
Focusing on the Cell Type Specific Regulatory Actions of NLRX1



Abstract
:1. Introduction
1.1. NLRs: A Large Receptor Family with Versatile Roles
- (1)
- The most thoroughly characterized NLRs form large multiprotein complexes, termed inflammasomes, the activation of which leads to the cleavage of pro-IL-1ß and pro-IL-18 pro-inflammatory cytokines into their mature, biologically active form. Among NLRs, NLRP1, NLRP3, NLRP6 and NLRC4 are known to form inflammasomes.
- (2)
- Several NLRs act as positive or negative regulators of signal transduction. The so-called regulatory NLRs control various intracellular signaling cascades such as the nuclear factor-kappa B (NF-κB), the type I interferon (IFN) and the mitogen-activated protein kinase (MAPK) pathways initiated by other PRRs. This group is composed of several members such as NOD1, NOD2, NLRC5, nucleotide-binding domain and leucine-rich repeat–containing protein X1 (NLRX1), NLRP12 and NLRC3.
- (3)
- A specific subgroup of NLRs, including NLRC5 and the class II major histocompatibility complex transactivator (CIITA), can function as enhanceosomes and control the transcription of MHC I and II genes, respectively.
- (4)
- Upon bacterial sensing, some NLRs, such as NOD1 and NOD2, are able to recruit the ATGL16L1 autophagy modulating protein to the cell membrane to initiate the formation of autophagosomes [4].
- (5)
- Reproductive NLRs (NLRP2, NLRP5, NLRP7) control embryogenesis and reproduction. The function of these NLRs has been extensively reviewed in reference [5].
- (1)
- The NLRA subfamily possesses an acidic transactivation domain (TA) and consists of only one member, the CIITA.
- (2)
- The NLRB subfamily has a baculoviral inhibition of apoptosis protein repeat (BIR) domain. Similar to NLRA subfamily, NLRB subfamily has also only one member, the NLR family apoptosis inhibitory protein (NAIP).
- (3)
- The NLRC subfamily owns a caspase recruitment and activation domain (CARD), and in contrast to the above mentioned two families, the NLRC subfamily consists of six receptors: NOD1 (or NLRC1), NOD2 (or NLRC2), NLRC3, NLRC4, NLRC5, and NLRX1.
- (4)
- The NLRP subfamily has a pyrin domain (PYD), and comprising 14 members represents the largest subfamily of NLRs: NLRP1, NLRP2, NLRP3, NLRP4, NLRP7, NLRP12, NLRP5, NLRP6, NLRP8, NLRP9, NLRP10, NLRP11, NLRP13, and finally NLRP14 (reviewed in reference [4]).
1.2. Beyond the Inflammasomes: Regulatory NLRs
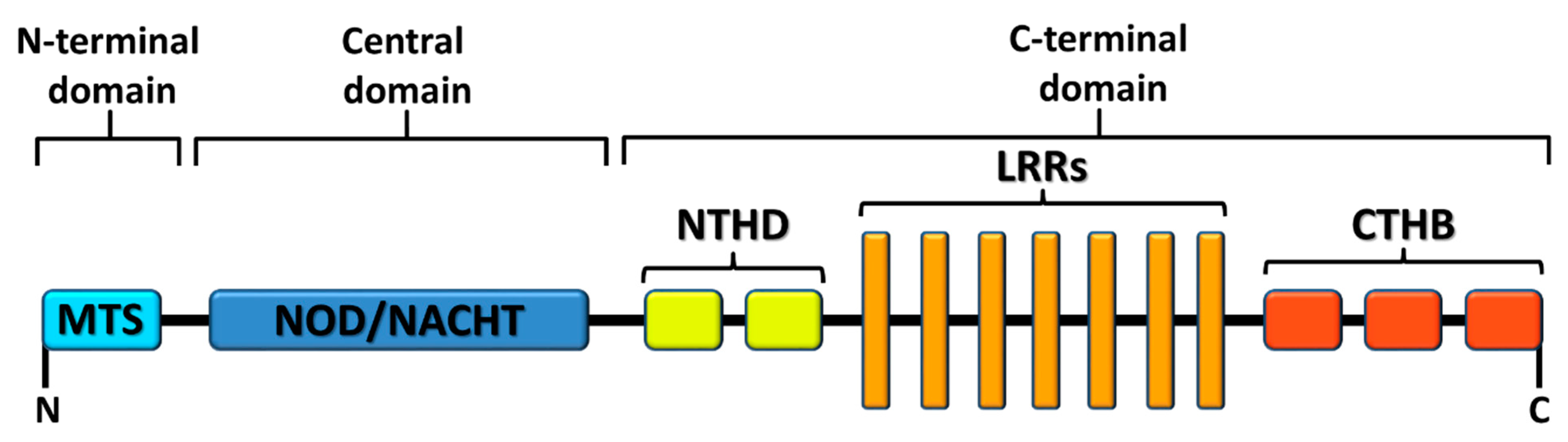
1.3. X Marks the Spot: NLRX1, a Mitochondria-Associated Regulatory NLR
2. Regulatory Mechanism behind the Multiple Actions of NLRX1
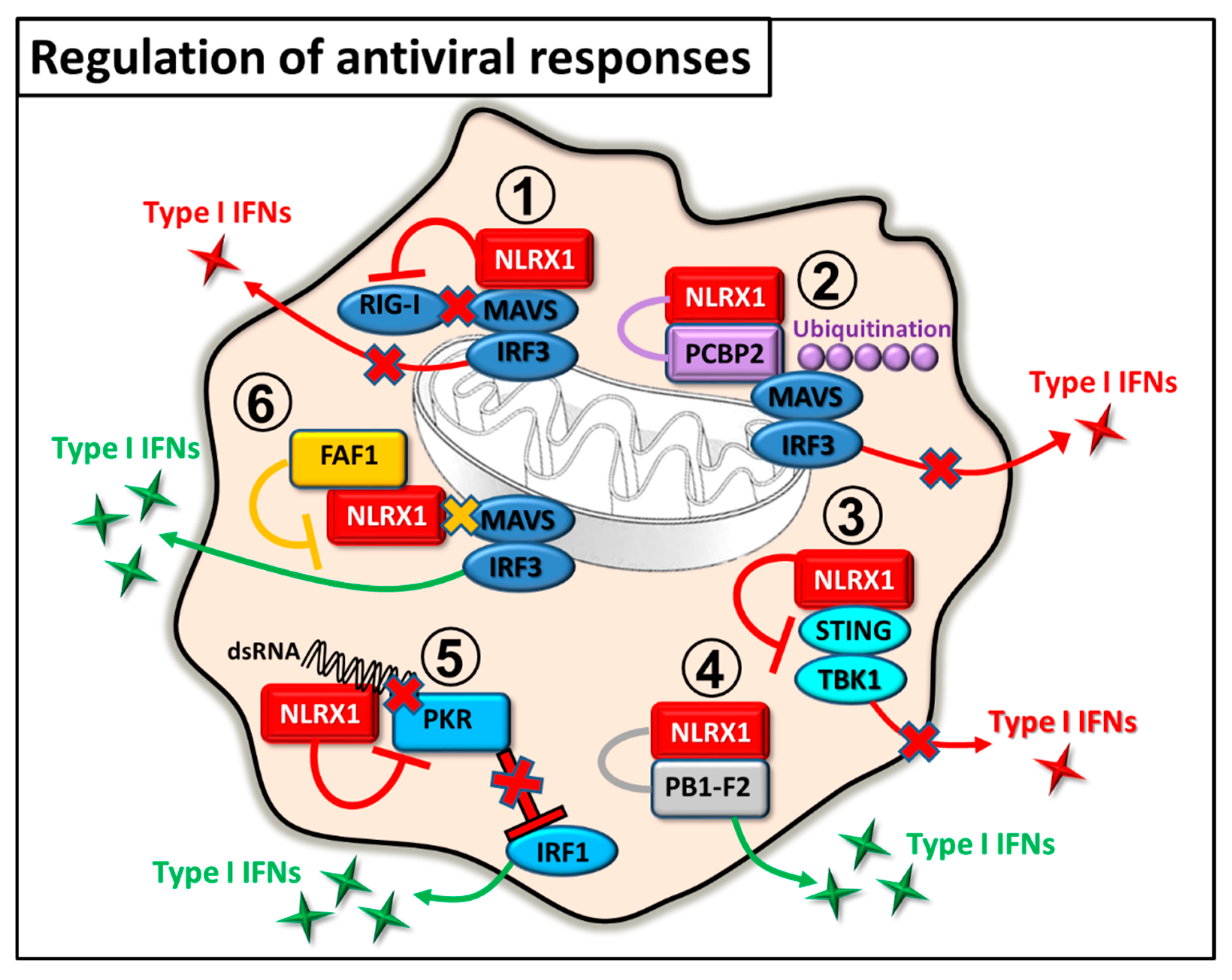
2.1. Regulation of Antiviral Immunity
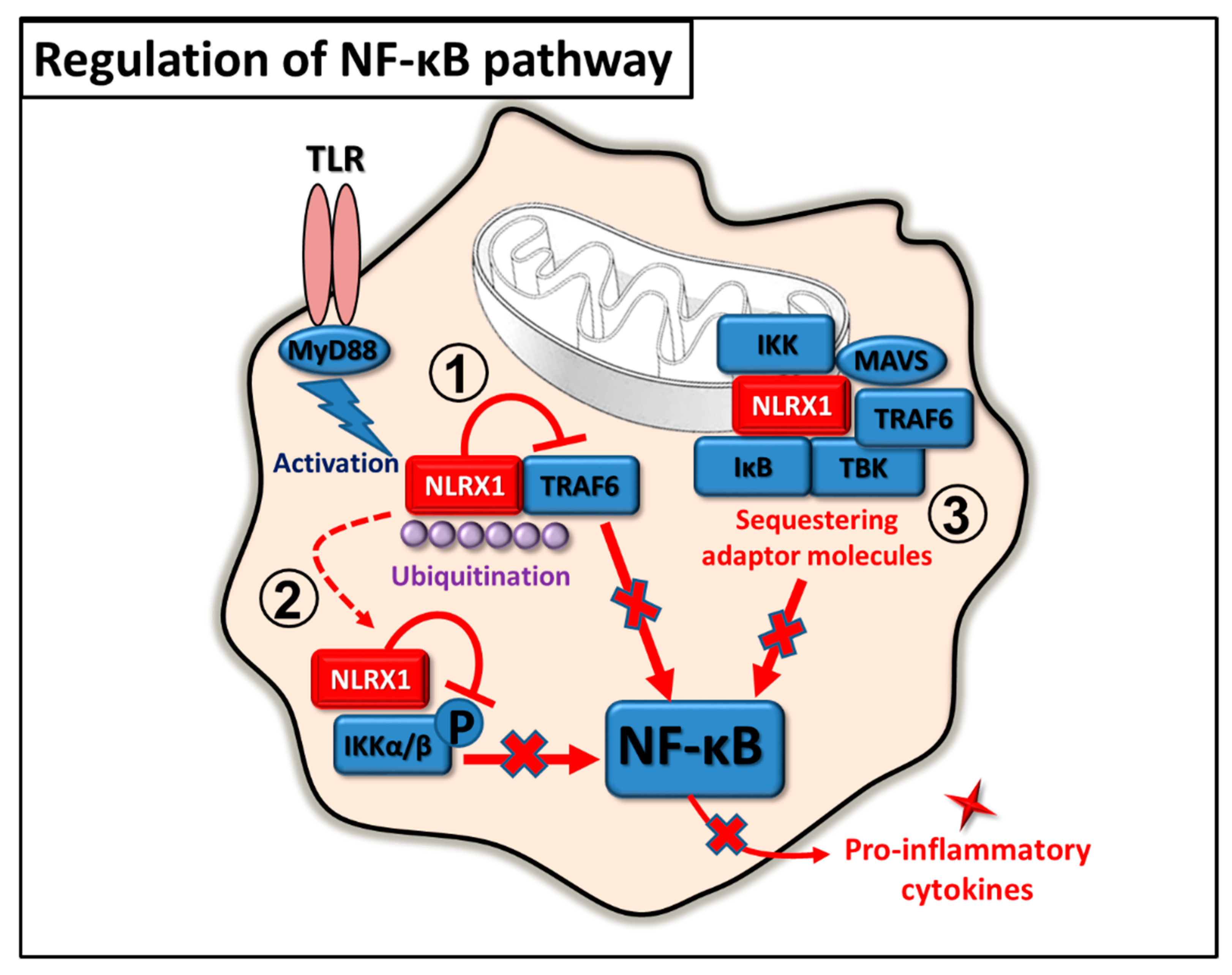
2.2. Regulation of NF-κB Pathway
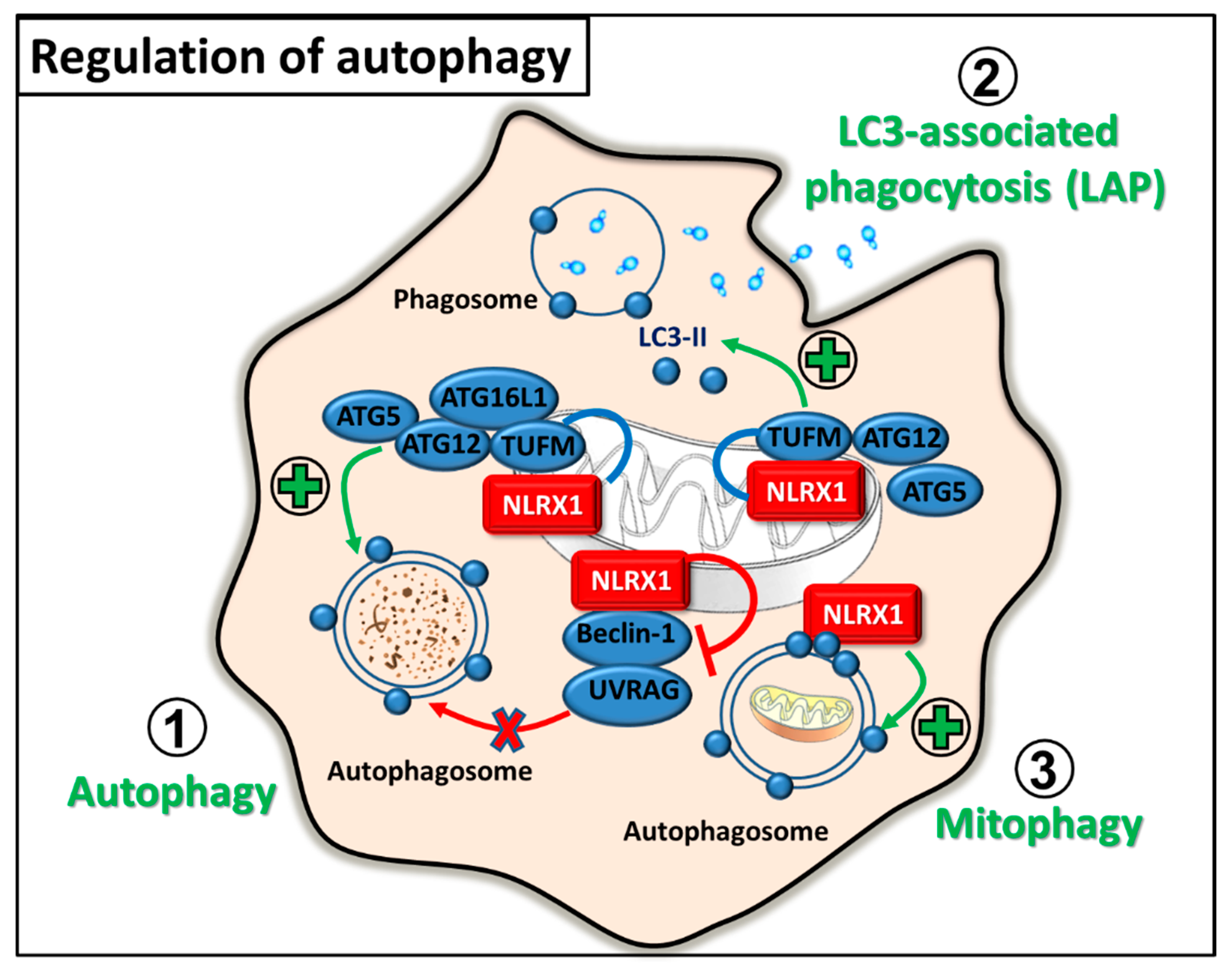
2.3. Regulation of Autophagy
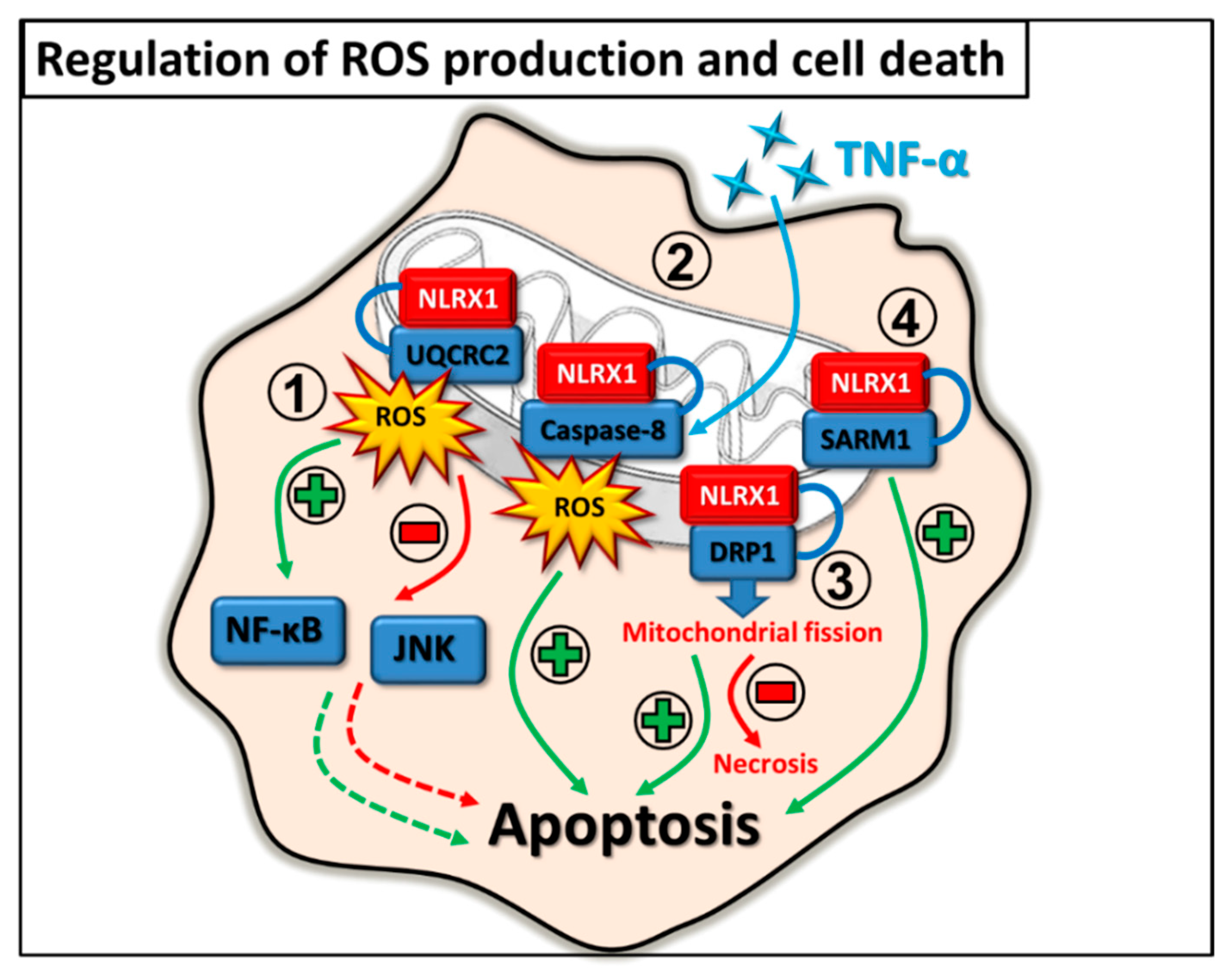
2.4. Regulation of ROS Production and Cell Death
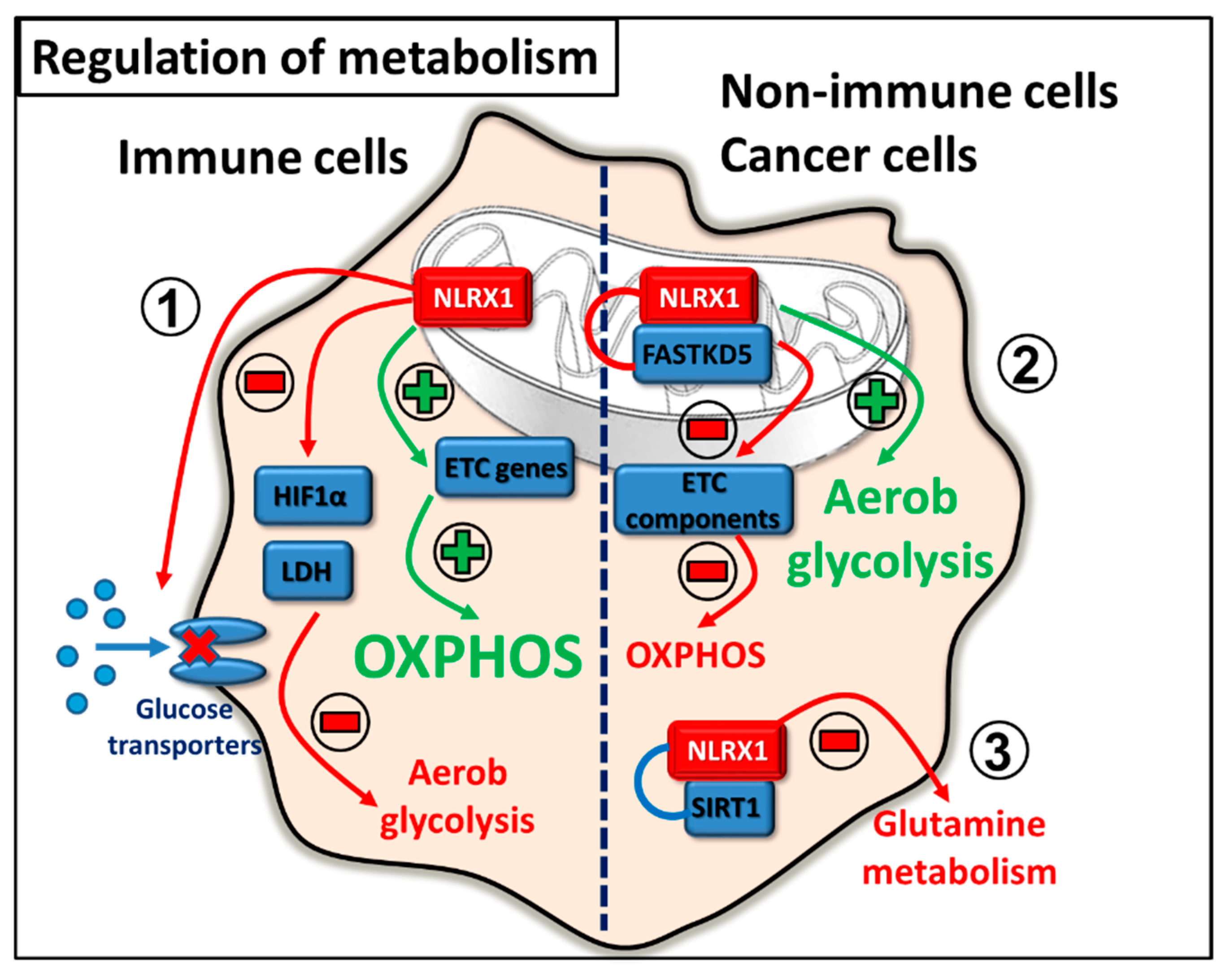
2.5. Regulation of Metabolism
3. Immune Cell Related Actions of NLRX1
3.1. Regulation of Myeloid Cell Functions by NLRX1
3.1.1. Macrophages
3.1.2. Dendritic Cells (DC)
3.1.3. Other Myeloid Cells
3.2. Regulation of Lymphoid Cell Functions by NLRX1
T Cells
3.3. Regulation of Peripheral Blood Mononuclear Cell (PBMC) Function by NLRX1
3.4. Regulation of Immune-Related Structural Cell Functions by NLRX1
Epithelial Cells and Fibroblasts
4. Discussion
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| APC | antigen presenting cell |
| ATG | autophagy-related protein |
| BBB | blood-brain barrier |
| BIR | baculoviral inhibition of apoptosis protein repeat |
| BMDCs | bone-marrow derived DCs |
| BMDM | bone marrow-derived macrophage |
| CARD | caspase recruitment and activation domain |
| cART | combined antiretroviral therapy |
| cDC | conventional DC |
| CIITA | Class II Major Histocompatibility Complex Transactivator |
| CLR | C-type lectin receptor |
| CNS | central nervous system |
| DAMP | damage-associated molecular pattern |
| DCs | dendritic cells |
| DHA | docosahexaenoic acid |
| DRP1 | dynamin-related protein 1 |
| DSS | dextran sodium sulfate |
| EAE | experimental autoimmune encephalomyelitis |
| EMCV | Encephalomyocarditis virus |
| ERK | extracellular signal-regulated kinase |
| ETC | electron transport chain |
| EYA | Eyes absent |
| FAF1 | FAS-associated factor-1 |
| FASTKD5 | Fas-activated serine-threonine kinase family protein-5 |
| GAS | Group A Streptococcus |
| GC | gastric cancer |
| G-CSF | granulocyte colony-stimulating factor |
| HCV | hepatitis C virus |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| HIV | human immunodeficiency virus |
| HPV16 | human papillomavirus 16 |
| IBD | inflammatory bowel disease |
| IFN | interferon |
| IRF | IFN regulatory factor |
| JNK | c-Jun N-terminal kinase |
| LAP | LC3-associated phagocytosis |
| LC3 | microtubule-associated protein 1 light chain 3 |
| LDH | lactate dehydrogenase |
| LDL | low-density lipoprotein |
| LIR | LC3-interacting region |
| LLO | listeriolysin O |
| LPS | lipopolysaccharide |
| LRR | leucine-rich repeat |
| MAPK | mitogen-activated protein kinase |
| MAVS | mitochondrial antiviral signaling protein |
| MCP1 | monocyte chemoattractant protein 1 |
| MDA5 | melanoma differentiation-associated protein 5, |
| MEF | mouse embryonic fibroblast |
| moDC | monocyte-derived DC |
| MS | multiple sclerosis |
| mtROS | mitochondrial reactive oxygen species |
| MTS | mitochondrial targeting sequence |
