. The Replicon Conundrum and a Putative Nucleopore
8. Viral Proteins–Structures, Expression and Assembly
8.1. The Nucleocapsid (N) Protein: Genome Packaging
8.2. The Envelope (E) Protein: Viral Assembly
8.3. The Spike (S) Protein: The Primary Receptor and Membrane Fusion Mediator
8.4. The Membrane Matrix (M) Protein, the Virion Scaffold
9. Viroporin Activities: The E, 3a and 4a Proteins, and the Ability of Mutated M to Substitute for E
10. Post Translational Modifications (PTMs) to Coronaviral Structural Proteins
10.1. Glycosylation
10.2. Palmitoylation
10.3. Ubiquitination
10.4. A Focus on S Protein PTMs
10.5. A Focus on M Protein PTMs
10.6. A Focus on E Protein PTMs
11. Viral Responses to and Interference with Normal Cellular Function
11.1. Interference with Host Immunological Responses by Interferon (IFN) Antagonism
11.2. The M Protein
11.3. The N Protein
12. Nonstructural Protein Interference with IFN Gene Expression
12.1. nsp1
12.2. nsp3
13. Accessory Protein Interference with IFN Expression
13.1. ORF6
13.2. ORF3b
14. Complement Activation by CoV Structural Proteins
15. Induction of Endoplasmic Reticulum (ER) Stress and the Unfolded Protein Response (UPR)
16. Coronavirus-Induced Host Cell Cycle Arrest
17. Coronavirus-Induced Autophagy and Abortive Apoptosis
18. Structural Proteins as Protective Antigens in Survivors, and Vaccine Development
18.1. S Protein as a Protective Antigen
18.2. N-Protein as a Protective Antigen
18.3. M-Protein as a Protective Antigen
19. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Viruses and associated abb’ns | |
| α β γ δ | Coronavirus (CoV) types |
| ACoV | avian CoV |
| BCoV | bovine respiratory CoV |
| CoV | coronavirus |
| CCoV | canine CoV |
| CoV HKU1 | a species of CoV that infects humans |
| HCoV-OC43 | another species of CoV that infects humans |
| Covid-19 | the disease caused by CoV-2 |
| FCoV | feline CoV |
| FIPV | feline infectious peritonitis virus |
| HAV, HBV, HCV | hepatitis A, B or C coronavirus |
| HCoV | human CoV |
| H1N1 | an influenza A viral subtype; swine flu |
| H5N1 | an influenza A viral subtype; asian avian flu |
| IBV or AIBV | avian infectious bronchitis coronavirus (avian CoV) |
| MERS | middle east respiratory syndrome |
| MHV | murine (mouse) hepatitis coronavirus |
| NDV | Newcastle disease virus |
| PEDV or PDCoV | porcine epidemic diarrhea coronavirus |
| PHEV | porcine hemagglutinating encephalomyelitis virus |
| RSV | respiratory syncytial virus |
| RV | Rhino virus |
| SARS, SARS-CoV or SARS-CoV-1 | severe acute respiratory syndrome coronavirus-1 |
| SARS-CoV-2 or CoV-2 | severe acute respiratory syndrome coronavirus-2 |
| SFV | semliki forest virus |
| TGEV | transmissible gastroenteritis coronavirus |
| VSV | vesicular stomatitis virus |
| VIRAL PROTEINS and RNAs | |
| E | envelope protein |
| H | helicase |
| HE | hemagglutinin esterase |
| M | membrane matrix protein |
| N | nucleocapsid protein |
| nsp or NSP | (viral) non-structural protein (e.g., nsp3a 3b, 4a, 5, 6, 7a, etc.) |
| P | protease |
| PLP | PLpro, papain-like protease |
| Pp | polyprotein (i.e., pp1a (Orf1a) is spliced to give nsp1–11 pp1ab (Orf1b) is spliced to give nsp12 - 16. |
| RdRp | RNA-dependent RNA polymerase |
| S | spike protein (the S1 domain binds ACE2, receptor for several CoVs the S2 domain induces membrane fusion) |
| gRNA | guide RNA |
| gsRNA | single guide RNA |
| ssRNA | single stranded RNA |
| dsRNA | double stranded RNA |
| TRS | transcription regulating sequence |
| HOST ENZYMES AND OTHER PROTEINS | |
| ACE2 | angiotensin-converting enzyme-2, receptor for several CoVs |
| ATG5 | an autophagy (ATG) protein - promotes fusion of phagocytic vesicles with lysosomes |
| Bcl-2 | an apoptosis regulator it contains tandemly repeated PDZ domains that bind the cytoplasmic, C-terminal domains of a variety of transmembrane proteins. |
| Beclin1 | a regulator of autophagy, ATG. |
| CCAAT | enhancer-binding proteins transcription factors |
| CCL1, 2, 3, 4 | CxCL2, proinflammatory chemokines |
| CDRH1, 2, 3 | the first, second, third complementarity-determining variable region of antibody heavy chains |
| Cdk | PKR-like endoplasmic reticulum (ER) kinase |
| C/EBP | CCAAT-enhancer binding protein |
| CHOP | C/EBP homologous protein (transcription factor) |
| Cyclin | protein involved in the cell cycle (e.g., cyclin D3, cyclin E) |
| DC-SIGN | dendritic cell-specific intercellular adhesion molecule grabbing nonintegrin also, CD209 |
| DUB | deubiquitinating enzyme |
| DPP4 | dipeptidyl peptidase 4 (adenosine deaminase complexing protein 2) |
| DUSP | dual specificity phosphatase |
| eIF2 | eukaryotic initiation factor 2 |
| GRP | glucose regulated protein (e.g., GPR78, GRP94) |
| HSP | heat shock protein |
| IFN | interferon |
| IL | interleukin (i.e., IL-1β) |
| IPS-1 | INFβ promoter stimulator |
| IRE1 | inositol-requiring enzyme (protein kinase a sensor) |
| IRF3 | interferon regulatory factor 3 |
| ISG | INF stimulated gene |
| JNK | cJUN amino terminal kinase |
| LC3 | cytosolic ubiquitin-like protein (regulates macroautophagy (autophagy, ATG)) |
| MAPK | MAP kinase, Mitogen-activated protein kinase |
| MBL | mannose binding lectin |
| MHCI and II | major histocompatibility complex, classes I and II |
| NF-κB or NFκB | a protein complex that controls transcription of DNA, cytokine production and cell survival. |
| NLRP3 | NLR family pyrin domain-containing protein-3 |
| PALS1 | protein associated with Lin seven 1, a Maguk PDZ guanylate kinase |
| PARP | poly ADP-ribose polymerase |
| PDI | protein disulfide isomerase |
| PERK | protein kinase RNA (PKR)-like ER kinase |
| p53 or TP53 | protein that regulates the cell cycle and hence functions as a tumor suppressor. |
| PIKfyve | phosphoinositol 3-kinase (makes PI-3,5-bisphosphate - a regulator of endosome sorting) |
| PKR | protein kinase R |
| Proteases | |
| CTP | cathepsin protease |
| DPP4 | dipeptidyl peptidase-4 |
| Furin | a host protease |
| HAT | human airway trypsin-like protease |
| MASP1 | MASP2, mannose-binding lectin (MBL)-associated serine proteases 1,2 |
| PLP or PLpro | papain-like protease |
| TMPRSS-2 or -4 | transmembrane serine protease-2 or -4 |
| PRR | pattern recognition receptor |
| Rb or pRb | retinoblastoma protein |
| STING | a key regulator of antiviral interferon (IFN) |
| Syntenin-1 | contains tandemly repeated PDZ domains that bind the cytoplasmic C-terminal domains of a variety of transmembrane proteins. |
| TBK1 | Tank-binding kinase-1 |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TPC | two pore calcium (cation) channel |
| TPC1 and 2 are in endosomes | |
| TRAF3 | TNF receptor-associated protein 3 |
| XBP | X-box binding protein - a transcription factor involved in endoplasmic reticulum (ER) stress |
| (UPR) | regulates the unfolded protein response |
| INTRACELLULAR MEMBRANE STRUCTURES AND PROCESSES | |
| CM | convoluted intracellular membrane |
| CTL | cytotoxic T lymphocyte involved in the T-cell immune response |
| DMS | double membrane spherule |
| DMV | double membrane vesicle |
| EM | electron microscopy |
| ER | endoplasmic reticulum |
| ERB | ER body |
| ERGIC | ER-golgi intermediate compartment (a mobile complex that delivers cargo from the ER to the golgi) |
| GV | giant vesicle; giant vesiculation |
| IRF3 | interferon (INF) regulatory factor 3 |
| ISR | interferon-stimulated response; |
| ISRE | ISR element |
| JNK | the C-Jun N-terminal kinase signaling pathway |
| LCVC | lysosomal virion-containing cisternae (golgi-derived) |
| MLB | multilamellar (maze-like) body |
| NMR | nuclear magnetic resonance |
| Omegasome | a cell membrane-enclosed compartment enriched for phosphatidylinositol-3-P |
| PPI | protein-protein interaction |
| PTM | post translational modification |
| RO | (viral) replication organelle |
| RTC | replication-transcription complex |
| RVN | reticulovesicular network |
| TRS | transcription regulating sequence (at the beginning of a gene) |
| UPR | unfolded protein response |
| VLP | vesicle-like particle |
| VP, | vesicle packet or vesicle particle |
| zipped membranes | ER membranes that fold to form spherules |
| TERMS | |
| aa | amino acyl residue |
| AP-MS | affinity purification-mass spectrometry |
| ARDS | acute respiratory distress syndrome |
| BrU | bromouridine (U uridine) |
| CDRH | complementarity-determining region of heavy chain antibody (variable region) |
| Click chemistry | simple chemistry involving the joining of two molecules |
| CPZ | chlorpromazine (a clathrin inhibitor) |
| Cryo EM | cryogenic electron microscopy |
| CTD | C-terminal domain |
| dpi | days post infection |
| DPUP | domain preceding Ubi2 and PL2pro |
| ESR | electron spin resonance |
| Fab | antigen-binding fragment of an antibody |
| FP | fusion peptide |
| GI (tract) | gastrointestinal (tract) |
| HD | hydrophobic domain (in a protein) |
| usually forms TMSs | |
| HMA | hexamethylene amiloride |
| hpi | hours post infection |
| HR1 HR2 | heptad repeat 1, 2… |
| IC | ion channel across a membrane |
| ISRE | interferon-stimulated response element |
| Mac | macrodomain |
| MβCD | methyl-β-cyclodextrins |
| Mitophagy | autophagy of mitochondria |
| NTD | N-terminal domain |
| ORF | open reading frame (a protein encoding gene) |
| PAMP | pathogen-associated molecular pattern |
| PBM | PDZ binding motif |
| PDZ | a protein domain in proteins that recognizes motifs in other proteins and is therefore a protein-protein interaction (PPI) domain |
| perinuclear | the cytoplasm immediately surrounding the nucleus |
| PPI | protein-protein interaction |
| PTM | post translational modification |
| RBD | receptor binding domain |
| RNP | ribonuclear protein |
| ROS | reactive oxygen species |
| RTC | replication-transcription complex |
| SUD | SARS unique domain |
| TM | transmembrane |
| TMD | transmembrane domain |
| TMS | transmembrane (α-helical) segment |
| TRS | transcription-regulating sequence |
| Ubl1 | ubiquitin-like (domain) 1 |
| VLP | virus-like particle |
References
- Gabutti, G.; d’Anchera, E.; Sandri, F.; Savio, M.; Stefanati, A. Coronavirus: Update related to the current outbreak of covid-19. Infect. Dis. Ther. 2020, 9, 1–13. [Google Scholar] [CrossRef]
- Wang, N.; Shang, J.; Jiang, S.; Du, L. Subunit vaccines against emerging pathogenic human coronaviruses. Front. Microbiol. 2020, 11, 298. [Google Scholar] [CrossRef]
- Park, M.; Cook, A.R.; Lim, J.T.; Sun, Y.; Dickens, B.L. A systematic review of covid-19 epidemiology based on current evidence. J. Clin. Med. 2020, 9, 967. [Google Scholar] [CrossRef] [Green Version]
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. Covid-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef]
- Amanat, F.; Krammer, F. Sars-cov-2 vaccines: Status report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef]
- Rabi, F.A.; Al Zoubi, M.S.; Kasasbeh, G.A.; Salameh, D.M.; Al-Nasser, A.D. Sars-cov-2 and coronavirus disease 2019: What we know so far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef]
- Kakodkar, P.; Kaka, N.; Baig, M.N. A comprehensive literature review on the clinical presentation, and management of the pandemic coronavirus disease 2019 (covid-19). Cureus 2020, 12, e7560. [Google Scholar] [CrossRef] [Green Version]
- Rokni, M.; Ghasemi, V.; Tavakoli, Z. Immune responses and pathogenesis of sars-cov-2 during an outbreak in iran: Comparison with sars and mers. Rev. Med. Virol. 2020, 30, e2107. [Google Scholar] [CrossRef] [Green Version]
- Emami, A.; Javanmardi, F.; Pirbonyeh, N.; Akbari, A. Prevalence of underlying diseases in hospitalized patients with covid-19: A systematic review and meta-analysis. Arch. Acad. Emerg. Med. 2020, 8, e35. [Google Scholar]
- Shahid, Z.; Kalayanamitra, R.; McClafferty, B.; Kepko, D.; Ramgobin, D.; Patel, R.; Aggarwal, C.S.; Vunnam, R.; Sahu, N.; Bhatt, D.; et al. Covid-19 and older adults: What we know. J. Am. Geriatr. Soc. 2020, 68, 926–929. [Google Scholar] [CrossRef] [Green Version]
- Smyk, W.; Janik, M.K.; Portincasa, P.; Milkiewicz, P.; Lammert, F.; Krawczyk, M. Covid-19: Focus on the lungs but do not forget the gastrointestinal tract. Eur. J. Clin. Investig. 2020, 50, e13276. [Google Scholar] [CrossRef]
