
Transgenic Mice Overexpressing PG1 Display Corneal Opacity and Severe Inflammation in the Eye

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Generation of PG1-Overexpressing Transgenic Mice
2.2. PG1 Expression Patterns under the Control of the Putative Porcine MUC1 Promoter
2.3. Analysis of Immune Cell Populations in PG1 Transgenic Mice
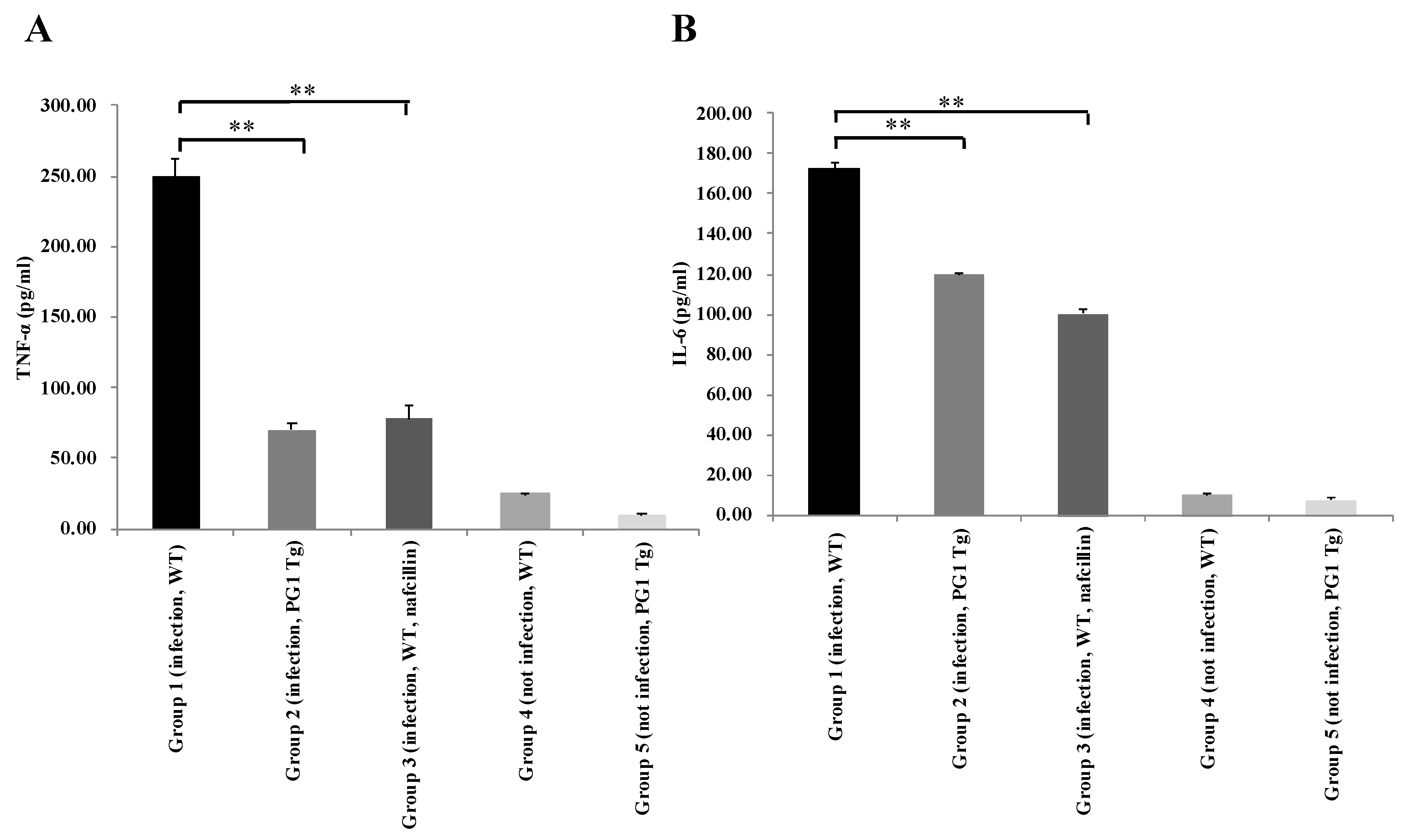
2.4. Enhanced Respiratory Resistance to Experimental Intranasal Infection with Staphylococcus aureus in PG1 Transgenic Mice
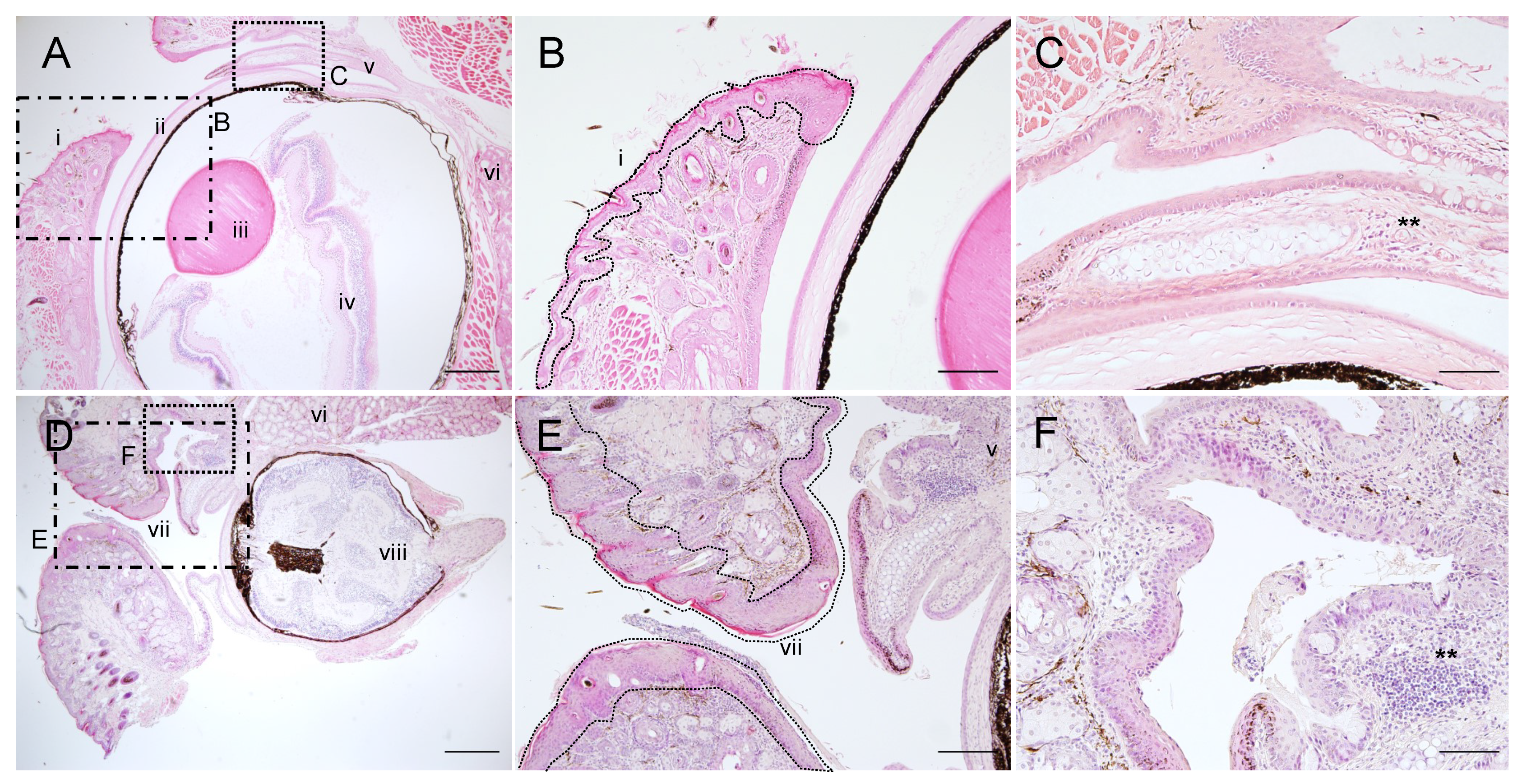
2.5. Corneal Opacity and Subsequent Inflammation in the Eyes of PG1 Transgenic Mice
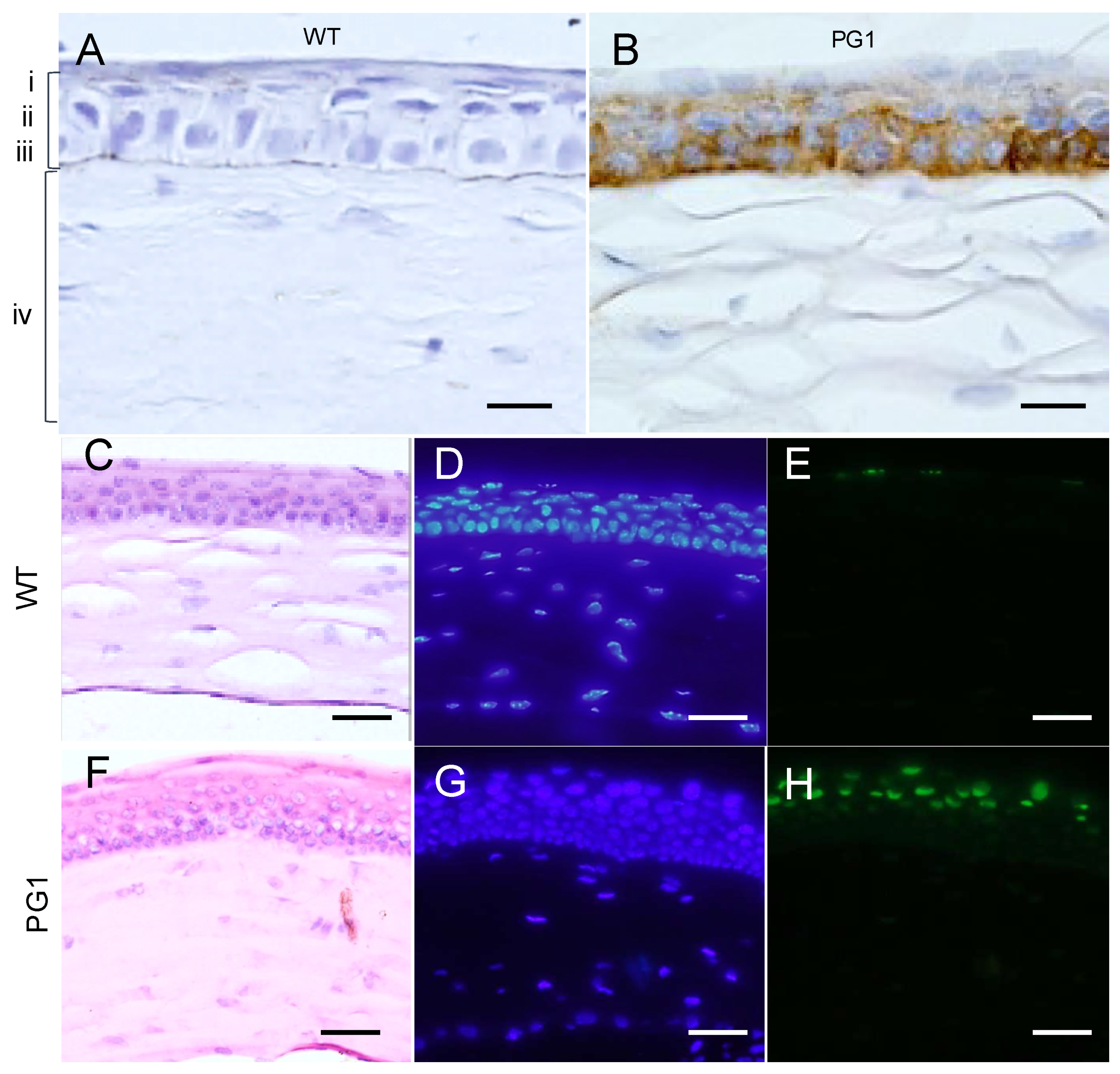
2.6. Immunohistochemical Analyses of PG1 Expression in the Eyes of PG1 Transgenic Mice
2.7. PG1 Treatment on Eyes Results in Corneal Opacity
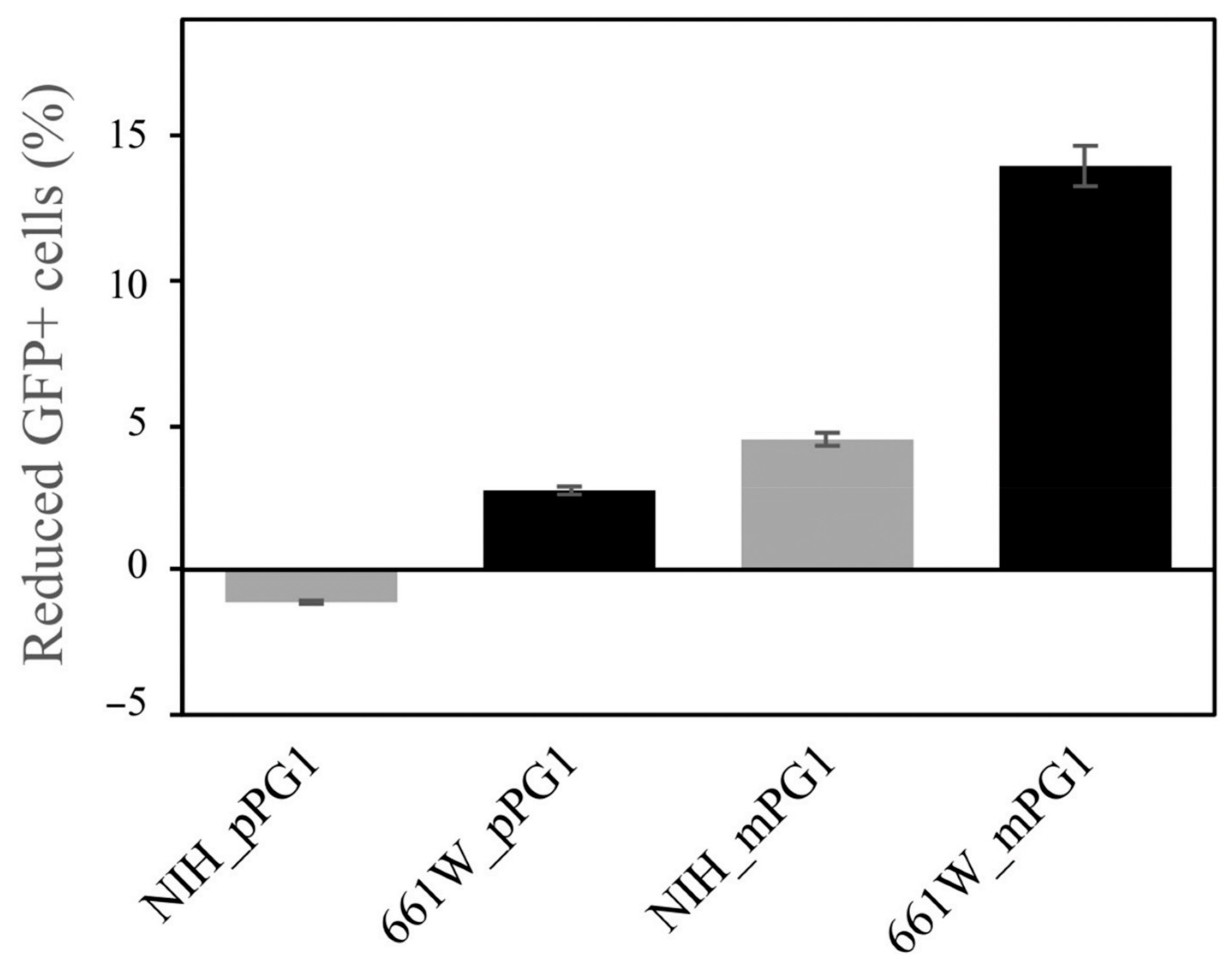
2.8. Comparison of Cytotoxicity between Pro- and Mature Peptides of PG1 in Mammalian Cells
2.9. Expression of Murine Neutrophil Elastase (ELANE) in the Eyes
3. Discussion
4. Materials and Methods
4.1. Preparation of the PG1 Expression Construct
4.2. Generation of Transgenic Mice
4.3. RNA Isolation and Semi-Quantitative RT-PCR
4.4. Immunohistochemical Analysis
4.5. TUNEL Assay
4.6. Analysis of Immune Cell Populations
4.7. Analysis of PG1 Cytotoxicity to Mammalian Cells
4.8. Corneal Opacity Induction and Analysis
4.9. Respiratory Infection and Post-Mortem Analysis
4.10. Measurement of Bacterial Load
4.11. Microscopic Evaluation of Lung Tissues
4.12. Cytokine Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gennaro, R.; Zanetti, M. Structural features and biological activities of the cathelicidin-derived antimicrobial peptides. Pept. Sci. 2000, 55, 31–49. [Google Scholar] [CrossRef]
- Zanetti, M. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 2004, 75, 39–48. [Google Scholar] [CrossRef]