| NEMO | NF-kappa-B essential modulator |
| NF-κB | nuclear factor-kappa B |
| NK | natural killer |
| NLR | NOD-like receptors |
| NOD | nucleotide-binding oligomerization domain |
| OXPHOS | oxidative phosphorylation |
| PAMP | pathogen-associated molecular patterns |
| PB1-F2 | polymerase basic protein 1-frame 2 |
| PBMC | peripheral blood mononuclear cell |
| PCBP2 | poly(rC) binding protein 2 |
| pDC | plasmacytoid DCs |
| PKR | protein kinase R |
| poly (I:C) | polyinosinic:polycytidylic acid |
| PRR | pattern recognition receptor |
| PUA | punicic acid |
| PYD | pyrin domain |
| RIG-1 | retinoic acid-inducible gene-1 |
| RIP2 | receptor-interacting-serine/threonine-protein kinase 2 |
| RLR | RIG-I-like receptor |
| RRMS | relapsing-remitting MS |
| RV | rhinovirus |
| SARM1 | Sterile Alpha and Toll/interleukin-1 receptor (TIR) Motif Containing 1 |
| SIRT1 | sirtuin 1 |
| SIV | simian immunodeficiency virus |
| SLE | systemic lupus erythematosus |
| STING | stimulator of interferon genes |
| SV5 | Simian virus 5 |
| TA | acidic transactivation domain |
| TBK1 | TANK-binding kinase 1 |
| Th | helper T cells |
| TIR | Toll/interleukin-1 receptor |
| TLR | Toll like receptor |
| TRAF | TNF receptor-associated factor |
| Treg | regulatory T cell |
| TUFM | Tu translation elongation factor |
| UC | ulcerative colitis |
| UVRAG | UV radiation resistance-associated gene |
| VSV | vesicular stomatitis virus |
| XIAP | X-linked inhibitor of apoptosis |
References
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Suresh, R.; Mosser, D.M. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 2013, 37, 284–291. [Google Scholar] [CrossRef]
- Bryant, C.E.; Monie, T.P. Mice, men and the relatives: Cross-species studies underpin innate immunity. Open Biol. 2012, 2, 120015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-like receptors: Versatile cytosolic sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gorp, H.; Kuchmiy, A.; Van Hauwermeiren, F.; Lamkanfi, M. NOD-like receptors interfacing the immune and reproductive systems. FEBS J. 2014, 281, 4568–4582. [Google Scholar] [CrossRef] [PubMed]
- Coutermarsh-Ott, S.; Eden, K.; Allen, I.C. Beyond the inflammasome: Regulatory NOD-like receptor modulation of the host immune response following virus exposure. J. Gen. Virol. 2016, 97, 825–838. [Google Scholar] [CrossRef]
- Zhang, L.; Mo, J.; Swanson, K.V.; Wen, H.; Petrucelli, A.; Gregory, S.M.; Zhang, Z.; Schneider, M.; Jiang, Y.; Fitzgerald, K.A.; et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity 2014, 40, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.M.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Benko, S.; Magalhaes, J.G.; Philpott, D.J.; Girardin, S.E. NLRC5 limits the activation of inflammatory pathways. J. Immunol. 2010, 185, 1681–1691. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Zhu, L.; Xia, X.; Wang, H.Y.; Legras, X.; Hong, J.; Ji, J.; Shen, P.; Zheng, S.; Chen, Z.J.; et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 2010, 141, 483–496. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Cui, J.; Li, Q.; Zou, J.; Wang, H.Y.; Wang, R.F. Enhanced TLR-induced NF-kappaB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res. 2012, 22, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Fekete, T.; Bencze, D.; Szabo, A.; Csoma, E.; Biro, T.; Bacsi, A.; Pazmandi, K. Regulatory NLRs Control the RLR-Mediated Type I Interferon and Inflammatory Responses in Human Dendritic Cells. Front. Immunol. 2018, 9, 2314. [Google Scholar] [CrossRef] [Green Version]
- Arnoult, D.; Soares, F.; Tattoli, I.; Castanier, C.; Philpott, D.J.; Girardin, S.E. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J. Cell Sci. 2009, 122, 3161–3168. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.B.; Bergstralh, D.T.; Duncan, J.A.; Lei, Y.; Morrison, T.E.; Zimmermann, A.G.; Accavitti-Loper, M.A.; Madden, V.J.; Sun, L.; Ye, Z.; et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008, 451, 573–577. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Yoon, S.I.; Wilson, I.A. Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity 2012, 36, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Reubold, T.F.; Hahne, G.; Wohlgemuth, S.; Eschenburg, S. Crystal structure of the leucine-rich repeat domain of the NOD-like receptor NLRP1: Implications for binding of muramyl dipeptide. FEBS Lett. 2014, 588, 3327–3332. [Google Scholar] [CrossRef]
- Nagai-Singer, M.A.; Morrison, H.A.; Allen, I.C. NLRX1 Is a Multifaceted and Enigmatic Regulator of Immune System Function. Front. Immunol. 2019, 10, 2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, R.J.; Booty, L.M. NLR in eXile: Emerging roles of NLRX1 in immunity and human disease. Immunology 2020. [Google Scholar] [CrossRef]