- Tariku, M.; Hajure, M. Available evidence and ongoing hypothesis on corona virus (covid-19) and psychosis: Is corona virus and psychosis related? A narrative review. Psychol. Res. Behav. Manag. 2020, 13, 701–704. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-converting enzyme 2: Sars-cov-2 receptor and regulator of the renin-angiotensin system: Celebrating the 20th anniversary of the discovery of ace2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Md Insiat Islam, R. Current drugs with potential for treatment of covid-19: A literature review. J. Pharm. Pharm. Sci. 2020, 23, 58–64. [Google Scholar] [CrossRef]
- Luo, H.; Tang, Q.L.; Shang, Y.X.; Liang, S.B.; Yang, M.; Robinson, N.; Liu, J.P. Can chinese medicine be used for prevention of corona virus disease 2019 (covid-19)? A review of historical classics, research evidence and current prevention programs. Chin. J. Integr. Med. 2020, 26, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.A. Compounds with therapeutic potential against novel respiratory 2019 coronavirus. Antimicrob. Agents Chemother. 2020, 64, e00399-20. [Google Scholar] [CrossRef] [Green Version]
- Amirian, E.S.; Levy, J.K. Current knowledge about the antivirals remdesivir (gs-5734) and gs-441524 as therapeutic options for coronaviruses. One Health 2020, 9, 100128. [Google Scholar] [CrossRef]
- Gbinigie, K.; Frie, K. Should chloroquine and hydroxychloroquine be used to treat covid-19? A rapid review. BJGP Open 2020, 4, bjgpopen20X101069. [Google Scholar] [CrossRef]
- Li, R.; Yin, K.; Zhang, K.; Wang, Y.Y.; Wu, Q.P.; Tang, S.B.; Cheng, J.D. Application prospects of virtual autopsy in forensic pathological investigations on covid-19. Fa Yi Xue Za Zhi 2020, 36, 149–156. [Google Scholar]
- Barlow, A.; Landolf, K.M.; Barlow, B.; Yeung, S.Y.A.; Heavner, J.J.; Claassen, C.W.; Heavner, M.S. Review of emerging pharmacotherapy for the treatment of coronavirus disease 2019. Pharmacotherapy 2020, 40, 416–437. [Google Scholar] [CrossRef] [Green Version]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of covid-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2020. [Google Scholar] [CrossRef]
- Prajapat, M.; Sarma, P.; Shekhar, N.; Avti, P.; Sinha, S.; Kaur, H.; Kumar, S.; Bhattacharyya, A.; Kumar, H.; Bansal, S.; et al. Drug targets for corona virus: A systematic review. Indian J. Pharmacol. 2020, 52, 56–65. [Google Scholar]
- Deng, L.; Li, C.; Zeng, Q.; Liu, X.; Li, X.; Zhang, H.; Hong, Z.; Xia, J. Arbidol combined with lpv/r versus lpv/r alone against corona virus disease 2019: A retrospective cohort study. J. Infect. 2020, 81, e1–e5. [Google Scholar] [CrossRef]
- Li, S.; Yuan, L.; Dai, G.; Chen, R.A.; Liu, D.X.; Fung, T.S. Regulation of the er stress response by the ion channel activity of the infectious bronchitis coronavirus envelope protein modulates virion release, apoptosis, viral fitness, and pathogenesis. Front. Microbiol. 2019, 10, 3022. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Hofmann, H.; Pohlmann, S. Cellular entry of the sars coronavirus. Trends Microbiol. 2004, 12, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Perrotta, F.; Matera, M.G.; Cazzola, M.; Bianco, A. Severe respiratory sars-cov2 infection: Does ace2 receptor matter? Respir. Med. 2020, 168, 105996. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ace2 protein, the functional receptor for sars coronavirus. A first step in understanding sars pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Barlan, A.; Zhao, J.; Sarkar, M.K.; Li, K.; McCray, P.B., Jr.; Perlman, S.; Gallagher, T. Receptor variation and susceptibility to middle east respiratory syndrome coronavirus infection. J. Virol. 2014, 88, 4953–4961. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. Sars-cov-2 cell entry depends on ace2 and tmprss2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of sars-cov-2 on virus entry and its immune cross-reactivity with sars-cov. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic cleavage of the sars-cov-2 spike protein and the role of the novel s1/s2 site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. Sars coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Lakadamyali, M.; Rust, M.J.; Zhuang, X. Endocytosis of influenza viruses. Microbes Infect. 2004, 6, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Barrow, E.; Nicola, A.V.; Liu, J. Multiscale perspectives of virus entry via endocytosis. Virol. J. 2013, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Anderson, H.A.; Chen, Y.; Norkin, L.C. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell 1996, 7, 1825–1834. [Google Scholar] [CrossRef] [Green Version]
- Devadas, D.; Koithan, T.; Diestel, R.; Prank, U.; Sodeik, B.; Dohner, K. Herpes simplex virus internalization into epithelial cells requires na+/h+ exchangers and p21-activated kinases but neither clathrin- nor caveolin-mediated endocytosis. J. Virol. 2014, 88, 13378–13395. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Shen, H.M. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in covid-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef]
- Milewska, A.; Nowak, P.; Owczarek, K.; Szczepanski, A.; Zarebski, M.; Hoang, A.; Berniak, K.; Wojarski, J.; Zeglen, S.; Baster, Z.; et al. Entry of human coronavirus nl63 into the cell. J. Virol. 2018, 92, e01933-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human coronavirus-229e, -oc43, -nl63, and -hku1. Ref. Modul. Life Sci. 2020, 77, 4435–4438. [Google Scholar]
- Liu, H.; Liu, Y.; Liu, S.; Pang, D.W.; Xiao, G. Clathrin-mediated endocytosis in living host cells visualized through quantum dot labeling of infectious hematopoietic necrosis virus. J. Virol. 2011, 85, 6252–6262. [Google Scholar] [CrossRef] [Green Version]
- Cong, Y.; Hart, B.J.; Gross, R.; Zhou, H.; Frieman, M.; Bollinger, L.; Wada, J.; Hensley, L.E.; Jahrling, P.B.; Dyall, J.; et al. Mers-cov pathogenesis and antiviral efficacy of licensed drugs in human monocyte-derived antigen-presenting cells. PLoS ONE 2018, 13, e0194868. [Google Scholar] [CrossRef]
- Currinn, H.; Guscott, B.; Balklava, Z.; Rothnie, A.; Wassmer, T. App controls the formation of pi(3,5)p(2) vesicles through its binding of the pikfyve complex. Cell Mol. Life Sci. 2016, 73, 393–408. [Google Scholar] [CrossRef] [Green Version]
- McCartney, A.J.; Zhang, Y.; Weisman, L.S. Phosphatidylinositol 3,5-bisphosphate: Low abundance, high significance. Bioessays 2014, 36, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Pitt, S.J.; Reilly-O’Donnell, B.; Sitsapesan, R. Exploring the biophysical evidence that mammalian two-pore channels are naadp-activated calcium-permeable channels. J. Physiol. 2016, 594, 4171–4179. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.K.; Schludi, V.; Chen, C.C.; Butz, E.; Nguyen, O.N.P.; Muller, M.; Kruger, J.; Kammerbauer, C.; Ben-Johny, M.; Vollmar, A.M.; et al. Tpc2 polymorphisms associated with a hair pigmentation phenotype in humans result in gain of channel function by independent mechanisms. Proc. Natl. Acad. Sci. USA 2017, 114, E8595–E8602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.H.; Lu, Y.Y.; Yue, J. Two pore channel 2 differentially modulates neural differentiation of mouse embryonic stem cells. PLoS ONE 2013, 8, e66077. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.X.; Ma, J.; Parrington, J.; Galione, A.; Evans, A.M. Tpcs: Endolysosomal channels for ca2+ mobilization from acidic organelles triggered by naadp. FEBS Lett. 2010, 584, 1966–1974. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, B.S.; Eden, E.R.; Hockey, L.N.; Yates, E.; Futter, C.E.; Patel, S. An endosomal naadp-sensitive two-pore ca(2+) channel regulates er-endosome membrane contact sites to control growth factor signaling. Cell Rep. 2017, 18, 1636–1645. [Google Scholar] [CrossRef] [Green Version]
- Grimm, C.; Chen, C.C.; Wahl-Schott, C.; Biel, M. Two-pore channels: Catalyzers of endolysosomal transport and function. Front. Pharmacol. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Ebola virus. Two-pore channels control ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar]
- Gunaratne, G.S.; Yang, Y.; Li, F.; Walseth, T.F.; Marchant, J.S. Naadp-dependent ca(2+) signaling regulates middle east respiratory syndrome-coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium 2018, 75, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Pager, C.T.; Dutch, R.E. Cathepsin l is involved in proteolytic processing of the hendra virus fusion protein. J. Virol. 2005, 79, 12714–12720. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin l prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [Green Version]
- Te Velthuis, A.J.; van den Worm, S.H.; Snijder, E.J. The sars-coronavirus nsp7+nsp8 complex is a unique multimeric rna polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. 2012, 40, 1737–1747. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [Green Version]
- Yount, B.; Roberts, R.S.; Lindesmith, L.; Baric, R.S. Rewiring the severe acute respiratory syndrome coronavirus (sars-cov) transcription circuit: Engineering a recombination-resistant genome. Proc. Natl. Acad. Sci. USA 2006, 103, 12546–12551. [Google Scholar] [CrossRef] [Green Version]
- Hagemeijer, M.C.; Vonk, A.M.; Monastyrska, I.; Rottier, P.J.; de Haan, C.A. Visualizing coronavirus rna synthesis in time by using click chemistry. J. Virol. 2012, 86, 5808–5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A contemporary view of coronavirus transcription. J. Virol. 2007, 81, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoops, K.; Swett-Tapia, C.; van den Worm, S.H.; Velthuis, A.J.T.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J.; Kikkert, M. Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus rna synthesis and virus-induced remodeling of endoplasmic reticulum membranes. J. Virol. 2010, 84, 833–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, R.L.; Denison, M.R. Replication of murine hepatitis virus is regulated by papain-like proteinase 1 processing of nonstructural proteins 1, 2, and 3. J. Virol. 2006, 80, 11610–11620. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, T.; Takemoto, C.; Kim, Y.T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. Sars-cov 3cl protease cleaves its c-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef] [Green Version]
- Stobart, C.C.; Sexton, N.R.; Munjal, H.; Lu, X.; Molland, K.L.; Tomar, S.; Mesecar, A.D.; Denison, M.R. Chimeric exchange of coronavirus nsp5 proteases (3clpro) identifies common and divergent regulatory determinants of protease activity. J. Virol. 2013, 87, 12611–12618. [Google Scholar] [CrossRef] [Green Version]
- Plant, E.P.; Perez-Alvarado, G.C.; Jacobs, J.L.; Mukhopadhyay, B.; Hennig, M.; Dinman, J.D. A three-stemmed mrna pseudoknot in the sars coronavirus frameshift signal. PLoS Biol. 2005, 3, e172. [Google Scholar] [CrossRef] [Green Version]
- Irigoyen, N.; Firth, A.E.; Jones, J.D.; Chung, B.Y.; Siddell, S.G.; Brierley, I. High-resolution analysis of coronavirus gene expression by rna sequencing and ribosome profiling. PLoS Pathog. 2016, 12, e1005473. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, K.; Ramirez, S.I.; Lokugamage, K.G.; Makino, S. Coronavirus nonstructural protein 1: Common and distinct functions in the regulation of host and viral gene expression. Virus Res. 2015, 202, 89–100. [Google Scholar] [CrossRef]
- Hackbart, M.; Deng, X.; Baker, S.C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. USA 2020, 117, 8094–8103. [Google Scholar] [CrossRef] [Green Version]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. Covid-2019: The role of the nsp2 and nsp3 in its pathogenesis. J. Med. Virol. 2020, 92, 584–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menachery, V.D.; Debbink, K.; Baric, R.S. Coronavirus non-structural protein 16: Evasion, attenuation, and possible treatments. Virus Res. 2014, 194, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the sars-cov nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, R.; Fielding, B.C. The role of severe acute respiratory syndrome (sars)-coronavirus accessory proteins in virus pathogenesis. Viruses 2012, 4, 2902–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Li, G.; Vasilakis, N.; Zhang, Y.; Shi, Z.; Zhong, Y.; Wang, L.F.; Zhang, S. Differential stepwise evolution of sars coronavirus functional proteins in different host species. BMC Evol. Biol. 2009, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Egloff, M.P.; Ferron, F.; Campanacci, V.; Longhi, S.; Rancurel, C.; Dutartre, H.; Snijder, E.J.; Gorbalenya, A.E.; Cambillau, C.; Canard, B. The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a single-stranded rna-binding subunit unique in the rna virus world. Proc. Natl. Acad. Sci. USA 2004, 101, 3792–3796. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the sars coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef] [Green Version]