- Tossi, A.; Scocchi, M.; Zanetti, M.; Storici, P.; Gennaro, R. PMAP-37, a novel antibacterial peptide from pig myeloid cells. cDNA cloning, chemical synthesis and activity. Eur. J. Biochem. FEBS 1995, 228, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Zhao, C.; Wu, X.; Dong, R.; Li, G.; Zhu, Q.; Zheng, E.; Liu, D.; Yang, J.; Moisyadi, S.; et al. Bacteria-induced expression of the pig-derived protegrin-1 transgene specifically in the respiratory tract of mice enhances resistance to airway bacterial infection. Sci. Rep. 2020, 10, 16020. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Turner, J.S.; Dinh, N.N.; Lehrer, R.I. Activity of protegrins against yeast-phase Candida albicans. Infect. Immun. 1998, 66, 2486–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamamura, H.; Murakami, T.; Horiuchi, S.; Sugihara, K.; Otaka, A.; Takada, W.; Ibuka, T.; Waki, M.; Yamamoto, N.; Fujii, N. Synthesis of protegrin-related peptides and their antibacterial and anti-human immunodeficiency virus activity. Chem. Pharm. Bull. 1995, 43, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Steinstraesser, L.; Tippler, B.; Mertens, J.; Lamme, E.; Homann, H.-H.; Lehnhardt, M.; Wildner, O.; Steinau, H.-U.; Überla, K. Inhibition of early steps in the lentiviral replication cycle by cathelicidin host defense peptides. Retrovirology 2005, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Shamova, O.V.; Sakuta, G.A.; Orlov, D.S.; Zenin, V.V.; Stein, G.I.; Kolodkin, N.I.; Afonina, I.V.; Kokryakov, V.N. Effects of antimicrobial peptides of neutrophils on tumor and normal host cells in culture. Cell Tissue Biol. 2007, 1, 524–533. [Google Scholar] [CrossRef]
- Hase, K.; Eckmann, L.; Leopard, J.D.; Varki, N.; Kagnoff, M.F. Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect. Immun. 2002, 70, 953–963. [Google Scholar] [CrossRef] [Green Version]
- Carretero, M.; Escámez, M.J.; García, M.; Duarte, B.; Holguín, A.; Retamosa, L.; Jorcano, J.L.; del Río, M.; Larcher, F. In Vitro and In Vivo Wound Healing-Promoting Activities of Human Cathelicidin LL-37. J. Investig. Dermatol. 