- Allen, I.C.; Moore, C.B.; Schneider, M.; Lei, Y.; Davis, B.K.; Scull, M.A.; Gris, D.; Roney, K.E.; Zimmermann, A.G.; Bowzard, J.B.; et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity 2011, 34, 854–865. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Xue, B.; Liu, C.; Wang, X.; Tian, R.; Xie, Q.; Guo, M.; Li, G.; Yang, D.; Zhu, H. NLRX1 Mediates MAVS Degradation To Attenuate the Hepatitis C Virus-Induced Innate Immune Response through PCBP2. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Barouch, D.H.; Ghneim, K.; Bosche, W.J.; Li, Y.; Berkemeier, B.; Hull, M.; Bhattacharyya, S.; Cameron, M.; Liu, J.; Smith, K.; et al. Rapid Inflammasome Activation following Mucosal SIV Infection of Rhesus Monkeys. Cell 2016, 165, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, M.E.; Nikapitiya, C.; Kim, T.H.; Uddin, M.B.; Lee, H.C.; Kim, E.; Ma, J.Y.; Jung, J.U.; Kim, C.J.; et al. FAS-associated factor-1 positively regulates type I interferon response to RNA virus infection by targeting NLRX1. PLoS Pathog. 2017, 13, e1006398. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Konig, R.; Deng, M.; Riess, M.; Mo, J.; Zhang, L.; Petrucelli, A.; Yoh, S.M.; Barefoot, B.; Samo, M.; et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe 2016, 19, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Lenarcic, E.M.; Yamane, D.; Wauthier, E.; Mo, J.; Guo, H.; McGivern, D.R.; Gonzalez-Lopez, O.; Misumi, I.; Reid, L.M.; et al. NLRX1 promotes immediate IRF1-directed antiviral responses by limiting dsRNA-activated translational inhibition mediated by PKR. Nat. Immunol. 2017, 18, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Jaworska, J.; Coulombe, F.; Downey, J.; Tzelepis, F.; Shalaby, K.; Tattoli, I.; Berube, J.; Rousseau, S.; Martin, J.G.; Girardin, S.E.; et al. NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc. Natl. Acad. Sci. USA 2014, 111, E2110–E2119. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Hopcraft, S.E.; Yang, F.; Petrucelli, A.; Guo, H.; Ting, J.P.; Dittmer, D.P.; Damania, B. NLRX1 negatively modulates type I IFN to facilitate KSHV reactivation from latency. PLoS Pathog. 2017, 13, e1006350. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Cui, J.; Wang, H.Y.; Zhu, L.; Matsueda, S.; Wang, Q.; Yang, X.; Hong, J.; Songyang, Z.; Chen, Z.J.; et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity 2011, 34, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.; Wu, S.; Raju, R. NLRX1 Regulation Following Acute Mitochondrial Injury. Front. Immunol. 2019, 10, 2431. [Google Scholar] [CrossRef]
- Koblansky, A.A.; Truax, A.D.; Liu, R.; Montgomery, S.A.; Ding, S.; Wilson, J.E.; Brickey, W.J.; Muhlbauer, M.; McFadden, R.M.; Hu, P.; et al. The Innate Immune Receptor NLRX1 Functions as a Tumor Suppressor by Reducing Colon Tumorigenesis and Key Tumor-Promoting Signals. Cell Rep. 2016, 14, 2562–2575. [Google Scholar] [CrossRef] [Green Version]
- Coutermarsh-Ott, S.; Simmons, A.; Capria, V.; LeRoith, T.; Wilson, J.E.; Heid, B.; Philipson, C.W.; Qin, Q.; Hontecillas-Magarzo, R.; Bassaganya-Riera, J.; et al. NLRX1 suppresses tumorigenesis and attenuates histiocytic sarcoma through the negative regulation of NF-kappaB signaling. Oncotarget 2016, 7, 33096–33110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Wen, H.; Yu, Y.; Taxman, D.J.; Zhang, L.; Widman, D.G.; Swanson, K.V.; Wen, K.W.; Damania, B.; Moore, C.B.; et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 2012, 36, 933–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Kansy, B.A.; Li, J.; Cong, L.; Liu, Y.; Trivedi, S.; Wen, H.; Ting, J.P.; Ouyang, H.; Ferris, R.L. EGFR-targeted mAb therapy modulates autophagy in head and neck squamous cell carcinoma through NLRX1-TUFM protein complex. Oncogene 2016, 35, 4698–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Donnelly, C.R.; Gong, W.; Heath, B.R.; Hao, Y.; Donnelly, L.A.; Moghbeli, T.; Tan, Y.S.; Lin, X.; Bellile, E.; et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Investig. 2020, 130, 1635–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikawa, C.; Nakajima, S.; Karimine, M.; Nozawa, T.; Minowa-Nozawa, A.; Toh, H.; Yamada, S.; Nakagawa, I. NLRX1 Negatively Regulates Group A Streptococcus Invasion and Autophagy Induction by Interacting with the Beclin 1-UVRAG Complex. Front. Cell. Infect. Microbiol. 2018, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.H.; Liu, C.Y.; Wu, S.Y.; Chen, W.Y.; Chang, T.H.; Kan, H.W.; Hsieh, S.T.; Ting, J.P.; Wu-Hsieh, B.A. NLRX1 Facilitates Histoplasma capsulatum-Induced LC3-Associated Phagocytosis for Cytokine Production in Macrophages. Front. Immunol. 2018, 9, 2761. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, Y.; Qiu, X.; Wang, G.; Hu, Z.; Chen, S.; Wu, Z.; Yuan, N.; Gao, H.; Wang, J.; et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 2019, 20, 433–446. [Google Scholar] [CrossRef]
- Singh, K.; Roy, M.; Prajapati, P.; Lipatova, A.; Sripada, L.; Gohel, D.; Singh, A.; Mane, M.; Godbole, M.M.; Chumakov, P.M.; et al. NLRX1 regulates TNF-alpha-induced mitochondria-lysosomal crosstalk to maintain the invasive and metastatic potential of breast cancer cells. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 1460–1476. [Google Scholar] [CrossRef]
- Unger, B.L.; Ganesan, S.; Comstock, A.T.; Faris, A.N.; Hershenson, M.B.; Sajjan, U.S. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J. Virol. 2014, 88, 3705–3718. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Sater, A.A.; Said-Sadier, N.; Lam, V.M.; Singh, B.; Pettengill, M.A.; Soares, F.; Tattoli, I.; Lipinski, S.; Girardin, S.E.; Rosenstiel, P.; et al. Enhancement of reactive oxygen species production and chlamydial infection by the mitochondrial Nod-like family member NLRX1. J. Biol. Chem. 2010, 285, 41637–41645. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Sater, A.A.; Koo, E.; Hacker, G.; Ojcius, D.M. Inflammasome-dependent caspase-1 activation in cervical epithelial cells stimulates growth of the intracellular pathogen Chlamydia trachomatis. J. Biol. Chem. 2009, 284, 26789–26796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elwell, C.; Mirrashidi, K.; Engel, J. Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 2016, 14, 385–400. [Google Scholar] [CrossRef]