- Te Velthuis, A.J.; Arnold, J.J.; Cameron, C.E.; van den Worm, S.H.; Snijder, E.J. The rna polymerase activity of sars-coronavirus nsp12 is primer dependent. Nucleic Acids Res. 2010, 38, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Kruger, J.; Sramala, I.; Hsu, H.J.; Henklein, P.; Chen, Y.M.; Fischer, W.B. Orf8a of sars-cov forms an ion channel: Experiments and molecular dynamics simulations. Biochim. Biophys. Acta 2011, 1808, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, K.A.; Thiel, V.; Dobbe, J.C.; van der Meer, Y.; Snijder, E.J.; Ziebuhr, J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 2004, 78, 5619–5632. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. Sars-coronavirus open reading frame-8b triggers intracellular stress pathways and activates nlrp3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. Sars-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the mavs/traf3/traf6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Guo, D. Molecular mechanisms of coronavirus rna capping and methylation. Virol. Sin. 2016, 31, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, M.M.; Neuman, B.W.; Buchmeier, M.J. Untangling membrane rearrangement in the nidovirales. DNA Cell Biol. 2014, 33, 122–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P.D.; Strating, J.R.; van Kuppeveld, F.J. Building viral replication organelles: Close encounters of the membrane types. PLoS Pathog. 2016, 12, e1005912. [Google Scholar] [CrossRef] [PubMed]
- van der Hoeven, B.D.; Oudshoorn, D.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Barcena, M. Biogenesis and architecture of arterivirus replication organelles. Virus Res. 2016, 220, 70–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemeijer, M.C.; Monastyrska, I.; Griffith, J.; van der Sluijs, P.; Voortman, J.; van Bergen en Henegouwen, P.M.; Vonk, A.M.; Rottier, P.J.; Reggiori, F.; de Haan, C.A. Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 2014, 458–459, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Oudshoorn, D.; Rijs, K.; Limpens, R.; Groen, K.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Barcena, M. Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral rna replication. mBio 2017, 8, e01658-17. [Google Scholar] [CrossRef] [Green Version]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Sars-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef] [Green Version]
- Snijder, E.J.; Limpens, R.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.; Koster, A.J.; Barcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down rna synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef] [PubMed]
- Doyle, N.; Hawes, P.C.; Simpson, J.; Adams, L.H.; Maier, H.J. The porcine deltacoronavirus replication organelle comprises double-membrane vesicles and zippered endoplasmic reticulum with double-membrane spherules. Viruses 2019, 11, 1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, H.J.; Hawes, P.C.; Cottam, E.M.; Mantell, J.; Verkade, P.; Monaghan, P.; Wileman, T.; Britton, P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. mBio 2013, 4, e00801-13. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Cong, Y.; Veenendaal, T.; Klumperman, J.; Shi, D.; Mari, M.; Reggiori, F. Ultrastructural characterization of membrane rearrangements induced by porcine epidemic diarrhea virus infection. Viruses 2017, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, K.; Zhang, X.; Guo, C.; Chen, Y.; Liu, X. An integrated analysis of membrane remodeling during porcine reproductive and respiratory syndrome virus replication and assembly. PLoS ONE 2018, 13, e0200919. [Google Scholar] [CrossRef]
- Sakai, Y.; Kawachi, K.; Terada, Y.; Omori, H.; Matsuura, Y.; Kamitani, W. Two-amino acids change in the nsp4 of sars coronavirus abolishes viral replication. Virology 2017, 510, 165–174. [Google Scholar] [CrossRef]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Kainz, K.; Hofer, S.J.; Kroemer, G.; Madeo, F. Digesting the crisis: Autophagy and coronaviruses. Microb. Cell 2020, 7, 119–128. [Google Scholar] [CrossRef]
- Maier, H.J.; Neuman, B.W.; Bickerton, E.; Keep, S.M.; Alrashedi, H.; Hall, R.; Britton, P. Extensive coronavirus-induced membrane rearrangements are not a determinant of pathogenicity. Sci. Rep. 2016, 6, 27126. [Google Scholar] [CrossRef]
- Belov, G.A.; Sztul, E. Rewiring of cellular membrane homeostasis by picornaviruses. J. Virol. 2014, 88, 9478–9489. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, N.; Anathy, V. Pathological consequences of the unfolded protein response and downstream protein disulphide isomerases in pulmonary viral infection and disease. J. Biochem. 2020, 167, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Cali, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses hijack the lc3-i-positive edemosomes, er-derived vesicles exporting short-lived erad regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.N.; Denison, M.R.; Virgin, H.W., IV. Coronavirus replication does not require the autophagy gene atg5. Autophagy 2007, 3, 581–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schatzl, H.M.; Gilch, S. Severe acute respiratory syndrome coronavirus replication is severely impaired by mg132 due to proteasome-independent inhibition of m-calpain. J. Virol. 2012, 86, 10112–10122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, B.W. Bioinformatics and functional analyses of coronavirus nonstructural proteins involved in the formation of replicative organelles. Antivir. Res. 2016, 135, 97–107. [Google Scholar] [CrossRef]
- Kusov, Y.; Tan, J.; Alvarez, E.; Enjuanes, L.; Hilgenfeld, R. A g-quadruplex-binding macrodomain within the “sars-unique domain” is essential for the activity of the sars-coronavirus replication-transcription complex. Virology 2015, 484, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.A.; Chatterjee, A.; Neuman, B.W.; Wuthrich, K. Sars coronavirus unique domain: Three-domain molecular architecture in solution and rna binding. J. Mol. Biol. 2010, 400, 724–742. [Google Scholar] [CrossRef]
- Baez-Santos, Y.M.; John, S.E.S.; Mesecar, A.D. The sars-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Nakashima, H.; Nguyen, T.; Goins, W.F.; Chiocca, E.A. Interferon-stimulated gene 15 (isg15) and isg15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (hdac6) and sqstm1/p62. J. Biol. Chem. 2015, 290, 1485–1495. [Google Scholar] [CrossRef] [Green Version]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Muller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; von Brunn, B.; Bairad, D.R.; et al. P53 down-regulates sars coronavirus replication and is targeted by the sars-unique domain and plpro via e3 ubiquitin ligase rchy1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in cell death and human cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Grimley, P.M.; Berezesky, I.K.; Friedman, R.M. Cytoplasmic structures associated with an arbovirus infection: Loci of viral ribonucleic acid synthesis. J. Virol. 1968, 2, 1326–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. Rna replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, G.; Limpens, R.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grunewald, K.; Koster, A.J.; et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Imbert, I.; Snijder, E.J.; Dimitrova, M.; Guillemot, J.C.; Lecine, P.; Canard, B. The sars-coronavirus plnc domain of nsp3 as a replication/transcription scaffolding protein. Virus Res. 2008, 133, 136–148. [Google Scholar] [CrossRef]
- Hilbert, B.J.; Hayes, J.A.; Stone, N.P.; Duffy, C.M.; Sankaran, B.; Kelch, B.A. Structure and mechanism of the atpase that powers viral genome packaging. Proc. Natl. Acad. Sci USA 2015, 112, E3792–E3799. [Google Scholar] [CrossRef] [Green Version]
- Taraporewala, Z.F.; Patton, J.T. Nonstructural proteins involved in genome packaging and replication of rotaviruses and other members of the reoviridae. Virus Res. 2004, 101, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Trus, B.L.; Cheng, N.; Newcomb, W.W.; Homa, F.L.; Brown, J.C.; Steven, A.C. Structure and polymorphism of the ul6 portal protein of herpes simplex virus type 1. J. Virol. 2004, 78, 12668–12671. [Google Scholar] [CrossRef] [Green Version]
- Kannan, S.; Ali, P.S.S.; Sheeza, A.; Hemalatha, K. Covid-19 (novel coronavirus 2019)—Recent trends. Eur. Rev. Med. Pharm. Sci. 2020, 24, 2006–2011. [Google Scholar]
- Chang, C.K.; Hou, M.H.; Chang, C.F.; Hsiao, C.D.; Huang, T.H. The sars coronavirus nucleocapsid protein—Forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef]
- Satarker, S.; Nampoothiri, M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef] [PubMed]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, Y.A. Properties of coronavirus and sars-cov-2. Malays. J. Pathol. 2020, 42, 3–11. [Google Scholar] [PubMed]
- Zhang, X.; Shi, H.; Chen, J.; Shi, D.; Dong, H.; Feng, L. Identification of the interaction between vimentin and nucleocapsid protein of transmissible gastroenteritis virus. Virus Res. 2015, 200, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Lee, E.Y.; Lee, M.; Kim, T.; Cho, Y.E. An in vivo cell-based assay for investigating the specific interaction between the sars-cov n-protein and its viral rna packaging sequence. Biochem. Biophys. Res. Commun. 2019, 520, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. Genus-specific pattern of intrinsically disordered central regions in the nucleocapsid protein of coronaviruses. Comput. Struct. Biotechnol. J. 2020, 18, 1884–1890. [Google Scholar] [CrossRef]
- Ye, Q.; West, A.M.V.; Silletti, S.; Corbett, K.D. Architecture and self-assembly of the sars-cov-2 nucleocapsid protein. Protein Sci. 2020. [Google Scholar] [CrossRef]
- Lu, S.; Ye, Q.; Singh, D.; Villa, E.; Cleveland, D.W.; Corbett, K.D. The sars-cov-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with rna and the membrane-associated m protein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Morgan, D.O. Phosphorylation modulates liquid-liquid phase separation of the sars-cov-2 n protein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’Kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.M.; Reggiori, F. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Zlotnick, A. Theoretical aspects of virus capsid assembly. J. Mol. Recognit. 2005, 18, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Liu, B.; Kumar, P.; Chow, V.T.; Lal, S.K. The nucleocapsid protein of the sars coronavirus is capable of self-association through a c-terminal 209 amino acid interaction domain. Biochem. Biophys. Res. Commun. 2004, 317, 1030–1036. [Google Scholar] [CrossRef]
- Zuniga, S.; Cruz, J.L.; Sola, I.; Mateos-Gomez, P.A.; Palacio, L.; Enjuanes, L. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 2010, 84, 2169–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaram, H.; Fan, H.; Bowman, B.R.; Ooi, A.; Jayaram, J.; Collisson, E.W.; Lescar, J.; Prasad, B.V. X-ray structures of the n- and c-terminal domains of a coronavirus nucleocapsid protein: Implications for nucleocapsid formation. J. Virol. 2006, 80, 6612–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, M.; Chang, C.K.; Ikeya, T.; Guntert, P.; Chang, Y.H.; Hsu, Y.L.; Huang, T.H.; Kainosho, M. Solution structure of the c-terminal dimerization domain of sars coronavirus nucleocapsid protein solved by the sail-nmr method. J. Mol. Biol. 2008, 380, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.J.; Yuann, J.M.; Chang, Y.M.; Lin, S.Y.; Zhao, J.; Perlman, S.; Shen, Y.Y.; Huang, T.H.; Hou, M.H. Crystal structure-based exploration of the important role of arg106 in the rna-binding domain of human coronavirus oc43 nucleocapsid protein. Biochim. Biophys. Acta 2013, 1834, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.M.; Lin, S.C.; Hsu, J.N.; Chang, C.K.; Chien, C.M.; Wang, Y.S.; Wu, H.Y.; Jeng, U.S.; Kehn-Hall, K.; Hou, M.H. Structure-based stabilization of non-native protein-protein interactions of coronavirus nucleocapsid proteins in antiviral drug design. J. Med. Chem. 2020, 63, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chang, C.K.; Chang, Y.W.; Sue, S.C.; Bai, H.I.; Riang, L.; Hsiao, C.D.; Huang, T.H. Structure of the sars coronavirus nucleocapsid protein rna-binding dimerization domain suggests a mechanism for helical packaging of viral rna. J. Mol. Biol. 2007, 368, 1075–1086. [Google Scholar] [CrossRef]
