
Long, Noncoding RNA SRA Induces Apoptosis of β-Cells by Promoting the IRAK1/LDHA/Lactate Pathway

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Demographic Characteristics of T1D Patients
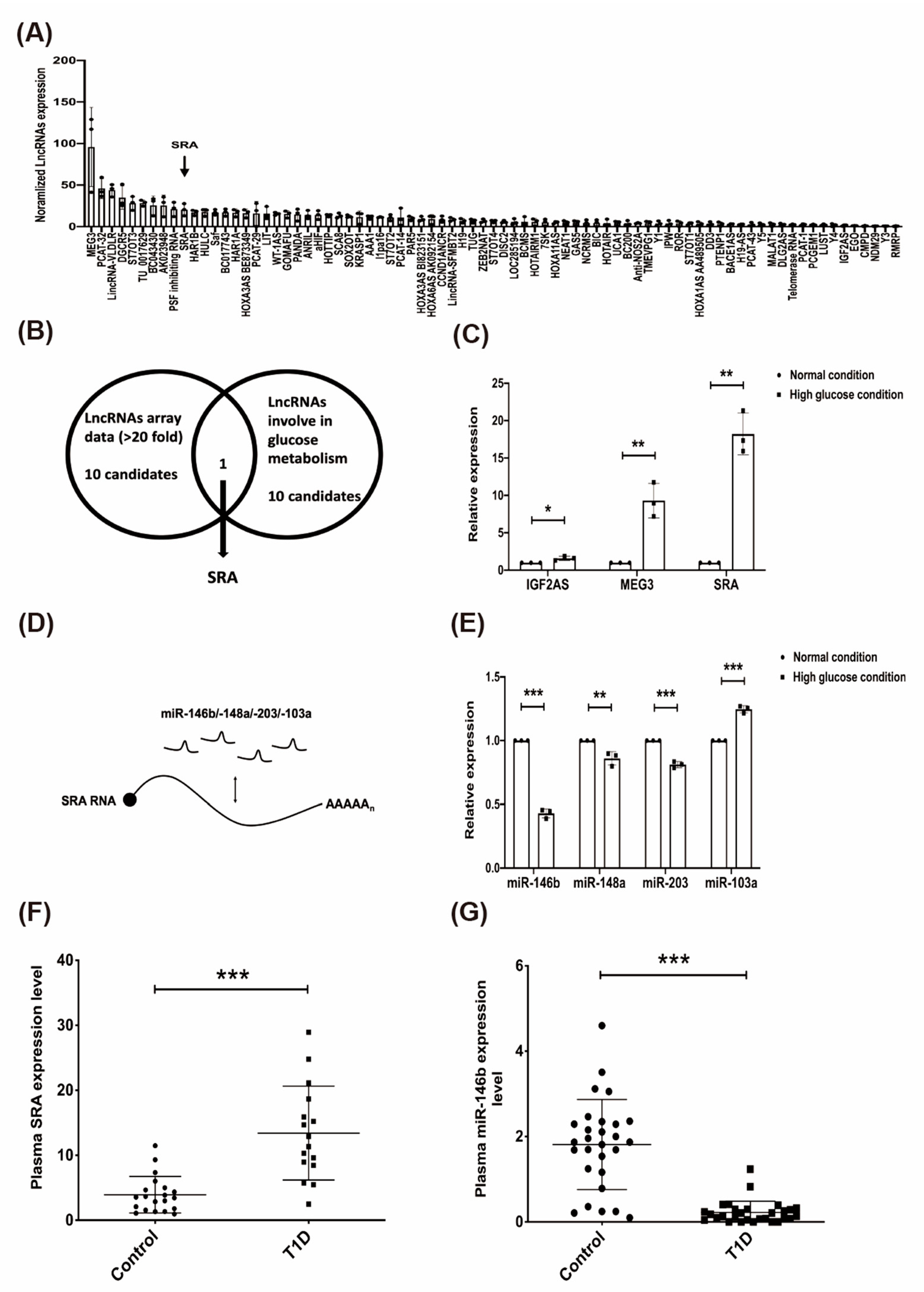