- Tattoli, I.; Carneiro, L.A.; Jehanno, M.; Magalhaes, J.G.; Shu, Y.; Philpott, D.J.; Arnoult, D.; Girardin, S.E. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. Embo Rep. 2008, 9, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Sun, G.; Yang, Q.; Chen, C.; Qi, Q.; Wang, H.; Li, J. NLRX1 accelerates cisplatin-induced ototoxity in HEI-OC1 cells via promoting generation of ROS and activation of JNK signaling pathway. Sci. Rep. 2017, 7, 44311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Yang, Q.; Cao, Z.; Li, H.; Yu, Z.; Zhang, G.; Sun, G.; Man, R.; Wang, H.; Li, J. Activation of NLRX1-mediated autophagy accelerates the ototoxic potential of cisplatin in auditory cells. Toxicol. Appl. Pharmacol. 2018, 343, 16–28. [Google Scholar] [CrossRef]
- Singh, K.; Poteryakhina, A.; Zheltukhin, A.; Bhatelia, K.; Prajapati, P.; Sripada, L.; Tomar, D.; Singh, R.; Singh, A.K.; Chumakov, P.M.; et al. NLRX1 acts as tumor suppressor by regulating TNF-alpha induced apoptosis and metabolism in cancer cells. Biochim. Et Biophys. Acta 2015, 1853, 1073–1086. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Zhao, Y.; She, J.; Zhu, Y.; Zhao, Y.; Liu, L.; Zhang, Y. NLRX1 alleviates lipopolysaccharide-induced apoptosis and inflammation in chondrocytes by suppressing the activation of NF-kappaB signaling. Int. Immunopharmacol. 2019, 71, 7–13. [Google Scholar] [CrossRef]
- Stokman, G.; Kors, L.; Bakker, P.J.; Rampanelli, E.; Claessen, N.; Teske, G.J.D.; Butter, L.; van Andel, H.; van den Bergh Weerman, M.A.; Larsen, P.W.B.; et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J. Exp. Med. 2017, 214, 2405–2420. [Google Scholar] [CrossRef]
- Imbeault, E.; Mahvelati, T.M.; Braun, R.; Gris, P.; Gris, D. Nlrx1 regulates neuronal cell death. Mol. Brain 2014, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Killackey, S.A.; Rahman, M.A.; Soares, F.; Zhang, A.B.; Abdel-Nour, M.; Philpott, D.J.; Girardin, S.E. The mitochondrial Nod-like receptor NLRX1 modifies apoptosis through SARM1. Mol. Cell. Biochem. 2019, 453, 187–196. [Google Scholar] [CrossRef]
- Leber, A.; Hontecillas, R.; Tubau-Juni, N.; Zoccoli-Rodriguez, V.; Hulver, M.; McMillan, R.; Eden, K.; Allen, I.C.; Bassaganya-Riera, J. NLRX1 Regulates Effector and Metabolic Functions of CD4(+) T Cells. J. Immunol. 2017, 198, 2260–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leber, A.; Hontecillas, R.; Zoccoli-Rodriguez, V.; Bienert, C.; Chauhan, J.; Bassaganya-Riera, J. Activation of NLRX1 by NX-13 Alleviates Inflammatory Bowel Disease through Immunometabolic Mechanisms in CD4(+) T Cells. J. Immunol. 2019, 203, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Soares, F.; Tattoli, I.; Rahman, M.A.; Robertson, S.J.; Belcheva, A.; Liu, D.; Streutker, C.; Winer, S.; Winer, D.A.; Martin, A.; et al. The mitochondrial protein NLRX1 controls the balance between extrinsic and intrinsic apoptosis. J. Biol. Chem. 2014, 289, 19317–19330. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Sripada, L.; Lipatova, A.; Roy, M.; Prajapati, P.; Gohel, D.; Bhatelia, K.; Chumakov, P.M.; Singh, R. NLRX1 resides in mitochondrial RNA granules and regulates mitochondrial RNA processing and bioenergetic adaptation. Biochim. Et Biophys. Acta. Mol. Cell Res. 2018, 1865, 1260–1276. [Google Scholar] [CrossRef]
- Kors, L.; Rampanelli, E.; Stokman, G.; Butter, L.M.; Held, N.M.; Claessen, N.; Larsen, P.W.B.; Verheij, J.; Zuurbier, C.J.; Liebisch, G.; et al. Deletion of NLRX1 increases fatty acid metabolism and prevents diet-induced hepatic steatosis and metabolic syndrome. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 1883–1895. [Google Scholar] [CrossRef]
- Leber, A.; Hontecillas, R.; Tubau-Juni, N.; Zoccoli-Rodriguez, V.; Abedi, V.; Bassaganya-Riera, J. NLRX1 Modulates Immunometabolic Mechanisms Controlling the Host-Gut Microbiota Interactions during Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 363. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Amrani, A.; Gris, D. NLRX1 Enhances Glutamate Uptake and Inhibits Glutamate Release by Astrocytes. Cells 2019, 8, 400. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Mosser, D.M.; Hamidzadeh, K.; Goncalves, R. Macrophages and the maintenance of homeostasis. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Edholm, E.S.; Rhoo, K.H.; Robert, J. Evolutionary Aspects of Macrophages Polarization. Results Probl. Cell Differ. 2017, 62, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Vazquez, J.; Tardivel, A.; Guarda, G.; Curran, J.; Tschopp, J. NLRX1/NOD5 deficiency does not affect MAVS signalling. Cell Death Differ. 2011, 18, 1387. [Google Scholar] [CrossRef] [PubMed]
- Soares, F.; Tattoli, I.; Wortzman, M.E.; Arnoult, D.; Philpott, D.J.; Girardin, S.E. NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate Immun. 2013, 19, 438–448. [Google Scholar] [CrossRef]
- Sun, Q.; Sun, L.; Liu, H.H.; Chen, X.; Seth, R.B.; Forman, J.; Chen, Z.J. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 2006, 24, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castano-Rodriguez, N.; Kaakoush, N.O.; Goh, K.L.; Fock, K.M.; Mitchell, H.M. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: A case-control study and gene expression analyses. PLoS ONE 2014, 9, e98899. [Google Scholar] [CrossRef] [PubMed]