- Barcena, M.; Oostergetel, G.T.; Bartelink, W.; Faas, F.G.; Verkleij, A.; Rottier, P.J.; Koster, A.J.; Bosch, B.J. Cryo-electron tomography of mouse hepatitis virus: Insights into the structure of the coronavirion. Proc. Natl. Acad. Sci. USA 2009, 106, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.; Imran, M.; Dhamija, P.; Suchal, K.; Handu, S. Virtual screening and dynamics of potential inhibitors targeting rna binding domain of nucleocapsid phosphoprotein from sars-cov-2. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Chang, C.K.; Lo, S.C.; Wang, Y.S.; Hou, M.H. Recent insights into the development of therapeutics against coronavirus diseases by targeting n protein. Drug Discov. Today 2016, 21, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Ma-Lauer, Y.; Zheng, Y.; Malesevic, M.; von Brunn, B.; Fischer, G.; von Brunn, A. Influences of cyclosporin a and non-immunosuppressive derivatives on cellular cyclophilins and viral nucleocapsid protein during human coronavirus 229e replication. Antivir. Res. 2020, 173, 104620. [Google Scholar] [CrossRef] [PubMed]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of crispr as an antiviral strategy to combat sars-cov-2 and influenza. Cell 2020, 181, 865–876.e12. [Google Scholar] [CrossRef] [PubMed]
- Venkatagopalan, P.; Daskalova, S.M.; Lopez, L.A.; Dolezal, K.A.; Hogue, B.G. Coronavirus envelope (e) protein remains at the site of assembly. Virology 2015, 478, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Benvenuto, D.; Giovanetti, M.; Angeletti, S.; Ciccozzi, M.; Pascarella, S. Sars-cov-2 envelope and membrane proteins: Structural differences linked to virus characteristics? Biomed. Res. Int. 2020, 2020, 4389089. [Google Scholar] [CrossRef] [PubMed]
- Arbely, E.; Khattari, Z.; Brotons, G.; Akkawi, M.; Salditt, T.; Arkin, I.T. A highly unusual palindromic transmembrane helical hairpin formed by sars coronavirus e protein. J. Mol. Biol. 2004, 341, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, Y.T.; Wang, S.M.; Huang, K.J.; Wang, C.T. Sars-cov envelope protein palmitoylation or nucleocapid association is not required for promoting virus-like particle production. J. Biomed. Sci. 2014, 21, 34. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, E.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Marcos-Villar, L.; Enjuanes, L. The envelope protein of severe acute respiratory syndrome coronavirus interacts with the non-structural protein 3 and is ubiquitinated. Virology 2010, 402, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Khattari, Z.; Brotons, G.; Akkawi, M.; Arbely, E.; Arkin, I.T.; Salditt, T. Sars coronavirus e protein in phospholipid bilayers: An x-ray study. Biophys. J. 2006, 90, 2038–2050. [Google Scholar] [CrossRef] [Green Version]
- Manor, J.; Arbely, E.; Beerlink, A.; Akkawi, M.; Arkin, I.T. Use of isotope-edited ftir to derive a backbone structure of a transmembrane protein. J. Phys. Chem. Lett. 2014, 5, 2573–2579. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; He, X.; Yang, F.; Liu, X.; Li, Y.; Liu, Y.; Yang, Z.; Yu, J.; Zhang, B.; Zhao, W. Analysis of the genome sequence and prediction of b-cell epitopes of the envelope protein of middle east respiratory syndrome-coronavirus. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Alvarez, E.; Almazan, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the e gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stodola, J.K.; Dubois, G.; le Coupanec, A.; Desforges, M.; Talbot, P.J. The oc43 human coronavirus envelope protein is critical for infectious virus production and propagation in neuronal cells and is a determinant of neurovirulence and cns pathology. Virology 2018, 515, 134–149. [Google Scholar] [CrossRef]
- Ortego, J.; Ceriani, J.E.; Patino, C.; Plana, J.; Enjuanes, L. Absence of e protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 2007, 368, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Almazan, F.; DeDiego, M.L.; Sola, I.; Zuniga, S.; Nieto-Torres, J.L.; Marquez-Jurado, S.; Andres, G.; Enjuanes, L. Engineering a replication-competent, propagation-defective middle east respiratory syndrome coronavirus as a vaccine candidate. mBio 2013, 4, e00650-13. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Usera, F.; Enjuanes, L. Coronavirus virulence genes with main focus on sars-cov envelope gene. Virus Res. 2014, 194, 124–137. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘cytokine storm’ in covid-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Castano-Rodriguez, C.; Honrubia, J.M.; Gutierrez-Alvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of severe acute respiratory syndrome coronavirus viroporins e, 3a, and 8a in replication and pathogenesis. mBio 2018, 9, e02325-17. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.; Hurst, K.R.; Masters, P.S. Exceptional flexibility in the sequence requirements for coronavirus small envelope protein function. J. Virol. 2007, 81, 2249–2262. [Google Scholar] [CrossRef] [Green Version]
- Corse, E.; Machamer, C.E. The cytoplasmic tail of infectious bronchitis virus e protein directs golgi targeting. J. Virol. 2002, 76, 1273–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.R.; Lin, L.D.; Machamer, C.E. Identification of a golgi complex-targeting signal in the cytoplasmic tail of the severe acute respiratory syndrome coronavirus envelope protein. J. Virol. 2011, 85, 5794–5803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, L.; Masters, P.S. The small envelope protein e is not essential for murine coronavirus replication. J. Virol. 2003, 77, 4597–4608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerbeck, J.W.; Machamer, C.E. A coronavirus e protein is present in two distinct pools with different effects on assembly and the secretory pathway. J. Virol. 2015, 89, 9313–9323. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathy, K.; Lu, H.; Surya, W.; Vararattanavech, A.; Pervushin, K.; Torres, J. Expression and purification of coronavirus envelope proteins using a modified beta-barrel construct. Protein Expr. Purif. 2012, 85, 133–141. [Google Scholar] [CrossRef]
- Pervushin, K.; Tan, E.; Parthasarathy, K.; Lin, X.; Jiang, F.L.; Yu, D.; Vararattanavech, A.; Soong, T.W.; Liu, D.X.; Torres, J. Structure and inhibition of the sars coronavirus envelope protein ion channel. PLoS Pathog. 2009, 5, e1000511. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus e protein transports calcium ions and activates the nlrp3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Verdia-Baguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; DeDiego, M.L.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Coronavirus e protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 2012, 432, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castano-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef]
- Ruch, T.R.; Machamer, C.E. A single polar residue and distinct membrane topologies impact the function of the infectious bronchitis coronavirus e protein. PLoS Pathog. 2012, 8, e1002674. [Google Scholar] [CrossRef] [Green Version]
- To, J.; Surya, W.; Fung, T.S.; Li, Y.; Verdia-Baguena, C.; Queralt-Martin, M.; Aguilella, V.M.; Liu, D.X.; Torres, J. Channel-inactivating mutations and their revertant mutants in the envelope protein of infectious bronchitis virus. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, L.; Gage, P.; Ewart, G. Hexamethylene amiloride blocks e protein ion channels and inhibits coronavirus replication. Virology 2006, 353, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Krahling, V.; Stein, D.A.; Spiegel, M.; Weber, F.; Muhlberger, E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase r but is resistant to its antiviral activity. J. Virol. 2009, 83, 2298–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.; Wong, C.K.; Li, P.; Xie, Y. A sars-cov protein, orf-6, induces caspase-3 mediated, er stress and jnk-dependent apoptosis. Biochim. Biophys. Acta 2008, 1780, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.P.; Siu, K.L.; Chin, K.T.; Yuen, K.Y.; Zheng, B.; Jin, D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, T.S.; Huang, M.; Liu, D.X. Coronavirus-induced er stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 2014, 194, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, H.; Zhang, Q.; Dong, J.; Liang, Y.; Huang, Y.; Liu, H.J.; Tong, D. Porcine epidemic diarrhea virus e protein causes endoplasmic reticulum stress and up-regulates interleukin-8 expression. Virol. J. 2013, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Alvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P. The nlrp3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Chen, X.; Guo, X.; Ge, Q.; Zhao, Y.; Mu, H.; Zhang, J. Er stress activates the nlrp3 inflammasome: A novel mechanism of atherosclerosis. Oxid Med. Cell Longev. 2019, 2019, 3462530. [Google Scholar] [CrossRef] [PubMed]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. Beta-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 2020, 183, 1520–1535.e14. [Google Scholar] [CrossRef] [PubMed]
- Westerbeck, J.W.; Machamer, C.E. The infectious bronchitis coronavirus envelope protein alters golgi ph to protect the spike protein and promote the release of infectious virus. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Alonso, M.; Rocha-Perugini, V.; Alvarez, S.; Moreno-Gonzalo, O.; Ursa, A.; Lopez-Martin, S.; Izquierdo-Useros, N.; Martinez-Picado, J.; Munoz-Fernandez, M.A.; Yanez-Mo, M.; et al. The pdz-adaptor protein syntenin-1 regulates hiv-1 entry. Mol. Biol. Cell 2012, 23, 2253–2263. [Google Scholar] [CrossRef] [Green Version]
- Pieczynski, J.; Margolis, B. Protein complexes that control renal epithelial polarity. Am. J. Physiol. Renal Physiol. 2011, 300, F589–F601. [Google Scholar] [CrossRef] [Green Version]
- Teoh, K.T.; Siu, Y.L.; Chan, W.L.; Schluter, M.A.; Liu, C.J.; Peiris, J.S.; Bruzzone, R.; Margolis, B.; Nal, B. The sars coronavirus e protein interacts with pals1 and alters tight junction formation and epithelial morphogenesis. Mol. Biol. Cell 2010, 21, 3838–3852. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Guardeno, J.M.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castano-Rodriguez, C.; Enjuanes, L. The pdz-binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef] [Green Version]
- Regla-Nava, J.A.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Fernandez-Delgado, R.; Fett, C.; Castano-Rodriguez, C.; Perlman, S.; Enjuanes, L.; DeDiego, M.L. Severe acute respiratory syndrome coronaviruses with mutations in the e protein are attenuated and promising vaccine candidates. J. Virol. 2015, 89, 3870–3887. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Zhao, J.; Perlman, S. T cell-mediated immune response to respiratory coronaviruses. Immunol. Res. 2014, 59, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Fischer, F.; Stegen, C.F.; Masters, P.S.; Samsonoff, W.A. Analysis of constructed e gene mutants of mouse hepatitis virus confirms a pivotal role for e protein in coronavirus assembly. J. Virol. 1998, 72, 7885–7894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, W.E.; Chuang, C.K.; Yeh, S.H.; Chang, M.S.; Chen, S.S. Functional characterization of heptad repeat 1 and 2 mutants of the spike protein of severe acute respiratory syndrome coronavirus. J. Virol. 2006, 80, 3225–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the sars-cov-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-em structure of the 2019-ncov spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiga, O.; Bernini, A.; Ciutti, A.; Chiellini, S.; Menciassi, N.; Finetti, F.; Causarono, V.; Anselmi, F.; Prischi, F.; Niccolai, N. Molecular modelling of s1 and s2 subunits of sars coronavirus spike glycoprotein. Biochem. Biophys. Res. Commun. 2003, 310, 78–83. [Google Scholar] [CrossRef]