2008, 128, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kościuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzałkowska, N.; Jóźwik, A.; Horbańczuk, J.; Krzyżewski, J.; Zwierzchowski, L.; Bagnicka, E. Cathelicidins: Family of antimicrobial peptides. A review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.M. Inhibition of neutrophil elastase prevents cathelicidin activation and impairs clearance of bacteria from wounds. Blood 2001, 97, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Dombrowski, Y.; Schauber, J. Cathelicidin LL-37: A defense molecule with a potential role in psoriasis pathogenesis. Exp. Dermatol. 2012, 21, 327–330. [Google Scholar] [CrossRef]
- Morizane, S.; Yamasaki, K.; Mühleisen, B.; Kotol, P.F.; Murakami, M.; Aoyama, Y.; Iwatsuki, K.; Hata, T.; Gallo, R.L. Cathelicidin Antimicrobial Peptide LL-37 in Psoriasis Enables Keratinocyte Reactivity against TLR9 Ligands. J. Investig. Dermatol. 2012, 132, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.-K.; Le, M.T.; Cho, H.; Soundrarajan, N.; Jeon, H.; Park, C.K.; Cha, S.Y.; Kim, J.-H.; Seo, K.; Park, C. Defining the genetic relationship of protegrin-related sequences and the in vivo expression of protegrins. FEBS J. 2014, 281, 5420–5431. [Google Scholar] [CrossRef]
- Pathak, N.; Salas-Auvert, R.; Ruche, G.; Janna, M.-H.; McCarthy, D.; Harrison, R.G. Comparison of the effects of hydrophobicity, amphiphilicity, and α-helicity on the activities of antimicrobial peptides. Proteins Struct. Funct. Bioinform. 1995, 22, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Schumann, M.; Wieprecht, T.; Winkler, A.; Beyermann, M.; Krause, E.; Matsuzaki, K.; Murase, O.; Bienert, M. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry 1996, 35, 12612–12622. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Wieprecht, T.; Nikolenko, H.; Handel, L.; Maloy, W.L.; MacDonald, D.L.; Beyermann, M.; Bienert, M. Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 1997, 403, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Gidalevitz, D.; Ishitsuka, Y.; Muresan, A.S.; Konovalov, O.; Waring, A.J.; Lehrer, R.I.; Lee, K.Y. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA 2003, 100, 6302–6307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, S.N.; Bishop, B.M.; van Hoek, M.L. Natural and synthetic cathelicidin peptides with anti-microbial and anti-biofilm activity against Staphylococcus aureus. BMC Microbiol. 2011, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar] [PubMed]