- Philipson, C.W.; Bassaganya-Riera, J.; Viladomiu, M.; Kronsteiner, B.; Abedi, V.; Hoops, S.; Michalak, P.; Kang, L.; Girardin, S.E.; Hontecillas, R. Modeling the Regulatory Mechanisms by Which NLRX1 Modulates Innate Immune Responses to Helicobacter pylori Infection. PLoS ONE 2015, 10, e0137839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Hontecillas, R.; Abedi, V.; Kale, S.; Leber, A.; Heltzel, C.; Langowski, M.; Godfrey, V.; Philipson, C.; Tubau-Juni, N.; et al. Modeling-Enabled Characterization of Novel NLRX1 Ligands. PLoS ONE 2015, 10, e0145420. [Google Scholar] [CrossRef]
- Okabe, Y.; Sano, T.; Nagata, S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature 2009, 460, 520–524. [Google Scholar] [CrossRef]
- Nicola, A.M.; Albuquerque, P.; Martinez, L.R.; Dal-Rosso, R.A.; Saylor, C.; De Jesus, M.; Nosanchuk, J.D.; Casadevall, A. Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infect. Immun. 2012, 80, 3065–3076. [Google Scholar] [CrossRef] [Green Version]
- Kyrmizi, I.; Gresnigt, M.S.; Akoumianaki, T.; Samonis, G.; Sidiropoulos, P.; Boumpas, D.; Netea, M.G.; van de Veerdonk, F.L.; Kontoyiannis, D.P.; Chamilos, G. Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting dectin-1/Syk kinase signaling. J. Immunol. 2013, 191, 1287–1299. [Google Scholar] [CrossRef] [Green Version]
- Eitas, T.K.; Chou, W.C.; Wen, H.; Gris, D.; Robbins, G.R.; Brickey, J.; Oyama, Y.; Ting, J.P. The nucleotide-binding leucine-rich repeat (NLR) family member NLRX1 mediates protection against experimental autoimmune encephalomyelitis and represses macrophage/microglia-induced inflammation. J. Biol. Chem. 2014, 289, 4173–4179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2018, 9, 3176. [Google Scholar] [CrossRef] [PubMed]
- Kastelberg, B.; Tubau-Juni, N.; Ayubi, T.; Leung, A.; Leber, A.; Hontecillas, R.; Bassaganya-Riera, J.; Kale, S.D. NLRX1 is a key regulator of immune signaling during invasive pulmonary aspergillosis. PLoS Pathog. 2020, 16, e1008854. [Google Scholar] [CrossRef] [PubMed]
- Kors, L.; Butter, L.M.; Claessen, N.; Teske, G.J.D.; Girardin, S.E.; Florquin, S.; Leemans, J.C. NLRX1 is not involved in the host defense against Escherichia coli induced pyelonephritis. F1000Research 2019, 7. [Google Scholar] [CrossRef]
- Theus, M.H.; Brickler, T.; Meza, A.L.; Coutermarsh-Ott, S.; Hazy, A.; Gris, D.; Allen, I.C. Loss of NLRX1 Exacerbates Neural Tissue Damage and NF-kappaB Signaling following Brain Injury. J. Immunol. 2017, 199, 3547–3558. [Google Scholar] [CrossRef] [Green Version]
- Geering, B.; Stoeckle, C.; Conus, S.; Simon, H.U. Living and dying for inflammation: Neutrophils, eosinophils, basophils. Trends Immunol. 2013, 34, 398–409. [Google Scholar] [CrossRef]
- Zhao, Y.; Marion, T.N.; Wang, Q. Multifaceted Roles of Neutrophils in Autoimmune Diseases. J. Immunol. Res. 2019, 2019, 7896738. [Google Scholar] [CrossRef] [Green Version]
- Stone, K.D.; Prussin, C.; Metcalfe, D.D. IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Immunol. 2010, 125, S73–S80. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Gharagozloo, M.; Mahmoud, S.; Simard, C.; Yamamoto, K.; Bobbala, D.; Ilangumaran, S.; Smith, M.D.; Lamontagne, A.; Jarjoura, S.; Denault, J.B.; et al. NLRX1 inhibits the early stages of CNS inflammation and prevents the onset of spontaneous autoimmunity. PLoS Biol. 2019, 17, e3000451. [Google Scholar] [CrossRef] [Green Version]
- Koo, J.H.; Kim, D.H.; Cha, D.; Kang, M.J.; Choi, J.M. LRR domain of NLRX1 protein delivery by dNP2 inhibits T cell functions and alleviates autoimmune encephalomyelitis. Theranostics 2020, 10, 3138–3150. [Google Scholar] [CrossRef]
- Lim, S.; Kim, W.J.; Kim, Y.H.; Lee, S.; Koo, J.H.; Lee, J.A.; Yoon, H.; Kim, D.H.; Park, H.J.; Kim, H.M.; et al. dNP2 is a blood-brain barrier-permeable peptide enabling ctCTLA-4 protein delivery to ameliorate experimental autoimmune encephalomyelitis. Nat. Commun. 2015, 6, 8244. [Google Scholar] [CrossRef] [Green Version]
- Kleiveland, C.R. Peripheral Blood Mononuclear Cells. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., Lopez-Exposito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, Switzerland, 2015; Chapter 15; pp. 161–167. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.H.; Shu, D.H.; Zhen, Y.; Hilliard, B.; Priest, S.O.; Cesaroni, M.; Ting, J.P.; Cohen, P.L. Prion-like Aggregation of Mitochondrial Antiviral Signaling Protein in Lupus Patients Is Associated With Increased Levels of Type I Interferon. Arthritis Rheumatol. 2016, 68, 2697–2707. [Google Scholar] [CrossRef]
- Nasi, M.; De Biasi, S.; Bianchini, E.; Digaetano, M.; Pinti, M.; Gibellini, L.; Pecorini, S.; Carnevale, G.; Guaraldi, G.; Borghi, V.; et al. Analysis of inflammasomes and antiviral sensing components reveals decreased expression of NLRX1 in HIV-positive patients assuming efficient antiretroviral therapy. Aids 2015, 29, 1937–1941. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.B.; Cowley, C.J.; Fuchs, E. Epithelial cells: Liaisons of immunity. Curr. Opin. Immunol. 2020, 62, 45–53. [Google Scholar] [CrossRef]
- Bautista-Hernandez, L.A.; Gomez-Olivares, J.L.; Buentello-Volante, B.; Bautista-de Lucio, V.M. Fibroblasts: The Unknown Sentinels Eliciting Immune Responses Against Microorganisms. Eur. J. Microbiol. Immunol. 2017, 7, 151–157. [Google Scholar] [CrossRef]
- Ling, A.; Soares, F.; Croitoru, D.O.; Tattoli, I.; Carneiro, L.A.; Boniotto, M.; Benko, S.; Philpott, D.J.; Girardin, S.E. Post-transcriptional inhibition of luciferase reporter assays by the Nod-like receptor proteins NLRX1 and NLRC3. J. Biol. Chem. 2012, 287, 28705–28716. [Google Scholar] [CrossRef] [Green Version]