- Watanabe, R.; Matsuyama, S.; Shirato, K.; Maejima, M.; Fukushi, S.; Morikawa, S.; Taguchi, F. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved s protein as revealed by pseudotype virus bearing cleaved s protein. J. Virol. 2008, 82, 11985–11991. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of sars-cov-2 entry by using human ace2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of sars-cov-2 (previously 2019-ncov) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Hofmann-Winkler, H.; Pöhlmann, S. Priming time: How cellular proteases arm coronavirus spike proteins. In Activation of Viruses by Host Proteases; Böttcher-Friebertshäuser, E., Garten, W., Klenk, H.D., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 71–98. [Google Scholar]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed sars-cov-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of sars-cov-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Marzi, A.; Gramberg, T.; Simmons, G.; Moller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. Dc-sign and dc-signr interact with the glycoprotein of marburg virus and the s protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesova, O.; Pavlik, A. Perinatal treatment with glucocorticoids and the risk of maldevelopment of the brain. Neuropharmacology 1989, 28, 89–97. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Physiological and molecular triggers for sars-cov membrane fusion and entry into host cells. Virology 2018, 517, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Martina, B.E.; van der Zee, R.; Lepault, J.; Haijema, B.J.; Versluis, C.; Heck, A.J.; de Groot, R.; Osterhaus, A.D.; Rottier, P.J. Severe acute respiratory syndrome coronavirus (sars-cov) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madu, I.G.; Roth, S.L.; Belouzard, S.; Whittaker, G.R. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein s2 domain with characteristics of a viral fusion peptide. J. Virol. 2009, 83, 7411–7421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.L.; Millet, J.K.; Daniel, S.; Freed, J.H.; Whittaker, G.R. The sars-cov fusion peptide forms an extended bipartite fusion platform that perturbs membrane order in a calcium-dependent manner. J. Mol. Biol. 2017, 429, 3875–3892. [Google Scholar] [CrossRef]
- Follis, K.E.; York, J.; Nunberg, J.H. Furin cleavage of the sars coronavirus spike glycoprotein enhances cell-cell fusion but does not affect virion entry. Virology 2006, 350, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Freeman, M.C.; Peek, C.T.; Becker, M.M.; Smith, E.C.; Denison, M.R. Coronaviruses induce entry-independent, continuous macropinocytosis. mBio 2014, 5, e01340-14. [Google Scholar] [CrossRef] [Green Version]
- McBride, C.E.; Li, J.; Machamer, C.E. The cytoplasmic tail of the severe acute respiratory syndrome coronavirus spike protein contains a novel endoplasmic reticulum retrieval signal that binds copi and promotes interaction with membrane protein. J. Virol. 2007, 81, 2418–2428. [Google Scholar] [CrossRef] [Green Version]
- de Wilde, A.H.; Raj, V.S.; Oudshoorn, D.; Bestebroer, T.M.; van Nieuwkoop, S.; Limpens, R.; Posthuma, C.C.; van der Meer, Y.; Barcena, M.; Haagmans, B.L.; et al. Mers-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin a or interferon-alpha treatment. J. Gen. Virol. 2013, 94, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of sars-cov-2 by tmprss2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease tmprss2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. Sars-cov-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, S.; Dijkman, R.; Habjan, M.; Heurich, A.; Gierer, S.; Glowacka, I.; Welsch, K.; Winkler, M.; Schneider, H.; Hofmann-Winkler, H.; et al. Tmprss2 activates the human coronavirus 229e for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 2013, 87, 6150–6160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, C.E.; Machamer, C.E. Palmitoylation of sars-cov s protein is necessary for partitioning into detergent-resistant membranes and cell-cell fusion but not interaction with m protein. Virology 2010, 405, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.M.; Li, Y.G.; Yamate, M.; Li, S.M.; Ikuta, K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect. 2007, 9, 96–102. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in sars-cov entry into vero e6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef]
- Glende, J.; Schwegmann-Wessels, C.; Al-Falah, M.; Pfefferle, S.; Qu, X.; Deng, H.; Drosten, C.; Naim, H.Y.; Herrler, G. Importance of cholesterol-rich membrane microdomains in the interaction of the s protein of sars-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology 2008, 381, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Yilla, M.; Harcourt, B.H.; Hickman, C.J.; McGrew, M.; Tamin, A.; Goldsmith, C.S.; Bellini, W.J.; Anderson, L.J. Sars-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 2005, 107, 93–101. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in covid-19: Lessons from sars and mers, and potential therapeutic interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wang, M.L.; Chien, C.S.; Yarmishyn, A.A.; Yang, Y.P.; Lai, W.Y.; Luo, Y.H.; Lin, Y.T.; Chen, Y.J.; Chang, P.C.; et al. Highlight of immune pathogenic response and hematopathologic effect in sars-cov, mers-cov, and sars-cov-2 infection. Front. Immunol. 2020, 11, 1022. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. Sars-cov-2 invades host cells via a novel route: Cd147-spike protein. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lu, K.; Pfefferle, S.; Bertram, S.; Glowacka, I.; Drosten, C.; Pohlmann, S.; Simmons, G. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J. Virol. 2010, 84, 8753–8764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ip, W.K.; Chan, K.H.; Law, H.K.; Tso, G.H.; Kong, E.K.; Wong, W.H.; To, Y.F.; Yung, R.W.; Chow, E.Y.; Au, K.L.; et al. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J. Infect. Dis. 2005, 191, 1697–1704. [Google Scholar] [CrossRef] [Green Version]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef] [Green Version]
- Noris, M.; Benigni, A.; Remuzzi, G. The case of complement activation in covid-19 multiorgan impact. Kidney Int. 2020, 98, 314–322. [Google Scholar] [CrossRef]
- Cagliani, R.; Forni, D.; Clerici, M.; Sironi, M. Computational inference of selection underlying the evolution of the novel coronavirus, severe acute respiratory syndrome coronavirus 2. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liu, D.X. Post-translational modifications of coronavirus proteins: Roles and function. Future Virol. 2018, 13, 405–430. [Google Scholar] [CrossRef] [Green Version]
- Holmes, K.V.; Doller, E.W.; Sturman, L.S. Tunicamycin resistant glycosylation of coronavirus glycoprotein: Demonstration of a novel type of viral glycoprotein. Virology 1981, 115, 334–344. [Google Scholar] [CrossRef]
- Stern, D.F.; Sefton, B.M. Coronavirus proteins: Biogenesis of avian infectious bronchitis virus virion proteins. J. Virol. 1982, 44, 794–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemann, H.; Geyer, R.; Klenk, H.D.; Linder, D.; Stirm, S.; Wirth, M. The carbohydrates of mouse hepatitis virus (mhv) a59: Structures of the o-glycosidically linked oligosaccharides of glycoprotein e1. EMBO J. 1984, 3, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Locker, J.K.; Rose, J.K.; Horzinek, M.C.; Rottier, P.J. Membrane assembly of the triple-spanning coronavirus m protein. Individual transmembrane domains show preferred orientation. J. Biol. Chem. 1992, 267, 21911–21918. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of m protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.T.; Chang, C.H.; Wang, S.M.; Huang, K.J.; Wang, C.T. Identifying sars-cov membrane protein amino acid residues linked to virus-like particle assembly. PLoS ONE 2013, 8, e64013. [Google Scholar] [CrossRef]
- Perrier, A.; Bonnin, A.; Desmarets, L.; Danneels, A.; Goffard, A.; Rouille, Y.; Dubuisson, J.; Belouzard, S. The c-terminal domain of the mers coronavirus m protein contains a trans-golgi network localization signal. J. Biol. Chem. 2019, 294, 14406–14421. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.; Hurst-Hess, K.R.; Koetzner, C.A.; Masters, P.S. “Analyses of coronavirus assembly interactions with interspecies membrane and nucleocapsid protein chimeras. J. Virol. 2016, 90, 4357–4368. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A sars-cov-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wang, K.; Lv, W.; Yu, W.; Xie, S.; Xu, K.; Schwarz, W.; Xiong, S.; Sun, B. The orf4a protein of human coronavirus 229e functions as a viroporin that regulates viral production. Biochim. Biophys. Acta 2014, 1838, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.; Masters, P.S. Evolved variants of the membrane protein can partially replace the envelope protein in murine coronavirus assembly. J. Virol. 2010, 84, 12872–12885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, V.; Mde, J.G.; Sanz, M.A.; Carrasco, L. Viroporin activity of murine hepatitis virus e protein. FEBS Lett. 2005, 579, 3607–3612. [Google Scholar] [CrossRef] [Green Version]
- Verdia-Baguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; Dediego, M.L.; Enjuanes, L.; Aguilella, V.M. Analysis of sars-cov e protein ion channel activity by tuning the protein and lipid charge. Biochim. Biophys. Acta 2013, 1828, 2026–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. Trimal: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Saier, M.H., Jr. A web-based program for the prediction of average hydropathy, average amphipathicity and average similarity of multiply aligned homologous proteins. J. Mol. Microbiol. Biotechnol. 2001, 3, 285–286. [Google Scholar] [PubMed]
- Wang, S.C.; Davejan, P.; Hendargo, K.J.; Javadi-Razaz, I.; Chou, A.; Yee, D.C.; Ghazi, F.; Lam, K.J.K.; Conn, A.M.; Madrigal, A.; et al. Expansion of the major facilitator superfamily (mfs) to include novel transporters as well as transmembrane-acting enzymes. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183277. [Google Scholar] [CrossRef]
- Medrano-Soto, A.; Ghazi, F.; Hendargo, K.J.; Moreno-Hagelsieb, G.; Myers, S.; Saier, M.H., Jr. Expansion of the transporter-opsin-g protein-coupled receptor superfamily with five new protein families. PLoS ONE 2020, 15, e0231085. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1480–1497. [Google Scholar] [CrossRef]
- Ferreira, I.G.; Pucci, M.; Venturi, G.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F. Glycosylation as a main regulator of growth and death factor receptors signaling. Int. J. Mol. Sci. 2018, 19, 580. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Carbohydrates Can Be Attached to Proteins to Form Glycoproteins; W. H. Freeman and Company: New York, NY, USA, 2002. [Google Scholar]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Fierke, C.A. Understanding protein palmitoylation: Biological significance and enzymology. Sci. China Chem. 2011, 54, 1888–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, J.; Salaun, C.; Fukata, Y.; Fukata, M.; Chamberlain, L.H. Palmitoylation and membrane interactions of the neuroprotective chaperone cysteine-string protein. J. Biol. Chem. 2008, 283, 25014–25026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boscarino, J.A.; Logan, H.L.; Lacny, J.J.; Gallagher, T.M. Envelope protein palmitoylations are crucial for murine coronavirus assembly. J. Virol. 2008, 82, 2989–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, H.A. Deubiquitination in virus infection. Virology 2007, 362, 245–256. [Google Scholar] [CrossRef]
- Murin, C.D.; Wilson, I.A.; Ward, A.B. Antibody responses to viral infections: A structural perspective across three different enveloped viruses. Nat. Microbiol. 2019, 4, 734–747. [Google Scholar] [CrossRef]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent binding of 2019 novel coronavirus spike protein by a sars coronavirus-specific human monoclonal antibody. Emerg Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [Green Version]
- Radosevich, M.; Burnouf, T. Intravenous immunoglobulin g: Trends in production methods, quality control and quality assurance. Vox Sang. 2010, 98, 12–28. [Google Scholar] [CrossRef]
- Qiu, M.; Shi, Y.; Guo, Z.; Chen, Z.; He, R.; Chen, R.; Zhou, D.; Dai, E.; Wang, X.; Si, B.; et al. Antibody responses to individual proteins of sars coronavirus and their neutralization activities. Microbes Infect. 2005, 7, 882–889. [Google Scholar] [CrossRef]