- Soundrarajan, N.; Park, S.; Le Van Chanh, Q.; Cho, H.-S.; Raghunathan, G.; Ahn, B.; Song, H.; Kim, J.-H.; Park, C. Protegrin-1 cytotoxicity towards mammalian cells positively correlates with the magnitude of conformational changes of the unfolded form upon cell interaction. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panyutich, A.; Shi, J.; Boutz, P.L.; Zhao, C.; Ganz, T. Porcine polymorphonuclear leukocytes generate extracellular microbicidal activity by elastase-mediated activation of secreted proprotegrins. Infect. Immun. 1997, 65, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, M.A.; Swanson, B.J. Mucins in cancer: Protection and control of the cell surface. Nat. Rev. Cancer 2004, 4, 45–60. [Google Scholar] [CrossRef]
- Kim, C.H.; Oh, Y.; Ha, Y.; Ahn, Q.; Kim, S.-H.; Cho, K.-D.; Lee, B.-H.; Chae, C. Expression of mucins in the mucosal surface of small intestines in 1 week-old pigs. J. Vet. Med. Sci. 2010, 72, 245–247. [Google Scholar] [CrossRef] [Green Version]
- Mookherjee, N.; Brown, K.L.; Bowdish, D.M.E.; Doria, S.; Falsafi, R.; Hokamp, K.; Roche, F.M.; Mu, R.; Doho, G.H.; Pistolic, J.; et al. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J. Immunol. 2006, 176, 2455–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinstraesser, L.; Kraneburg, U.; Jacobsen, F.; Benna, S.A. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 2011, 216, 322–333. [Google Scholar] [CrossRef]
- Van Winkle, T.J.; Balk, M.W. Spontaneous corneal opacities in laboratory mice. Lab. Anim. Sci. 1986, 36, 248–255. [Google Scholar]