- Hung, S.C.; Huang, P.R.; Almeida-da-Silva, C.L.C.; Atanasova, K.R.; Yilmaz, O.; Ojcius, D.M. NLRX1 modulates differentially NLRP3 inflammasome activation and NF-kappaB signaling during Fusobacterium nucleatum infection. Microbes Infect. 2018, 20, 615–625. [Google Scholar] [CrossRef]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, V.; Rivera, A. First Line of Defense: Innate Cell-Mediated Control of Pulmonary Aspergillosis. Front. Microbiol. 2016, 7, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Amarante, M.K.; Watanabe, M.A. Toll-like receptor 3: Involvement with exogenous and endogenous RNA. Int. Rev. Immunol. 2010, 29, 557–573. [Google Scholar] [CrossRef]
- Tang, F.; Du, Q.; Liu, Y.J. Plasmacytoid dendritic cells in antiviral immunity and autoimmunity. Sci. China Life Sci. 2010, 53, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leber, A.; Hontecillas, R.; Zoccoli-Rodriguez, V.; Ehrich, M.; Chauhan, J.; Bassaganya-Riera, J. Exploratory studies with NX-13: Oral toxicity and pharmacokinetics in rodents of an orally active, gut-restricted first-in-class therapeutic for IBD that targets NLRX1. Drug Chem. Toxicol. 2019. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, S.; Li, F.; Qin, L. NLRX1 attenuates apoptosis and inflammatory responses in myocardial ischemia by inhibiting MAVS-dependent NLRP3 inflammasome activation. Mol. Immunol. 2016, 76, 90–97. [Google Scholar] [CrossRef]
- Peng, L.; Zhou, Y.; Jiang, N.; Wang, T.; Zhu, J.; Chen, Y.; Li, L.; Zhang, J.; Yu, S.; Zhao, Y. DJ-1 exerts anti-inflammatory effects and regulates NLRX1-TRAF6 via SHP-1 in stroke. J. Neuroinflamm. 2020, 17, 81. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Wang, X.; Edwards, S.; Dai, M.; Li, J.; Wu, L.; Xu, R.; Han, J.; Yuan, H. NLRX1 knockout aggravates lipopolysaccharide (LPS)-induced heart injury and attenuates the anti-LPS cardioprotective effect of CYP2J2/11,12-EET by enhancing activation of NF-kappaB and NLRP3 inflammasome. Eur. J. Pharmacol. 2020, 881, 173276. [Google Scholar] [CrossRef]
- Kang, M.J.; Yoon, C.M.; Kim, B.H.; Lee, C.M.; Zhou, Y.; Sauler, M.; Homer, R.; Dhamija, A.; Boffa, D.; West, A.P.; et al. Suppression of NLRX1 in chronic obstructive pulmonary disease. J. Clin. Investig. 2015, 125, 2458–2462. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Zhou, Z.; Han, Y.; Wen, Z.; Guo, C.; Huang, S.; Xiao, D.; Ye, X.; Ou, M.; Huang, C.; et al. Interactions of TRAF6 and NLRX1 gene polymorphisms with environmental factors on the susceptibility of type 2 diabetes mellitus vascular complications in a southern Han Chinese population. J. Diabetes Its Complicat. 2017, 31, 1652–1657. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I. Regulation of Myeloid Cell Functions by NLRX1 | ||||
|---|---|---|---|---|
| Macrophages | ||||
| Cell Type | Model | Observed Effects | Mechanism | Ref. |
| NLRX1−/− mouse BMDM | poly (I:C) challenge, Legionella pneumophila or Listeria monocytogenes infection | NLRX1 deficiency has no effect on IFNβ secretion | - | [19] |
| NLRX1−/− mouse BMDM | Sendai virus infection | NLRX1 deficiency has no effect on IFNβ gene expression mediated by MAVS dependent signaling | - | [62] |
| NLRX1−/− mouse BMDM | Sendai virus infection | NLRX1 deficiency has no effect on IFNα/β, CXCL10, STAT2, IRF7, IL-6 and KC gene expression mediated by MAVS dependent signaling | - | [63] |
| NLRX1−/− mouse BMDM | LPS treatment | NLRX1 deficiency increases IFNβ, IL-6 and IL-1β production mediated by NF-kB dependent signaling | NLRX1 binds to TRAF6 and inhibits NF-kB signaling (demonstrated on MEFs) | [19] |
| human THP-1 | Helicobacter pylori infection | Downregulation of NLRX1 expression upon infection, which is associated with enhanced NF-κB signaling | NLRX1 interferes with NF-κB pathway | [65] |
| mouse BMDM (in silico) | Helicobacter pylori infection | Infection induced downregulation of NLRX1 is associated with increased NF-κB signaling | NLRX1 interferes with NF-κB pathway | [66] |
| NLRX1−/− mouse BMDM | Helicobacter pylori infection | NLRX1 deficiency increases IFNγ and ROS production | NLRX1 interferes with NF-κB pathway | [66] |
| mouse BMDM | PUA or DHA treatments together with LPS | PUA and DHA suppresses the LPS-activated NF-κB signaling | PUA or DHA binds to NLRX1 and inhibits NF-κB signaling | [67] |
| mouse fetal liver macrophages | EYA4 overexpression and poly(I:C) challenge | Overexpression of EYA4 enhances IFNβ expression | EYA4 binds to NLRX1 and enhances IRF3 signaling | [68] |
| NLRX1−/− mouse BMDM | Influenza A virus infection | NLRX1 deficiency lowers IFNβ secretion | NLRX1 binds to viral PB1-F2 | [26] |
| NLRX1−/− mouse BMDM | Influenza A virus infection | NLRX1 deficiency enhances mitochondrial damages and apoptosis | NLRX1 binds to viral PB1-F2 | [26] |
| FAF1−/− mouse BMDM, mouse RAW264.7, human THP-1 | VSV or H1N1 infections | FAF1 deficiency decreases IFNβ and IL-6 secretion and increases virus replication | FAF1 binds to NLRX1 and competes with MAVS | [23] |
| NLRX1−/− human monocytes derived macrophages, THP-1 | HIV or DNA virus infection | NLRX1 deficiency increases STING dependent type I IFN responses | NLRX1 binds to STING and inhibits STING dependent TBK1 activation | [24] |
| NLRX1−/− mouse peritoneal macrophages | VSV infection | NLRX1 deficiency enhances IFNβ secretion | NLRX1 binds to TUFM/ATG5-ATG12 complex (demonstrated on MEFs) | [32] |