- Sun, B.; Feng, Y.; Mo, X.; Zheng, P.; Wang, Q.; Li, P.; Peng, P.; Liu, X.; Chen, Z.; Huang, H.; et al. Kinetics of sars-cov-2 specific igm and igg responses in covid-19 patients. Emerg. Microbes Infect. 2020, 9, 940–948. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Reynders, M. Igg antibody response to sars-cov-2 infection and viral rna persistence in patients on maintenance hemodialysis. Am. J. Kidney Dis. 2020, 76, 440–441. [Google Scholar] [CrossRef] [PubMed]
- Wagh, K.; Kreider, E.F.; Li, Y.; Barbian, H.J.; Learn, G.H.; Giorgi, E.; Hraber, P.T.; Decker, T.G.; Smith, A.G.; Gondim, M.V.; et al. Completeness of hiv-1 envelope glycan shield at transmission determines neutralization breadth. Cell Rep. 2018, 25, 893–908.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Berndsen, Z.T.; Raghwani, J.; Seabright, G.E.; Allen, J.D.; Pybus, O.G.; McLellan, J.S.; Wilson, I.A.; Bowden, T.A.; Ward, A.B.; et al. Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nat. Commun. 2020, 11, 2688. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, G.; Harvey, D.J.; Feldmann, F.; Stroeher, U.; Feldmann, H.; Royle, L.; Dwek, R.A.; Rudd, P.M. Identification of n-linked carbohydrates from severe acute respiratory syndrome (sars) spike glycoprotein. Virology 2010, 399, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the sars-cov-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Song, H.C.; Seo, M.Y.; Stadler, K.; Yoo, B.J.; Choo, Q.L.; Coates, S.R.; Uematsu, Y.; Harada, T.; Greer, C.E.; Polo, J.M.; et al. Synthesis and characterization of a native, oligomeric form of recombinant severe acute respiratory syndrome coronavirus spike glycoprotein. J. Virol. 2004, 78, 10328–10335. [Google Scholar] [CrossRef] [Green Version]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the n- and o- glycosylation profile of the spike protein of novel coronavirus sars-cov-2. Glycobiology 2020, 30, 981–988. [Google Scholar] [CrossRef]
- Zimmermann, S.; Lepenies, B. Glycans as vaccine antigens and adjuvants: Immunological considerations. Methods Mol. Biol. 2015, 1331, 11–26. [Google Scholar]
- Petit, C.M.; Chouljenko, V.N.; Iyer, A.; Colgrove, R.; Farzan, M.; Knipe, D.M.; Kousoulas, K.G. Palmitoylation of the cysteine-rich endodomain of the sars-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology 2007, 360, 264–274. [Google Scholar] [CrossRef] [Green Version]
- Vuoksimaa, E.; Eriksson, C.J.; Pulkkinen, L.; Rose, R.J.; Kaprio, J. Decreased prevalence of left-handedness among females with male co-twins: Evidence suggesting prenatal testosterone transfer in humans? Psychoneuroendocrinology 2010, 35, 1462–1472. [Google Scholar] [CrossRef] [Green Version]
- Ujike, M.; Huang, C.; Shirato, K.; Matsuyama, S.; Makino, S.; Taguchi, F. Two palmitylated cysteine residues of the severe acute respiratory syndrome coronavirus spike (s) protein are critical for s incorporation into virus-like particles, but not for m-s co-localization. J. Gen. Virol. 2012, 93, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Gelhaus, S.; Thaa, B.; Eschke, K.; Veit, M.; Schwegmann-Wessels, C. Palmitoylation of the alphacoronavirus tgev spike protein s is essential for incorporation into virus-like particles but dispensable for s-m interaction. Virology 2014, 464–465, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yuan, Z.; Pavel, M.A.; Hansen, S.B. The role of high cholesterol in age-related covid19 lethality. bioRxiv 2020. [Google Scholar] [CrossRef]
- Oostra, M.; de Haan, C.A.; de Groot, R.J.; Rottier, P.J. Glycosylation of the severe acute respiratory syndrome coronavirus triple-spanning membrane proteins 3a and m. J. Virol. 2006, 80, 2326–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Haan, C.A.; de Wit, M.; Kuo, L.; Montalto, C.; Masters, P.S.; Weiss, S.R.; Rottier, P.J. O-glycosylation of the mouse hepatitis coronavirus membrane protein. Virus Res. 2002, 82, 77–81. [Google Scholar] [CrossRef]
- Voss, D.; Pfefferle, S.; Drosten, C.; Stevermann, L.; Traggiai, E.; Lanzavecchia, A.; Becker, S. Studies on membrane topology, n-glycosylation and functionality of sars-cov membrane protein. Virol. J. 2009, 6, 79. [Google Scholar] [CrossRef] [Green Version]
- de Haan, C.A.; de Wit, M.; Kuo, L.; Montalto-Morrison, C.; Haagmans, B.L.; Weiss, S.R.; Masters, P.S.; Rottier, P.J. The glycosylation status of the murine hepatitis coronavirus m protein affects the interferogenic capacity of the virus in vitro and its ability to replicate in the liver but not the brain. Virology 2003, 312, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Liao, Y.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical evidence for the presence of mixed membrane topologies of the severe acute respiratory syndrome coronavirus envelope protein expressed in mammalian cells. FEBS Lett. 2006, 580, 3192–3200. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Torres, J.L.; Dediego, M.L.; Alvarez, E.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Llorente, M.; Kremer, L.; Shuo, S.; Enjuanes, L. Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 2011, 415, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Yuan, Q.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology 2006, 349, 264–275. [Google Scholar] [CrossRef]
- Liao, Y.; Lescar, J.; Tam, J.P.; Liu, D.X. Expression of sars-coronavirus envelope protein in escherichia coli cells alters membrane permeability. Biochem. Biophys. Res. Commun. 2004, 325, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H.; Staub, O. Ubiquitylation of ion channels. Physiology 2005, 20, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Raaben, M.; Posthuma, C.C.; Verheije, M.H.; te Lintelo, E.G.; Kikkert, M.; Drijfhout, J.W.; Snijder, E.J.; Rottier, P.J.; de Haan, C.A. The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol 2010, 84, 7869–7879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindler, E.; Thiel, V. Sars-cov and ifn: Too little, too late. Cell Host Microbe 2016, 19, 139–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, K.M.; Elliott, R.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weiss, S.R. Murine coronavirus delays expression of a subset of interferon-stimulated genes. J. Virol. 2010, 84, 5656–5669. [Google Scholar] [CrossRef] [Green Version]
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral activities of type i interferons to sars-cov-2 infection. Antivir. Res. 2020, 179, 104811. [Google Scholar] [CrossRef]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by rig-i-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type i interferon production by interfering with trim25-mediated rig-i ubiquitination. J. Virol. 2017, 91, e02143-16. [Google Scholar] [CrossRef] [Green Version]
- Ramos, H.J.; Gale, M., Jr. Rig-i like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr. Opin. Virol. 2011, 1, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. Nf-kappab signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosse, K.M.; Monson, E.A.; Beard, M.R.; Helbig, K.J. Interferon-stimulated genes as enhancers of antiviral innate immune signaling. J. Innate Immun. 2018, 10, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Au-Yeung, N.; Mandhana, R.; Horvath, C.M. Transcriptional regulation by stat1 and stat2 in the interferon jak-stat pathway. JAKSTAT 2013, 2, e23931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Dallagi, A.; Girouard, J.; Hamelin-Morrissette, J.; Dadzie, R.; Laurent, L.; Vaillancourt, C.; Lafond, J.; Carrier, C.; Reyes-Moreno, C. The activating effect of ifn-gamma on monocytes/macrophages is regulated by the lif-trophoblast-il-10 axis via stat1 inhibition and stat3 activation. Cell Mol. Immunol. 2015, 12, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Paolini, R.; Bernardini, G.; Molfetta, R.; Santoni, A. Nk cells and interferons. Cytokine Growth Factor Rev. 2015, 26, 113–120. [Google Scholar] [CrossRef]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral t cell responses by type i interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef]
- Swann, J.B.; Hayakawa, Y.; Zerafa, N.; Sheehan, K.C.; Scott, B.; Schreiber, R.D.; Hertzog, P.; Smyth, M.J. Type i ifn contributes to nk cell homeostasis, activation, and antitumor function. J. Immunol. 2007, 178, 7540–7549. [Google Scholar] [CrossRef]
- Oudshoorn, D.; van der Hoeven, B.; Limpens, R.W.; Beugeling, C.; Snijder, E.J.; Barcena, M.; Kikkert, M. Antiviral innate immune response interferes with the formation of replication-associated membrane structures induced by a positive-strand rna virus. mBio 2016, 7, e01991-16. [Google Scholar] [CrossRef] [Green Version]
- Fung, S.Y.; Yuen, K.S.; Ye, Z.W.; Chan, C.P.; Jin, D.Y. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: Lessons from other pathogenic viruses. Emerg. Microbes Infect. 2020, 9, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Kindler, E.; Thiel, V.; Weber, F. Interaction of sars and mers coronaviruses with the antiviral interferon response. Adv. Virus Res. 2016, 96, 219–243. [Google Scholar] [PubMed]
- Totura, A.L.; Baric, R.S. Sars coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.K.; Lam, J.Y.; Wong, W.M.; Mak, L.F.; Wang, X.; Chu, H.; Cai, J.P.; Jin, D.Y.; To, K.K.; Chan, J.F.; et al. Sars-cov-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type i interferon responses by sars-cov-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, L.; Geng, H.; Deng, Y.; Huang, B.; Guo, Y.; Zhao, Z.; Tan, W. The structural and accessory proteins m, orf 4a, orf 4b, and orf 5 of middle east respiratory syndrome coronavirus (mers-cov) are potent interferon antagonists. Protein Cell 2013, 4, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Comar, C.E.; Goldstein, S.A.; Li, Y.; Yount, B.; Baric, R.S.; Weiss, S.R. Antagonism of dsrna-induced innate immune pathways by ns4a and ns4b accessory proteins during mers coronavirus infection. mBio 2019, 10, e00319-19. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ye, F.; Zhu, N.; Wang, W.; Deng, Y.; Zhao, Z.; Tan, W. Middle east respiratory syndrome coronavirus orf4b protein inhibits type i interferon production through both cytoplasmic and nuclear targets. Sci. Rep. 2015, 5, 17554. [Google Scholar] [CrossRef] [Green Version]
- Siu, K.L.; Chan, C.P.; Kok, K.H.; Woo, P.C.; Jin, D.Y. Suppression of innate antiviral response by severe acute respiratory syndrome coronavirus m protein is mediated through the first transmembrane domain. Cell Mol. Immunol. 2014, 11, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Lui, P.Y.; Wong, L.Y.; Fung, C.L.; Siu, K.L.; Yeung, M.L.; Yuen, K.S.; Chan, C.P.; Woo, P.C.; Yuen, K.Y.; Jin, D.Y. Middle east respiratory syndrome coronavirus m protein suppresses type i interferon expression through the inhibition of tbk1-dependent phosphorylation of irf3. Emerg. Microbes Infect. 2016, 5, e39. [Google Scholar] [CrossRef] [Green Version]
- Di Palma, F.; Daino, G.L.; Ramaswamy, V.K.; Corona, A.; Frau, A.; Fanunza, E.; Vargiu, A.V.; Tramontano, E.; Ruggerone, P. Relevance of ebola virus vp35 homo-dimerization on the type i interferon cascade inhibition. Antivir. Chem. Chemother. 2019, 27, 2040206619889220. [Google Scholar] [CrossRef] [PubMed]
- Schwerk, J.; Negash, A.; Savan, R.; Gale, M., Jr. Innate immunity in hepatitis c virus infection. Cold Spring Harb. Perspect. Med. 2020, a036988. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, L. The membrane protein of severe acute respiratory syndrome coronavirus functions as a novel cytosolic pathogen-associated molecular pattern to promote beta interferon induction via a toll-like-receptor-related traf3-independent mechanism. mBio 2016, 7, e01872-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.Y.; Liu, H.M.; Chang, M.F.; Chang, S.C. Middle east respiratory syndrome coronavirus nucleocapsid protein suppresses type i and type iii interferon induction by targeting rig-i signaling. J. Virol. 2020, 94, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Likai, J.; Shasha, L.; Wenxian, Z.; Jingjiao, M.; Jianhe, S.; Hengan, W.; Yaxian, Y. Porcine deltacoronavirus nucleocapsid protein suppressed ifn-beta production by interfering porcine rig-i dsrna-binding and k63-linked polyubiquitination. Front. Immunol. 2019, 10, 1024. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shi, K.; Yoo, D. Suppression of type i interferon production by porcine epidemic diarrhea virus and degradation of creb-binding protein by nsp1. Virology 2016, 489, 252–268. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. Sars-cov nucleocapsid protein antagonizes ifn-beta response by targeting initial step of ifn-beta induction pathway, and its c-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Martin-Vicente, M.; Medrano, L.M.; Resino, S.; Garcia-Sastre, A.; Martinez, I. Trim25 in the regulation of the antiviral innate immunity. Front. Immunol. 2017, 8, 1187. [Google Scholar] [CrossRef] [Green Version]