- Paredes- Gamero, E.J.; Martins, M.N.C.; Cappabianco, F.A.M.; Ide, J.S.; Miranda, A. Characterization of dual effects induced by antimicrobial peptides: Regulated cell death or membrane disruption. BBA Gen. Subj. 2012, 1820, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streilein, J.W. Ocular immune privilege: Therapeutic opportunities from an experiment of nature. Nat. Rev. Immunol. 2003, 3, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.; Nagase, T.; Tomita, T.; Nakamura, K.; Fukuhara, S.; Amano, T.; Yamamoto, H.; Ide, Y.; Suzuki, M.; Teramoto, S.; et al. Beta-defensin overexpression induces progressive muscle degeneration in mice. AJP Cell Physiol. 2007, 292, C2141–C2149. [Google Scholar] [CrossRef]
- Tan, L.T.; Isa, H.; Lightman, S.; Taylor, S.R. Prevalence and causes of phthisis bulbi in a uveitis clinic. Acta Ophthalmol. 2012, 90, e417–e418. [Google Scholar] [CrossRef]
- Apple, D.J.; Jones, G.R.; Reidy, J.J.; Loftfield, K. Ocular perforation and phthisis bulbi secondary to strabismus surgery. J. Pediatric Ophthalmol. Strabismus 1985, 22, 184–187. [Google Scholar]
- Tripathy, K.; Chawla, R.; Temkar, S.; Sagar, P.; Kashyap, S.; Pushker, N.; Sharma, Y.R. Phthisis Bulbi—A Clinicopathological Perspective. Semin. Ophthalmol. 2018, 33, 788–803. [Google Scholar] [CrossRef]
- Khodadoust, A.A. The Allograft Rejection Reaction: The Leading Cause of Late Failure of Clinical Corneal Grafts; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2008; pp. 151–167. [Google Scholar]
- Spadea, L.; Maraone, G.; Verboschi, F.; Vingolo, E.M.; Tognetto, D. Effect of corneal light scatter on vision: A review of the literature. Int. J. Ophthalmol. 2016, 9, 459–464. [Google Scholar] [CrossRef]
- Bolintineanu, D.; Hazrati, E.; Davis, H.T.; Lehrer, R.I.; Kaznessis, Y.N. Antimicrobial mechanism of pore-forming protegrin peptides: 100 pores to kill E. coli. Peptides 2010, 31, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lazaridis, T.; He, Y.; Prieto, L. Membrane interactions and pore formation by the antimicrobial peptide protegrin. Biophys. J. 2013, 104, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Cheung, Q.C.; Turner, P.V.; Song, C.; Wu, D.; Cai, H.Y.; MacInnes, J.I.; Li, J. Enhanced resistance to bacterial infection in protegrin-1 transgenic mice. Antimicrob. Agents Chemother. 