| NLRX1−/− mouse peritoneal macrophages | VSV infection | NLRX1 deficiency reduces autophagy | NLRX1 binds to TUFM/ATG5-ATG12 complex (demonstrated on MEFs) | [32] |
| primary mouse peritoneal macrophages | Histoplasma capsulatum infection | Infection induces LC3-associated phagocytosis (LAP) via NLRX1 | NLRX1 binds to TUFM/ATG5-ATG12 complex and promotes LC3-II incorporation into the phagosomes | [36] |
| NLRX1−/− primary mouse peritoneal macrophages | Histoplasma capsulatum infection | NLRX1 deficiency decreases MAPK pathway dependent cytokine production | NLRX1 positively regulates MAPK pathway | [36] |
| NLRX1−/− primary mouse peritoneal macrophages | Histoplasma capsulatum infection | NLRX1 deficiency has no effect on NF-κB signaling | - | [36] |
| NLRX1−/− mouse peritoneal macrophages | Listeria monocytogenes infection | NLRX1 deficiency reduces mitophagy and inhibits Listeria growth | LLO of Listeria induces NLRX1 oligomerization and LC3 binding via the LIR domain of NLRX1 | [37] |
| NLRX1−/− mouse peritoneal macrophages | Listeria monocytogenes infection | NLRX1 deficiency increases mtROS production | undefined | [37] |
| NLRX1−/− mouse peritoneal macrophages | Listeria monocytogenes infection | NLRX1 deficiency has no effect on NF-kB signaling and IFNβ response | - | [37] |
| NLRX1−/− mouse microglia of spinal cord | EAE | NLRX1 deficiency elevates MHC II expression | undefined | [71] |
| NLRX1−/− microglia from neonatal mice | LPS/IFNγ treatment | NLRX1 deficiency elevates IL-6, CCL2 secretion, and increases NOS2 and MHCII expression | undefined | [71] |
| NLRX1−/− mouse BMDM | mouse model of urethane-induced tumorigenesis | NLRX1 deficiency increases CCL2 and G-CSF expression | undefined | [31] |
| Dendritic Cells | ||||
| Cell Type | Model | Observed Effects | Mechanism | Ref. |
| NLRX1 silenced human pDC | 5′ppp-dsRNA or poly (I:C) treatment, VSV infection | Silencing of NLRX1 enhances type I IFN secretion | NLRX1 interferes with RLR-mediated antiviral signaling | [12] |
| NLRX1 silenced human pDC | 5′ppp-dsRNA or poly (I:C) treatment, VSV infection, | Silencing of NLRX1 has no effect on pro-inflammatory cytokine production induced by RLR-mediated NF-κB activation | - | [12] |
| NLRX1 silenced human pDC | CpG-B, Imiquimod and PAM3CSK4 treatments | Silencing of NLRX1 has no effect on pro-inflammatory cytokine production induced by TLR-mediated NF-κB activation | - | [12] |
| NLRX1 silenced human moDC | 5′ppp-dsRNA or poly (I:C) treatment, VSV infection | Silencing of NLRX1 enhances type I IFN secretion | NLRX1 interferes with RLR-mediated antiviral signaling | [12] |
| NLRX1 silenced human moDC | 5′ppp-dsRNA or poly (I:C) treatment, VSV infection | Silencing of NLRX1 increases inflammatory cytokine production induced by RLR-mediated NF-κB activation | NLRX1 interferes with RLR-mediated NF-κB signaling | [12] |
| NLRX1−/− mouse pDC | R848, CpG and poly (I:C) treatments | NLRX1 deficiency has no effect on TLR-mediated pathways | - | [19] |
| FAF1−/− mouse BMDC | poly (I:C) treatment, VSV and influenza virus infections | FAF1 deficiency decreases IFNβ and IL-6 secretion and increases virus replication | FAF1 binds to NLRX1 and competes with MAVS | [23] |
| NLRX1−/− mouse CD103+ DC | mouse models of invasive pulmonary aspergillosis | NLRX1 deficiency increases the promotion of IL-4-producing DCs and Th2 responses | NLRX1 interferes with JNK/JunB signaling pathway | [73] |
| Other Myeloid Cells | ||||
| Cell Type | Model | Observed Effects | Mechanism | Ref. |
| NLRX1−/− mouse granulocytes, monocytes | Escherichia coli induced pyelonephritis | NLRX1 deficiency has no effects | - | [74] |
| NLRX1−/− mouse CD11b + cells | murine model of traumatic brain injury | NLRX1 deficiency results in larger cortical lesions, more infiltrating cells and upregulated NF-κB activity | NLRX1 interferes with NF-κB signaling | [75] |
| NLRX1−/− mouse CD11b+ cells | murine model of traumatic brain injury | NLRX1 deficiency enhances apoptosis | NLRX1 interferes with apoptotic pathway | [75] |
| II. Regulation of Lymphoid Cell Functions by NLRX1 | ||||
|---|---|---|---|---|
| T Cells | ||||
| Cell Type | Model | Observed Effects | Mechanism | Ref. |
| NLRX1−/− mouse CD4+ T cells | Cellular-, DSS-, and Citrobacter rodentium-induced IBD | NLRX1 deficiency enhances the proliferation and differentiation of Th1 and Th17 cells | NLRX1 deficiency promotes metabolic shift towards aerobic glycolysis | [51] |
| mouse CD4+ T cells | DSS-induced IBD, administration of the NLRX1 activator, NX-13 | NLRX1 activation decreases the differentiation of naïve CD4+ T cells into Th1 and Th17 cells | NLRX1 attenuates aerobic glycolysis and promotes OXPHOS | [52] |
| NLRX1−/− mouse CD4+ T cells | EAE (NLRX1 deficient myelin-specific TCR transgenic 2D2 mice) | NLRX1 deficiency promotes the differentiation of autoreactive Th1 and Th17 cells | NLRX inhibits the differentiation of autoreactive T-cells and the activation and migration of myeloid cells | [80] |
| mouse CD4+ T cells | EAE, administration of the dNP2-LRR fusion protein | Administration of NLRX1 fusion protein inhibits T cell activation or Th1 cell differentiation | NLRX1 interferes with NF-κB signaling via reducing the level of T-bet | [81] |
| III. Regulation of Peripheral Blood Mononuclear Cell (PBMC) Functions by NLRX1 | ||||
|---|---|---|---|---|
| Cell Type | Model | Observed Effects | Mechanism | Ref. |