- Mu, J.; Fang, Y.; Yang, Q.; Shu, T.; Wang, A.; Huang, M.; Jin, L.; Deng, F.; Qiu, Y.; Zhou, X. Sars-cov-2 n protein antagonizes type i interferon signaling by suppressing phosphorylation and nuclear translocation of stat1 and stat2. Cell Discov. 2020, 6, 65. [Google Scholar] [CrossRef]
- Narayanan, K.; Huang, C.; Lokugamage, K.; Kamitani, W.; Ikegami, T.; Tseng, C.T.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type i interferon, in infected cells. J. Virol. 2008, 82, 4471–4479. [Google Scholar] [CrossRef] [Green Version]
- Gaglia, M.M.; Covarrubias, S.; Wong, W.; Glaunsinger, B.A. A common strategy for host rna degradation by divergent viruses. J. Virol. 2012, 86, 9527–9530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, K.; Narayanan, K.; Wada, M.; Popov, V.L.; Cajimat, M.; Baric, R.S.; Makino, S. The endonucleolytic rna cleavage function of nsp1 of middle east respiratory syndrome coronavirus promotes the production of infectious virus particles in specific human cell lines. J. Virol. 2018, 92, e01157-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamitani, W.; Huang, C.; Narayanan, K.; Lokugamage, K.G.; Makino, S. A two-pronged strategy to suppress host protein synthesis by sars coronavirus nsp1 protein. Nat. Struct. Mol. Biol. 2009, 16, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Lokugamage, K.G.; Rozovics, J.M.; Narayanan, K.; Semler, B.L.; Makino, S. Sars coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mrnas: Viral mrnas are resistant to nsp1-induced rna cleavage. PLoS Pathog. 2011, 7, e1002433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wathelet, M.G.; Orr, M.; Frieman, M.B.; Baric, R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: Role of nsp1 and rational design of an attenuated strain. J. Virol. 2007, 81, 11620–11633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieman, M.; Ratia, K.; Johnston, R.E.; Mesecar, A.D.; Baric, R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of irf3 and nf-kappab signaling. J. Virol. 2009, 83, 6689–6705. [Google Scholar] [CrossRef] [Green Version]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.T.; Baker, S.C.; et al. Regulation of irf-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef] [Green Version]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef] [Green Version]
- Matthews, K.; Schafer, A.; Pham, A.; Frieman, M. The sars coronavirus papain like protease can inhibit irf3 at a post activation step that requires deubiquitination activity. Virol. J. 2014, 11, 209. [Google Scholar] [CrossRef]
- Li, J.Y.; Liao, C.H.; Wang, Q.; Tan, Y.J.; Luo, R.; Qiu, Y.; Ge, X.Y. The orf6, orf8 and nucleocapsid proteins of sars-cov-2 inhibit type i interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (orf) 3b, orf 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieman, M.; Yount, B.; Heise, M.; Kopecky-Bromberg, S.A.; Palese, P.; Baric, R.S. Severe acute respiratory syndrome coronavirus orf6 antagonizes stat1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/golgi membrane. J. Virol. 2007, 81, 9812–9824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Yao, Z.; Shan, Y.; Chen, B.; Yang, Z.; Wu, J.; Zhao, Z.; Chen, J.; Cong, Y. Nucleolar localization of non-structural protein 3b, a protein specifically encoded by the severe acute respiratory syndrome coronavirus. Virus Res. 2005, 114, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.; Consortium, U.-C.; Nakagawa, S.; et al. Sars-cov-2 orf3b is a Potent Interferon Antagonist Whose Activity is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef] [PubMed]
- Stoermer, K.A.; Morrison, T.E. Complement and viral pathogenesis. Virology 2011, 411, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Thorgersen, E.B.; Barratt-Due, A.; Haugaa, H.; Harboe, M.; Pischke, S.E.; Nilsson, P.H.; Mollnes, T.E. The role of complement in liver injury, regeneration, and transplantation. Hepatology 2019, 70, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Abenavoli, L.; Gentile, I.; Maraolo, A.E.; Negro, F. Sars-cov-2 and liver damage: A possible pathogenetic link. Hepatobiliary Surg. Nutr. 2020, 9, 322–324. [Google Scholar] [CrossRef]
- Keshari, R.S.; Silasi, R.; Popescu, N.I.; Patel, M.M.; Chaaban, H.; Lupu, C.; Coggeshall, K.M.; Mollnes, T.E.; DeMarco, S.J.; Lupu, F. Inhibition of complement c5 protects against organ failure and reduces mortality in a baboon model of escherichia coli sepsis. Proc. Natl. Acad. Sci. USA 2017, 114, E6390–E6399. [Google Scholar] [CrossRef] [Green Version]
- Garcia, C.C.; Weston-Davies, W.; Russo, R.C.; Tavares, L.P.; Rachid, M.A.; Alves-Filho, J.C.; Machado, A.V.; Ryffel, B.; Nunn, M.A.; Teixeira, M.M. Complement c5 activation during influenza a infection in mice contributes to neutrophil recruitment and lung injury. PLoS ONE 2013, 8, e64443. [Google Scholar] [CrossRef] [Green Version]
- Thurman, J.M.; Le Quintrec, M. Targeting the complement cascade: Novel treatments coming down the pike. Kidney Int. 2016, 90, 746–752. [Google Scholar] [CrossRef] [Green Version]
- Rittirsch, D.; Redl, H.; Huber-Lang, M. Role of complement in multiorgan failure. Clin. Dev. Immunol. 2012, 2012, 962927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, K.H.; Tsang, W.K.; Tang, C.S.; Lam, M.F.; Lai, F.M.; To, K.F.; Fung, K.S.; Tang, H.L.; Yan, W.W.; Chan, H.W.; et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. 2005, 67, 698–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, M.; Abel, B.; Gallimore, A.; Carroll, M.; Bachmann, M.F. Complement component c3 promotes t-cell priming and lung migration to control acute influenza virus infection. Nat. Med. 2002, 8, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, K.H.; Ko, E.J.; Kim, M.C.; Lee, Y.N.; Jung, Y.J.; Lee, Y.T.; Kwon, Y.M.; Song, J.M.; Kang, S.M. Complement c3 plays a key role in inducing humoral and cellular immune responses to influenza virus strain-specific hemagglutinin-based or cross-protective m2 extracellular domain-based vaccination. J. Virol. 2018, 92, e00969-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattan, A.; Pawar, S.D.; Nawadkar, R.; Kulkarni, N.; Lal, G.; Mullick, J.; Sahu, A. Synergy between the classical and alternative pathways of complement is essential for conferring effective protection against the pandemic influenza a(h1n1) 2009 virus infection. PLoS Pathog. 2017, 13, e1006248. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhao, G.; Song, N.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; Du, L.; Jiang, S.; Guo, R.; et al. Blockade of the c5a-c5ar axis alleviates lung damage in hdpp4-transgenic mice infected with mers-cov. Emerg. Microbes Infect. 2018, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, T.; Tsukamoto, H. Complement-targeted therapy: Development of c5- and c5a-targeted inhibition. Inflamm. Regen. 2016, 36, 11. [Google Scholar] [CrossRef] [Green Version]
- Bekker, P.; Dairaghi, D.; Seitz, L.; Leleti, M.; Wang, Y.; Ertl, L.; Baumgart, T.; Shugarts, S.; Lohr, L.; Dang, T.; et al. Characterization of pharmacologic and pharmacokinetic properties of ccx168, a potent and selective orally administered complement 5a receptor inhibitor, based on preclinical evaluation and randomized phase 1 clinical study. PLoS ONE 2016, 11, e0164646. [Google Scholar] [CrossRef]
- Versteeg, G.A.; van de Nes, P.S.; Bredenbeek, P.J.; Spaan, W.J. The coronavirus spike protein induces endoplasmic reticulum stress and upregulation of intracellular chemokine mrna concentrations. J. Virol. 2007, 81, 10981–10990. [Google Scholar] [CrossRef] [Green Version]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The sars coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhang, H.; Zhang, Q.; Huang, Y.; Dong, J.; Liang, Y.; Liu, H.J.; Tong, D. Porcine epidemic diarrhea virus n protein prolongs s-phase cell cycle, induces endoplasmic reticulum stress, and up-regulates interleukin-8 expression. Vet. Microbiol. 2013, 164, 212–221. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, Y.; Chang, R.; Tong, D.; Xu, X. Transmissible gastroenteritis virus n protein causes endoplasmic reticulum stress, up-regulates interleukin-8 expression and its subcellular localization in the porcine intestinal epithelial cell. Res. Vet. Sci. 2018, 119, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Xu, J.; Duan, X.; Xu, X.; Li, P.; Cheng, L.; Zheng, L.; Li, X.; Zhang, Y.; Wang, X.; et al. Porcine epidemic diarrhea virus orf3 protein causes endoplasmic reticulum stress to facilitate autophagy. Vet. Microbiol. 2019, 235, 209–219. [Google Scholar] [CrossRef]
- Liang, J.Q.; Fang, S.; Yuan, Q.; Huang, M.; Chen, R.A.; Fung, T.S.; Liu, D.X. N-linked glycosylation of the membrane protein ectodomain regulates infectious bronchitis virus-induced er stress response, apoptosis and pathogenesis. Virology 2019, 531, 48–56. [Google Scholar] [CrossRef]
- Yuan, X.; Yao, Z.; Wu, J.; Zhou, Y.; Shan, Y.; Dong, B.; Zhao, Z.; Hua, P.; Chen, J.; Cong, Y. G1 phase cell cycle arrest induced by sars-cov 3a protein via the cyclin d3/prb pathway. Am. J. Respir. Cell Mol. Biol. 2007, 37, 9–19. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, J.; Shan, Y.; Yao, Z.; Dong, B.; Chen, B.; Zhao, Z.; Wang, S.; Chen, J.; Cong, Y. Sars coronavirus 7a protein blocks cell cycle progression at g0/g1 phase via the cyclin d3/prb pathway. Virology 2006, 346, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.G.; Zhang, H.L.; Zhang, Q.; Dong, J.; Huang, Y.; Tong, D.W. Porcine epidemic diarrhea virus m protein blocks cell cycle progression at s-phase and its subcellular localization in the porcine intestinal epithelial cells. Acta Virol. 2015, 59, 265–275. [Google Scholar] [CrossRef]
- Sun, P.; Wu, H.; Huang, J.; Xu, Y.; Yang, F.; Zhang, Q.; Xu, X. Porcine epidemic diarrhea virus through p53-dependent pathway causes cell cycle arrest in the g0/g1 phase. Virus Res. 2018, 253, 1–11. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.; Liu, S.; Kou, Z.; Li, W.; Farzan, M.; Jiang, S. Receptor-binding domain of sars-cov spike protein induces highly potent neutralizing antibodies: Implication for developing subunit vaccine. Biochem. Biophys. Res. Commun. 2004, 324, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhao, G.; Chan, C.C.; Sun, S.; Chen, M.; Liu, Z.; Guo, H.; He, Y.; Zhou, Y.; Zheng, B.J.; et al. Recombinant receptor-binding domain of sars-cov spike protein expressed in mammalian, insect and e. Coli cells elicits potent neutralizing antibody and protective immunity. Virology 2009, 393, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, T.S.; Liu, D.X. Coronavirus infection, er stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Xiong, X.; Park, Y.J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell 2019, 176, 1026–1039.e15. [Google Scholar] [CrossRef] [Green Version]
- Li, C.K.; Wu, H.; Yan, H.; Ma, S.; Wang, L.; Zhang, M.; Tang, X.; Temperton, N.J.; Weiss, R.A.; Brenchley, J.M.; et al. T cell responses to whole sars coronavirus in humans. J. Immunol. 2008, 181, 5490–5500. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yuan, Z.; Matsumoto, M.; Hengge, U.R.; Chang, Y.F. Immune responses with DNA vaccines encoded different gene fragments of severe acute respiratory syndrome coronavirus in balb/c mice. Biochem. Biophys. Res. Commun. 2005, 327, 130–135. [Google Scholar] [CrossRef]
- Huang, J.; Ma, R.; Wu, C.Y. Immunization with sars-cov s DNA vaccine generates memory cd4+ and cd8+ t cell immune responses. Vaccine 2006, 24, 4905–4913. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Kong, W.P.; Huang, Y.; Roberts, A.; Murphy, B.R.; Subbarao, K.; Nabel, G.J. A DNA vaccine induces sars coronavirus neutralization and protective immunity in mice. Nature 2004, 428, 561–564. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Lu, X.; Tao, L.; Bai, B.; Zhang, Z.; Chen, Y.; Zheng, F.; Chen, J.; Chen, Z.; Wang, H. Induction of specific immune responses by severe acute respiratory syndrome coronavirus spike DNA vaccine with or without interleukin-2 immunization using different vaccination routes in mice. Clin. Vaccine Immunol 2007, 14, 894–901. [Google Scholar] [CrossRef] [Green Version]