2008, 52, 1812–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early local immune defences in the respiratory tract. Nat. Rev. Immunol. 2017, 17, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Selders, G.S.; Fetz, A.E.; Radic, M.Z.; Bowlin, G.L. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen. Biomater. 2017, 4, 55–68. [Google Scholar] [CrossRef]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 1999, 341, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.P.; Wu, C.; Yang, J.-L. Membranolytic selectivity of cystine-stabilized cyclic protegrins. Eur. J. Biochem. 2000, 267, 3289–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouri- Sorkhabi, M.H.; Wright, L.C.; Sullivan, D.R.; Kuchel, P.W. Quantitative31P nuclear magnetic resonance analysis of the phospholipids of erythrocyte membranes using detergent. Lipids 1996, 31, 765–770. [Google Scholar] [CrossRef]
- Dong, X.-X.; Wang, Y.; Qin, Z.-H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, E.; Ding, X.-Q.; Saadi, A.; Agarwal, N.; Naash, M.I.; Al-Ubaidi, M.R. Expression of Cone-Photoreceptor–Specific Antigens in a Cell Line Derived from Retinal Tumors in Transgenic Mice. Investig. Ophthalmol. Vis. Sci. 2004, 45, 764–768. [Google Scholar] [CrossRef] [Green Version]
- Wheway, G.; Nazlamova, L.; Turner, D.; Cross, S. 661W Photoreceptor Cell Line as a Cell Model for Studying Retinal Ciliopathies. Front. Genet. 2019, 10, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, R.; Halford, K.; Sokolov, B.P.; Khillan, J.S.; Prockop, D.J. Phenotypic variability and incomplete penetrance of spontaneous fractures in an inbred strain of transgenic mice expressing a mutated collagen gene (COL1A1). J. Clin. Investig. 1994, 93, 1765–1769. [Google Scholar] [CrossRef] [Green Version]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar] [PubMed]
- Graziani-Bowering, G.M.; Graham, J.M.; Filion, L.G. A quick, easy and inexpensive method for the isolation of human peripheral blood monocytes. J. Immunol. Methods 1997, 207, 157–168. [Google Scholar] [CrossRef]