| human PBMC | ulcerative colitis (UC), administration of the NLRX1 activator, NX-13 | NLRX1 activation decreases the number of IFNγ, TNFα and IL-4 positive cells and reduces pro-inflammatory cytokine and chemokine levels | NLRX1 interferes with NF-κB signaling | [52] |
| human PBMC | ulcerative colitis (UC), administration of the NLRX1 activator, NX-13 | NLRX1 activation decreases H2O2 triggered ROS production | undefined | [52] |
| human PBMC | relapsing-remitting MS (RRMS) disease | Increased NLRX1 expression is observed | Increased NLRX1 may serve as a negative feedback regulatory loop | [80] |
| human PBMC | SLE disease | No difference in NLRX1 expression between MAVS aggregate positive and negative patients | - | [84] |
| human PBMC | HIV infection | Decreased NLRX1 expression, which presumably serves as a viral escape mechanism | undefined | [85] |
| IV. Regulation of Immune-Related Structural Cell Functions by NLRX1 | ||||
|---|---|---|---|---|
| Epithelial Cells and Fibroblasts | ||||
| Cell Type | Model | Observed Effects | Mechanism | Ref. |
| NLRX1 silenced human airway epithelial cells | rhinovirus infection, poly (I:C) treatment | Silencing of NLRX1 abrogates virus induced epithelial barrier disruption and decreased ROS production | NLRX1 promotes mtROS production | [39] |
| NLRX1 silenced human HEK293T, HeLa | Sendai virus infection | Silencing of NLRX1 promotes type I IFN production and decreases viral replication | NLRX1 interferes with MAVS signaling and inhibits IRF3 dimer formation | [14] |
| NLRX1 silenced human HEK293T | Sendai virus infection | Silencing of NLRX1 has no effect on type I IFN response | - | [88] |
| NLRX1−/− mouse airway epithelial cells | mouse models of invasive pulmonary aspergillosis | NLRX1 deficiency enhances pulmonary inflammation and increases chemokine and cytokine production | NLRX1 impairs P38 phosphorylation | [73] |
| NLRX1−/− human BEAS-2B airway epithelial cells | Aspergillus fumigatus infection | NLRX1 deficiency enhances chemokine (CXCL8, CXCL1) and cytokine (IL-6) production | NLRX1 impairs P38 phosphorylation | [73] |
| human HEK293T | Overexpressed NLRX1, Shigella infection, TNFα treatment | Overexpression of NLRX1 facilitates NF-κB and JNK pathways | NLRX1 promotes ROS production | [43] |
| NLRX1 silenced human gingival epithelial cells | Fusobacterium nucleatum infection | Silencing of NLRX1 attenuates the NLRP3 inflammasome activity | NLRX1 promotes mtROS production | [89] |
| NLRX1 silenced human gingival epithelial cells | Fusobacterium nucleatum infection | Silencing of NLRX1 increases NF-κB activity and enhances IL-8 production | NLRX1 interferes with NF-κB pathway | [89] |
| NLRX1−/− mouse intestinal epithelial cells | DSS-induced colitis | NLRX1 deficiency increases proliferation, glutamine metabolism and pro-inflammatory cytokine production | NLRX1 interacts with SIRT1 and regulates glutamine metabolism | [56] |
| NLRX1−/− mouse MEFs | poly (I:C) stimulation | NLRX1 deficiency has no effect on MAVS-dependent IFNβ and IL-6 production | - | [62] |
| NLRX1−/− mouse MEFs | Sendai virus, EMCV and VSV infections | NLRX1 deficiency has no effect on MAVS-dependent IL-6, CXCL10, KC and IFNβ expression | - | [63] |
| NLRX1−/− mouse MEFs | EMCV infection | NLRX1 deficiency has no effect on EMCV induced IFN-β production mediated by MDA5 activation | - | [19] |
| NLRX1−/− mouse MEFs | Simian Virus, Sendai Virus, VSV and Influenza A virus infections | NLRX1 deficiency increases IFNβ and IL-6 production mediated by RIG-I activation | NLRX1 interferes with the RIG-I dependent MAVS signaling pathway | [19] |
| NLRX1−/− mouse MEFs | LPS treatment | NLRX1 deficiency enhances p65 phosphorylation and decreases IκBα level | NLRX1 binds to TRAF6 and interferes with NF-κB signaling | [19] |
| mouse MEFs | EYA4 overexpression, Escherichia coli infection, mammalian DNA or poly (I:C) treatments | Overexpression of EYA4 enhances IFNβ expression | EYA4 binds to NLRX1 and enhances IRF3 signaling | [68] |
| NLRX1−/− mouse MEFs | VSV infection | NLRX1 deficiency increases IL-6, TNF-α and type I IFN production | NLRX1 binds to TUFM/ATG5-ATG12 complex | [32] |
| NLRX1−/− mouse MEFs | VSV infection | NLRX1 deficiency decreases LC3B-II level which leads to defective autophagy | NLRX1 binds to TUFM/ATG5-ATG12 complex | [32] |
| NLRX1 silenced human HeLa cells, NLRX1−/− mouse MEFs | Chlamydia trachomatis infection | NLRX1 deficiency decreases the survival of the pathogen and the production of ROS | NLRX1 promotes ROS production and caspase-1 activation | [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fekete, T.; Bencze, D.; Bíró, E.; Benkő, S.; Pázmándi, K. Focusing on the Cell Type Specific Regulatory Actions of NLRX1. Int. J. Mol. Sci. 2021, 22, 1316. https://doi.org/10.3390/ijms22031316
Fekete T, Bencze D, Bíró E, Benkő S, Pázmándi K. Focusing on the Cell Type Specific Regulatory Actions of NLRX1. International Journal of Molecular Sciences. 2021; 22(3):1316. https://doi.org/10.3390/ijms22031316
Chicago/Turabian StyleFekete, Tünde, Dóra Bencze, Eduárd Bíró, Szilvia Benkő, and Kitti Pázmándi. 2021. "Focusing on the Cell Type Specific Regulatory Actions of NLRX1" International Journal of Molecular Sciences 22, no. 3: 1316. https://doi.org/10.3390/ijms22031316
APA StyleFekete, T., Bencze, D., Bíró, E., Benkő, S., & Pázmándi, K. (2021). Focusing on the Cell Type Specific Regulatory Actions of NLRX1. International Journal of Molecular Sciences, 22(3), 1316. https://doi.org/10.3390/ijms22031316