- Janice Oh, H.L.; Gan, S.K.; Bertoletti, A.; Tan, Y.J. Understanding the t cell immune response in sars coronavirus infection. Emerg. Microbes Infect. 2012, 1, e23. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.K.; Lam, C.W.; Wu, A.K.; Ip, W.K.; Lee, N.L.; Chan, I.H.; Lit, L.C.; Hui, D.S.; Chan, M.H.; Chung, S.S.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of t cell responses to sars-cov-2 coronavirus in humans with covid-19 disease and unexposed individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Huang, J.L.; Huang, J.; Duan, Z.H.; Wei, J.; Min, J.; Luo, X.H.; Li, J.G.; Tan, W.P.; Wu, L.Z.; Liu, R.Y.; et al. Th2 predominance and cd8+ memory t cell depletion in patients with severe acute respiratory syndrome. Microbes Infect. 2005, 7, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodriguez, L. Sars-cov-2 infection: The role of cytokines in covid-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.W.; Li, Y.; Zhang, H.N.; Wang, W.; Yang, X.; Qi, H.; Li, H.; Men, D.; Zhou, J.; Tao, S.C. Sars-cov-2 proteome microarray for global profiling of covid-19 specific igg and igm responses. Nat. Commun. 2020, 11, 3581. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.V.; Brown, O.R. Comparative analysis of coronaviridae nucleocapsid and surface glycoprotein sequences. Front. Biosci. (Landmark Ed.) 2020, 25, 1894–1900. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Koohy, H. In silico identification of vaccine targets for 2019-ncov. F1000Res 2020, 9, 145. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the covid-19 coronavirus (sars-cov-2) based on sars-cov immunological studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [Green Version]
- Ng, O.W.; Chia, A.; Tan, A.T.; Jadi, R.S.; Leong, H.N.; Bertoletti, A.; Tan, Y.J. Memory t cell responses targeting the sars coronavirus persist up to 11 years post-infection. Vaccine 2016, 34, 2008–2014. [Google Scholar] [CrossRef]
- Ong, E.; Wong, M.U.; Huffman, A.; He, Y. Covid-19 coronavirus vaccine design using reverse vaccinology and machine learning. Front. Immunol. 2020, 11, 1581. [Google Scholar] [CrossRef]
- de Haan, C.A.; Rottier, P.J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar] [PubMed]
- Liu, J.; Sun, Y.; Qi, J.; Chu, F.; Wu, H.; Gao, F.; Li, T.; Yan, J.; Gao, G.F. The membrane protein of severe acute respiratory syndrome coronavirus acts as a dominant immunogen revealed by a clustering region of novel functionally and structurally defined cytotoxic t-lymphocyte epitopes. J. Infect. Dis. 2010, 202, 1171–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.H.; Hu, C.Y.; Wu, N.P.; Yao, H.P.; Li, L.J. Molecular characteristics, functions, and related pathogenicity of mers-cov proteins. Engineering 2019, 5, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Ignjatovic, J.; Galli, L. Structural proteins of avian infectious bronchitis virus: Role in immunity and protection. Adv. Exp. Med. Biol. 1993, 342, 449–453. [Google Scholar] [PubMed]
- Ignjatovic, J.; Galli, L. The s1 glycoprotein but not the n or m proteins of avian infectious bronchitis virus induces protection in vaccinated chickens. Arch. Virol. 1994, 138, 117–134. [Google Scholar] [CrossRef]
- Saif, L.J. Coronavirus immunogens. Vet. Microbiol. 1993, 37, 285–297. [Google Scholar] [CrossRef]
- Okada, M.; Takemoto, Y.; Okuno, Y.; Hashimoto, S.; Yoshida, S.; Fukunaga, Y.; Tanaka, T.; Kita, Y.; Kuwayama, S.; Muraki, Y.; et al. The development of vaccines against sars corona virus in mice and scid-pbl/hu mice. Vaccine 2005, 23, 2269–2272. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, J.; Shi, H.; Chen, X.; Shi, D.; Feng, L.; Yang, B. Identification of a conserved linear b-cell epitope in the m protein of porcine epidemic diarrhea virus. Virol. J. 2012, 9, 225. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Zhao, Y.; Hu, Y.; Qiu, J.; Lei, W.; Ji, W.; Li, X.; Wu, Q.; Shi, X.; Li, Z. Protection of chickens against infectious bronchitis virus with a multivalent DNA vaccine and boosting with an inactivated vaccine. J. Vet. Sci. 2013, 14, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Takano, T.; Morioka, H.; Gomi, K.; Tomizawa, K.; Doki, T.; Hohdatsu, T. Screening and identification of t helper 1 and linear immunodominant antibody-binding epitopes in spike 1 domain and membrane protein of feline infectious peritonitis virus. Vaccine 2014, 32, 1834–1840. [Google Scholar] [CrossRef]
- Ventola, C.L. Immunization in the united states: Recommendations, barriers, and measures to improve compliance: Part 1: Childhood vaccinations. P T 2016, 41, 426–436. [Google Scholar] [PubMed]
- Duggan, N.M.; Ludy, S.M.; Shannon, B.C.; Reisner, A.T.; Wilcox, S.R. Is novel coronavirus 2019 reinfection possible? Interpreting dynamic sars-cov-2 test results through a case report. Am. J. Emerg. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gunthard, H.F.; Saag, M.S.; Benson, C.A.; del Rio, C.; Eron, J.J.; Gallant, J.E.; Hoy, J.F.; Mugavero, M.J.; Sax, P.E.; Thompson, M.A.; et al. Antiretroviral drugs for treatment and prevention of hiv infection in adults: 2016 recommendations of the international antiviral society-USA panel. JAMA 2016, 316, 191–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.U.; Vanmeert, M.; Ali, A.; Iman, K.; Froeyen, M.; Idrees, M. Perspectives towards antiviral drug discovery against ebola virus. J. Med. Virol. 2019, 91, 2029–2048. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Woo, P.C.; Yip, C.C.; Tse, H.; Tsoi, H.W.; Cheng, V.C.; Lee, P.; Tang, B.S.; Cheung, C.H.; Lee, R.A.; et al. Coronavirus hku1 and other coronavirus infections in hong kong. J. Clin. Microbiol. 2006, 44, 2063–2071. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, A.; Selvaraju, S.; Esper, F. Human coronavirus-hku1 infection among adults in cleveland, ohio. Open Forum. Infect. Dis. 2017, 4, ofx052. [Google Scholar] [CrossRef] [Green Version]
- Schulz, L.L.; Tonsor, G.T. Assessment of the economic impacts of porcine epidemic diarrhea virus in the united states. J. Anim. Sci. 2015, 93, 5111–5118. [Google Scholar] [CrossRef] [Green Version]
- Legnardi, M.; Tucciarone, C.M.; Franzo, G.; Cecchinato, M. Infectious bronchitis virus evolution, diagnosis and control. Vet. Sci. 2020, 7, 79. [Google Scholar] [CrossRef]
- Wu, T.; Perrings, C.; Kinzig, A.; Collins, J.P.; Minteer, B.A.; Daszak, P. Economic growth, urbanization, globalization, and the risks of emerging infectious diseases in china: A review. Ambio 2017, 46, 18–29. [Google Scholar] [CrossRef]
- Njume, C.; Goduka, N.I. Treatment of diarrhoea in rural african communities: An overview of measures to maximise the medicinal potentials of indigenous plants. Int. J. Environ. Res. Public Health 2012, 9, 3911–3933. [Google Scholar] [CrossRef] [Green Version]
- Levy, B.; Edholm, C.; Gaoue, O.; Kaondera-Shava, R.; Kgosimore, M.; Lenhart, S.; Lephodisa, B.; Lungu, E.; Marijani, T.; Nyabadza, F. Modeling the role of public health education in ebola virus disease outbreaks in sudan. Infect. Dis. Model. 2017, 2, 323–340. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
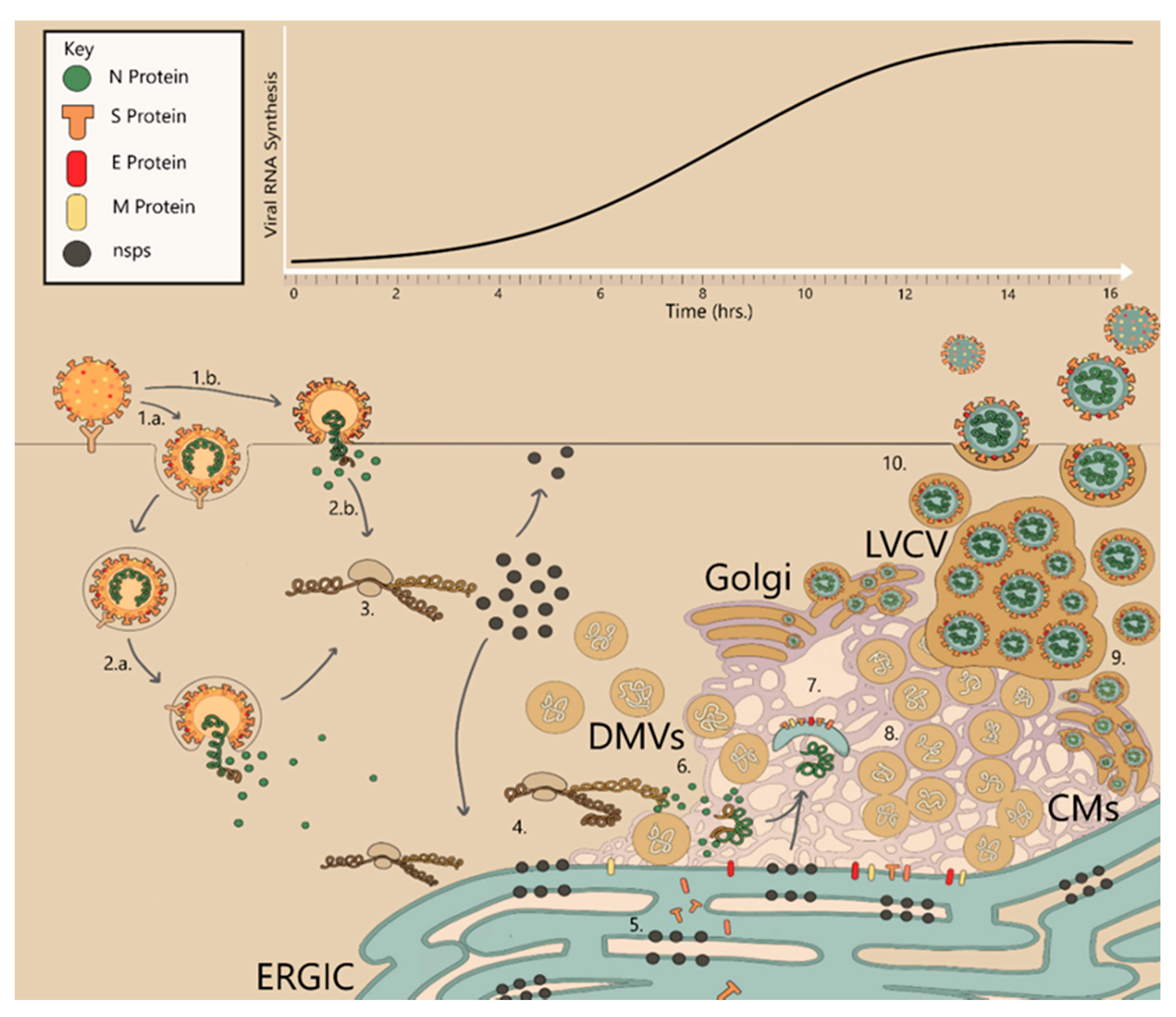{kind=link}
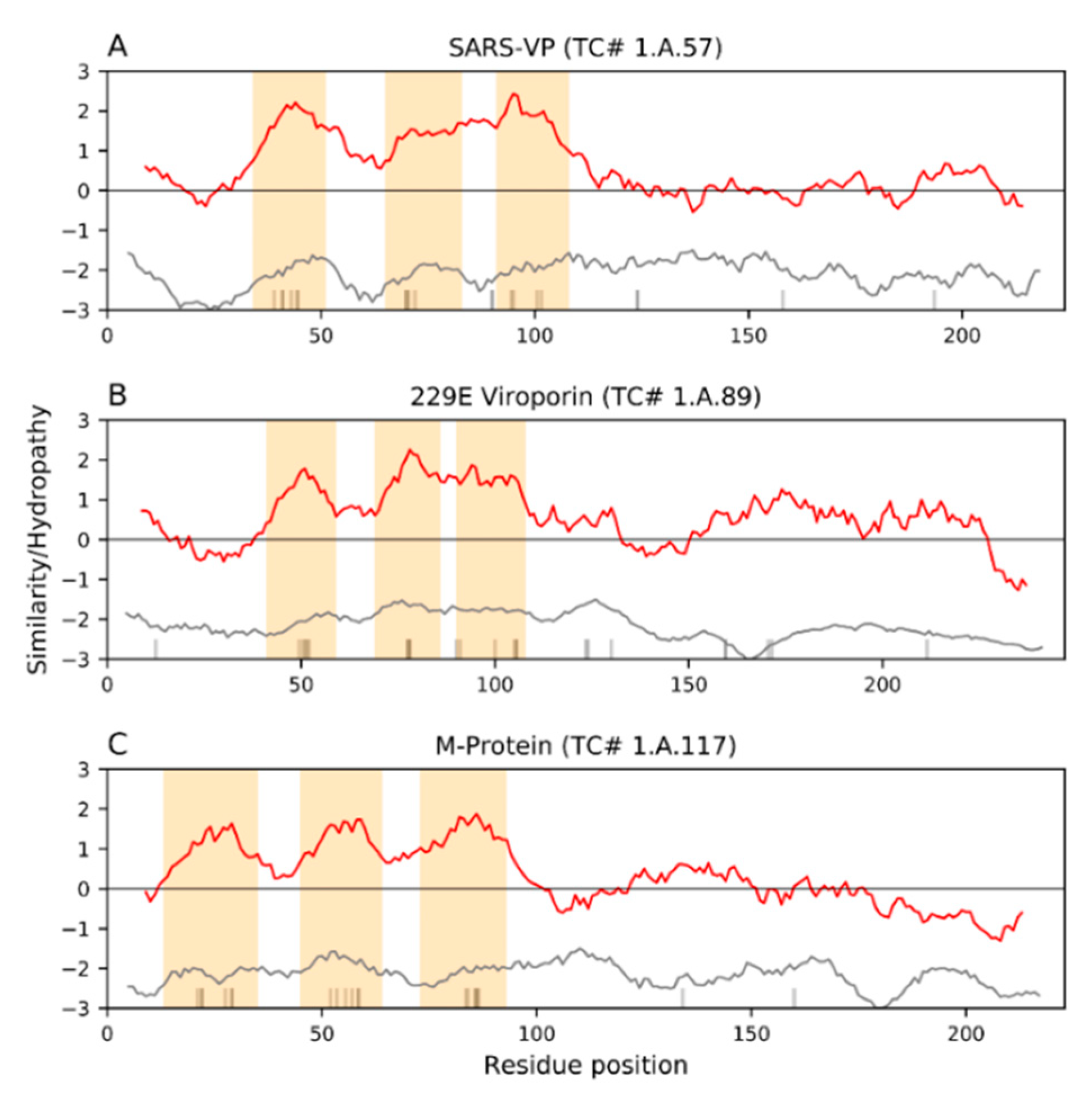{kind=link}
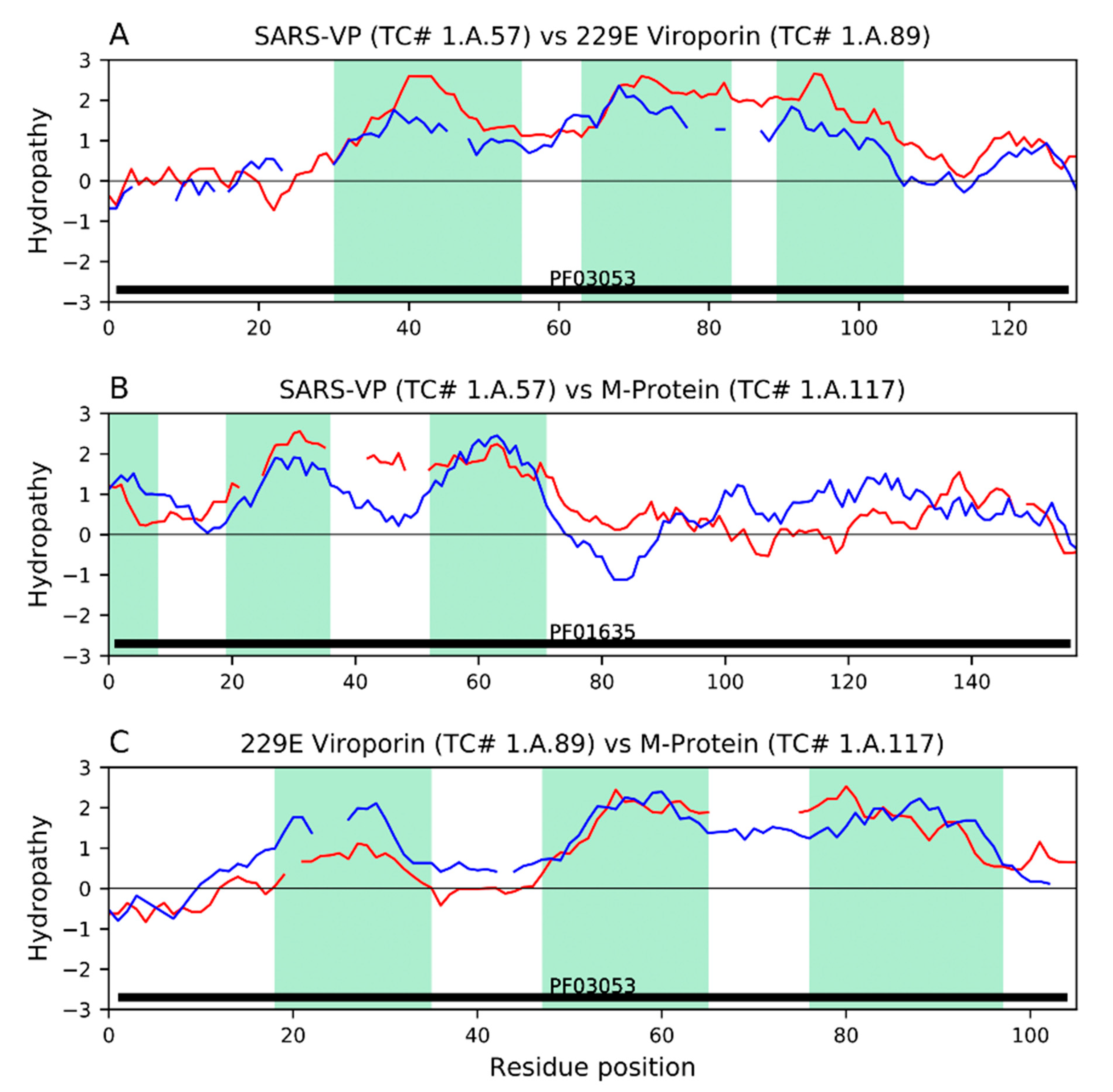{kind=link}
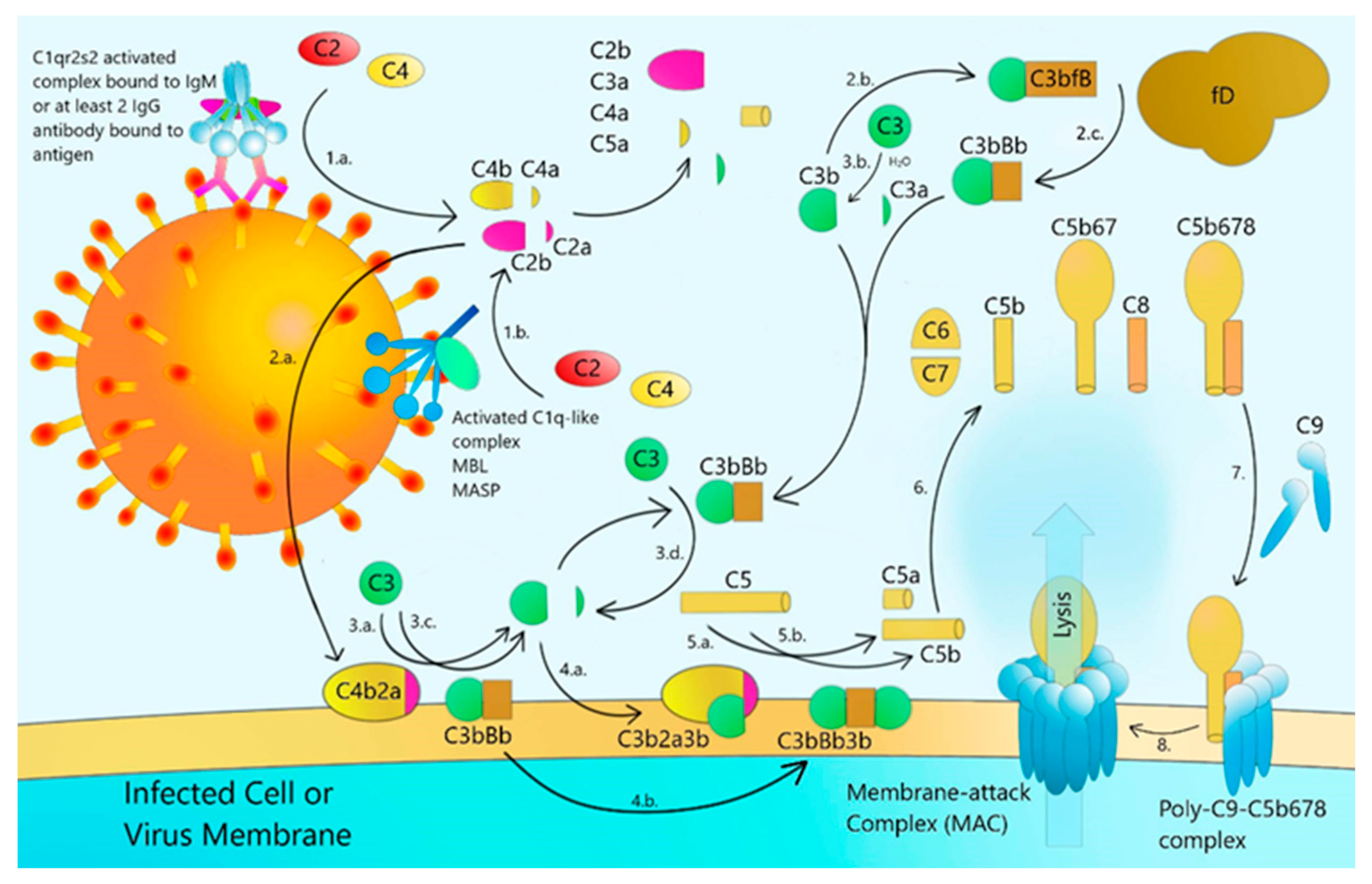{kind=link}
| Nsp1 | Likely induces cell arrest in the G0/G1 phase and interferes with type I IFNs [70]. | Nsp15 | Endoribonuclease activity; Immune evasion; degrades viral polyuridine sequences to prevent host antiviral detection [71]. |
| Nsp2 | Potential nonessential role in pathogenesis [72]. | Nsp16 | 2′O-methyl-transferase activity [73]. |
| Nsp3 | Membrane rearrangements for replication organelle formation. Viral proteolytic activity. Membrane anchoring of other viral proteins to perinuclear membranes; potential role in genome packaging. IFN antagonism. | S | Spike protein for binding/fusion/entry. Role in ER stress and syncytium formation. |
| Nsp4 | Membrane rearrangements for replication organelle formation. | E | Envelope protein essential for virion formation and exit. Viroporin and membrane rearranging activity. Role in NLRP3 inflammasome activation. |
| Nsp5 | Viral proteolytic activity. | M | Membrane/Matrix protein essential for virion formation. Binds to S, E and lipids to form the virial envelope-protein capsule. |
| Nsp6 | Essential for membrane rearrangements for SARS-CoV. May induce autophagy. | N | Nucleocapsid protein, binds to viral RNAs. Necessary for packaging genome and protection from host RNAases. |
| Nsp7 | Cofactor for RdRp complex [74]. | (ORF)3a | Viroporin activity similar to E. Interacts with S, E, and M. Activates NLRP3 inflammasomes [75,76]. |
| Nsp8 | RNA binding, RNA polymerase activity and essential RdRp complex cofactor protein [74]. | (ORF)3b | Putative function in upregulating cytokine secretion [75]. |
| Nsp9 | Novel ssRNA binding protein. May participate in RNA processing [77]. | (ORF)6a | Colocalizes with nsp3, nsp8 and RO-associated membranes. Upstream and downstream Type I IFN antagonist [75]. |
| Nsp10 | Replicative cofactor to nsp14 [78]. | (ORF)7a | Interacts with structural proteins M, E and S. May form a complex with 3a. Possibly essential for viral replication [75]. |
| Nsp11 | Unknown. | (ORF)7b | Possibly essential for viral replication. [75]. |
| Nsp12 | RNA-dependent RNA polymerase (RdRp) [79]. | (ORF)8a | Viroporin activity similar to 3a and E and activates NLRP3 inflammasome [80]. |
| Nsp13 | Cofactor for the RdRp complex. Viral helicase. Unwinding duplex RNA [81]. | (ORF)8b | Contributor to lysosomal stress, autophagy and inflammation; activation of NLRP3 inflammasome [82]. |
| Nsp14 | S-adenosyl methionine-dependent (N7-guanine)-methyl transferase, assembling cap1 structure at 5′ end of viral mRNA to promote translation and avoid antiviral detection. Proofreading of viral RNA transcripts [78]. | (ORF)9b | Suppresses innate immunity by usurping poly-C-binding protein 2 and HECT domain E3 ligase AIP4 to degrade MAVS/TRAF3/TRAF6 signalosome [83]. |
| Membrane Structures | SARS | MERS | MHV | HCoV-229E | PEDV | PDCov | IBV |
|---|---|---|---|---|---|---|---|
| DMV | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| DMS | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| CM | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| VP | ✓ | ✓ | ✓ | ||||
| Zippered ER | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| LVCV | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| GVP | ✓ | ✓ | ✓ | ||||
| TB | ✓ | ✓ | ✓ | ✓ | |||
| Interconnections | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ (Perinuclear DMVs) | ✓ (Perinuclear DMVs) |
| Association with Itself and All Other Structural Proteins to Assemble Virions | |
|---|---|
| 1. | Interference with the host immunological response by interferon (IFN) antagonism |
| 2. | Involvement of M in host cell cycle arrest |
| 3. | Induction of endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) |
| 4. | Coronavirus-induced autophagy and abortive apoptosis |
| 5. | Functioning of M as a protective antigen |
| 6. | Viroporin activities: The E and 3a proteins, and the ability of M to substitute for E |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, N.A.; Saier, M.H., Jr. The SARS-Coronavirus Infection Cycle: A Survey of Viral Membrane Proteins, Their Functional Interactions and Pathogenesis. Int. J. Mol. Sci. 2021, 22, 1308. https://doi.org/10.3390/ijms22031308
Wong NA, Saier MH Jr. The SARS-Coronavirus Infection Cycle: A Survey of Viral Membrane Proteins, Their Functional Interactions and Pathogenesis. International Journal of Molecular Sciences. 2021; 22(3):1308. https://doi.org/10.3390/ijms22031308
Chicago/Turabian StyleWong, Nicholas A., and Milton H. Saier, Jr. 2021. "The SARS-Coronavirus Infection Cycle: A Survey of Viral Membrane Proteins, Their Functional Interactions and Pathogenesis" International Journal of Molecular Sciences 22, no. 3: 1308. https://doi.org/10.3390/ijms22031308
APA StyleWong, N. A., & Saier, M. H., Jr. (2021). The SARS-Coronavirus Infection Cycle: A Survey of Viral Membrane Proteins, Their Functional Interactions and Pathogenesis. International Journal of Molecular Sciences, 22(3), 1308. https://doi.org/10.3390/ijms22031308