- Al-Ubaidi, M.R.; Font, R.L.; Quiambao, A.B.; Keener, M.J.; Liou, G.I.; Overbeek, P.A.; Baehr, W. Bilateral retinal and brain tumors in transgenic mice expressing simian virus 40 large T antigen under control of the human interphotoreceptor retinoid-binding protein promoter. J. Cell Biol. 1992, 119, 1681–1687. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
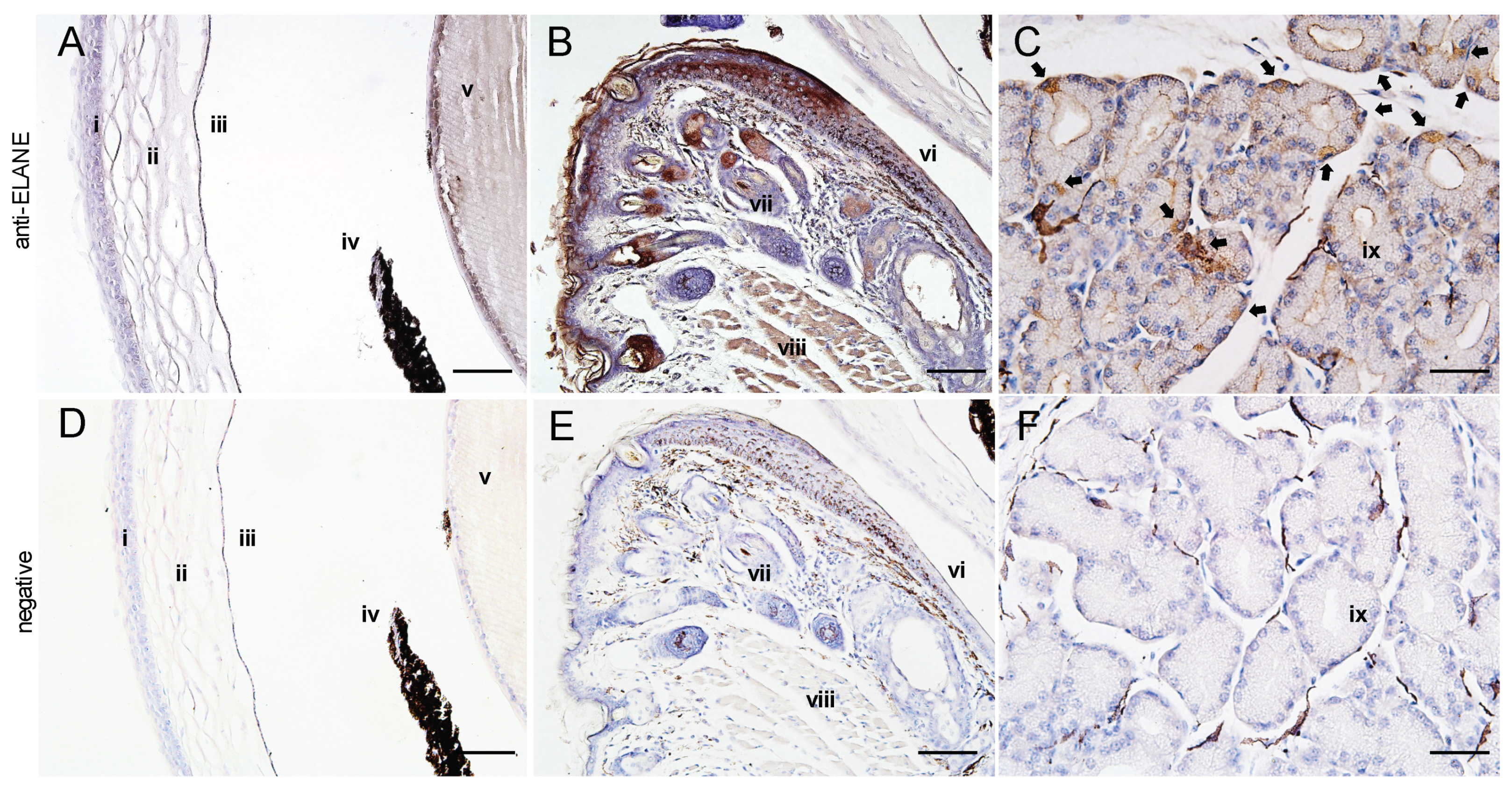{kind=link}
| Tissue | Population | Surface Phenotype | PG1 Transgenic | Wild-Type | p-Value |
|---|---|---|---|---|---|
| Spleen (×105) | T cell | TCR-beta+ | 317.04 ± 22.46 | 305.65 ± 33.82 | 0.79 |
| CD4+ T cell | CD4+ | 216.46 ± 15.34 | 202.04 ± 25.42 | 0.64 | |
| CD8+ T cell | CD8+ | 116.89 ± 8.04 | 122.96 ± 12.80 | 0.70 | |
| Activated CD4+ T cell | CD4+CD44+ | 55.51 ± 3.33 | 43.93 ± 4.14 | 0.06 | |
| CD4+CD69+ | 22.07 ± 2.47 | 21.40 ± 2.42 | 0.85 | ||
| CD4+CD62L- | 57.39 ± 7.26 | 52.17 ± 6.92 | 0.61 | ||
| Activated CD8+ T cell | CD8+CD44+ | 27.94 ± 1.00 | 25.86 ± 3.04 | 0.54 | |
| CD8+CD69+ | 4.04 ± 0.28 | 3.88 ± 0.24 | 0.67 | ||
| CD8+CD62L- | 15.24 ± 3.76 | 10.31 ± 2.81 | 0.32 | ||
| Regulatory T cell | CD4+CD25+ | 23.23 ± 1.21 | 21.60 ± 2.28 | 0.55 | |
| B cell | B220+ | 503.92 ± 36.70 | 466.04 ± 51.20 | 0.56 | |
| Dendritic cell | CD11b+ Cd11c+ | 105.42 ± 10.62 | 95.17 ± 8.57 | 0.47 | |
| Granulocyte | Gr1+ high | 11.09 ± 4.31 | 13.01 ± 4.08 | 0.75 | |
| Ly-6C+ monocytes | Gr1+low | 71.49 ± 4.46 | 72.85 ± 8.12 | 0.89 | |
| NK cell | NK1.1+ TCR-beta+ | 11.54 ± 0.58 | 11.54 ± 1.48 | 0.92 | |
| Mesenteric Lymph node (×105) | T cell | TCR-beta+ | 87.71 ± 14.83 | 102.27 ± 9.12 | 0.43 |
| CD4+ T cell | CD4+ | 373.90 ± 26.00 | 351.61 ± 41.75 | 0.66 | |
| CD8+ T cell | CD8+ | 250.44 ± 15.08 | 246.78 ± 23.92 | 0.90 | |
| Activated CD4+ T cell | CD4+CD44+ | 7.48 ± 1.28 | 8.82 ± 0.61 | 0.37 | |
| CD4+CD69+ | 9.23 ± 1.63 | 9.93 ± 0.95 | 0.72 | ||
| CD4+CD62L- | 11.54 ± 2.31 | 12.30 ± 0.95 | 0.77 | ||
| Activated CD8+ T cell | CD8+CD44+ | 4.73 ± 0.90 | 6.40 ± 0.71 | 0.18 | |
| CD8+CD69+ | 2.11 ± 0.43 | 2.54 ± 0.28 | 0.43 | ||
| CD8+CD62L- | 4.73 ± 1.80 | 3.95 ± 0.42 | 0.69 | ||
| Regulatory T cell | CD4+CD25+ | 5.97 ± 1.20 | 6.39 ± 0.64 | 0.77 | |
| B cell | B220+ | 36.34 ± 7.29 | 40.87 ± 4.86 | 0.62 | |
| Dendritic cell | CD11b+ Cd11c+ | 5.50 ± 1.02 | 6.82 ± 1.06 | 0.39 | |
| Granulocyte | Gr1+ | 0.08 ± 0.05 | 0.07 ± 0.03 | 0.88 | |
| Ly-6C+ monocytes | Gr1+ | 9.51 ± 1.76 | 10.94 ± 1.79 | 0.58 | |
| Thymus (×105) | CD4+ SP | CD4+ | 106.34 ± 11.57 | 85.44 ± 9.80 | 0.20 |
| CD8+ SP | CD8+ | 32.81 ± 2.57 | 26.78 ± 3.14 | 0.17 | |
| DN | CD4-CD8- | 35.27 ± 2.51 | 30.40 ± 3.26 | 0.27 | |
| DP | CD4+CD8+ | 1033.98 ± 111.15 | 941.16 ± 162.80 | 0.65 | |
| NK cell | NK1.1+ TCR-beta+ | 7.34 ± 0.69 | 6.36 ± 1.26 | 0.52 | |
| Liver (×105) | NK cell | NK1.1+ TCR-beta+ | 0.27 ± 0.08 | 0.36 ± 0.08 | 0.38 |
| Phenotypes | Total | ||
|---|---|---|---|
| No. of Normal | No. of Corneal Opacity | ||
| Total | 161 | 54 | 215 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, M.-K.; Le, M.T.; Cho, H.-S.; Lee, J.; Jeon, H.; Cha, S.-Y.; Na, M.; Chun, T.; Kim, J.-H.; Song, H.; et al. Transgenic Mice Overexpressing PG1 Display Corneal Opacity and Severe Inflammation in the Eye. Int. J. Mol. Sci. 2021, 22, 1586. https://doi.org/10.3390/ijms22041586
Choi M-K, Le MT, Cho H-S, Lee J, Jeon H, Cha S-Y, Na M, Chun T, Kim J-H, Song H, et al. Transgenic Mice Overexpressing PG1 Display Corneal Opacity and Severe Inflammation in the Eye. International Journal of Molecular Sciences. 2021; 22(4):1586. https://doi.org/10.3390/ijms22041586
Chicago/Turabian StyleChoi, Min-Kyeung, Minh Thong Le, Hye-Sun Cho, Juyoung Lee, Hyoim Jeon, Se-Yeoun Cha, Manheum Na, Taehoon Chun, Jin-Hoi Kim, Hyuk Song, and et al. 2021. "Transgenic Mice Overexpressing PG1 Display Corneal Opacity and Severe Inflammation in the Eye" International Journal of Molecular Sciences 22, no. 4: 1586. https://doi.org/10.3390/ijms22041586
APA StyleChoi, M. -K., Le, M. T., Cho, H. -S., Lee, J., Jeon, H., Cha, S. -Y., Na, M., Chun, T., Kim, J. -H., Song, H., & Park, C. (2021). Transgenic Mice Overexpressing PG1 Display Corneal Opacity and Severe Inflammation in the Eye. International Journal of Molecular Sciences, 22(4), 1586. https://doi.org/10.3390/ijms22041586