2.2. The Effect of High Glucose on Candidate lncRNAs/miRNAs in CD4+ MOLT4 Tregs and the Expression of lncRNA-SRA/miR-146b in the Plasma of Patients with T1D
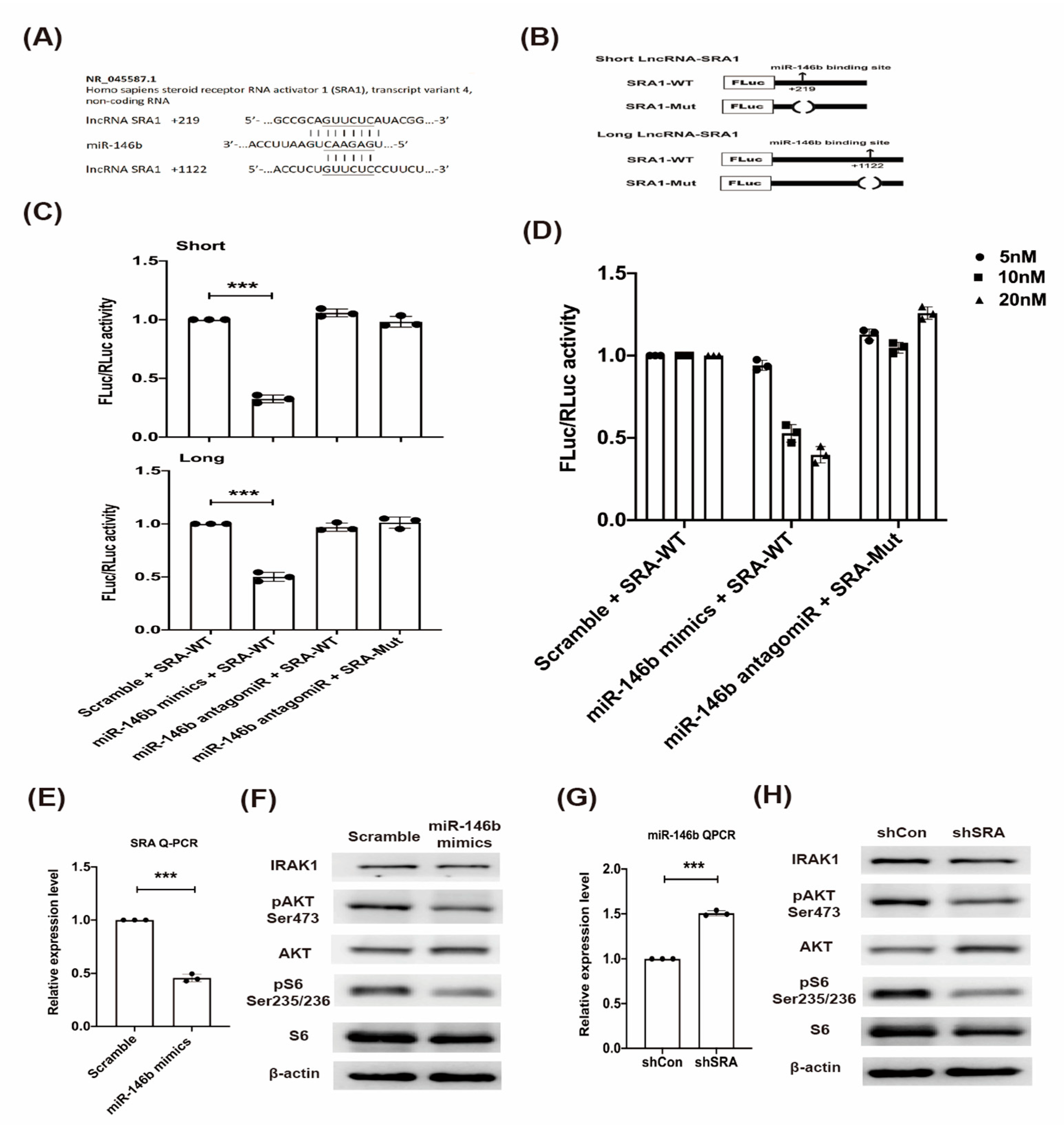
2.3. LncRNA SRA Is a Direct Target of miR-146b and Regulates IRAK1-AKT-S6K1 Signaling
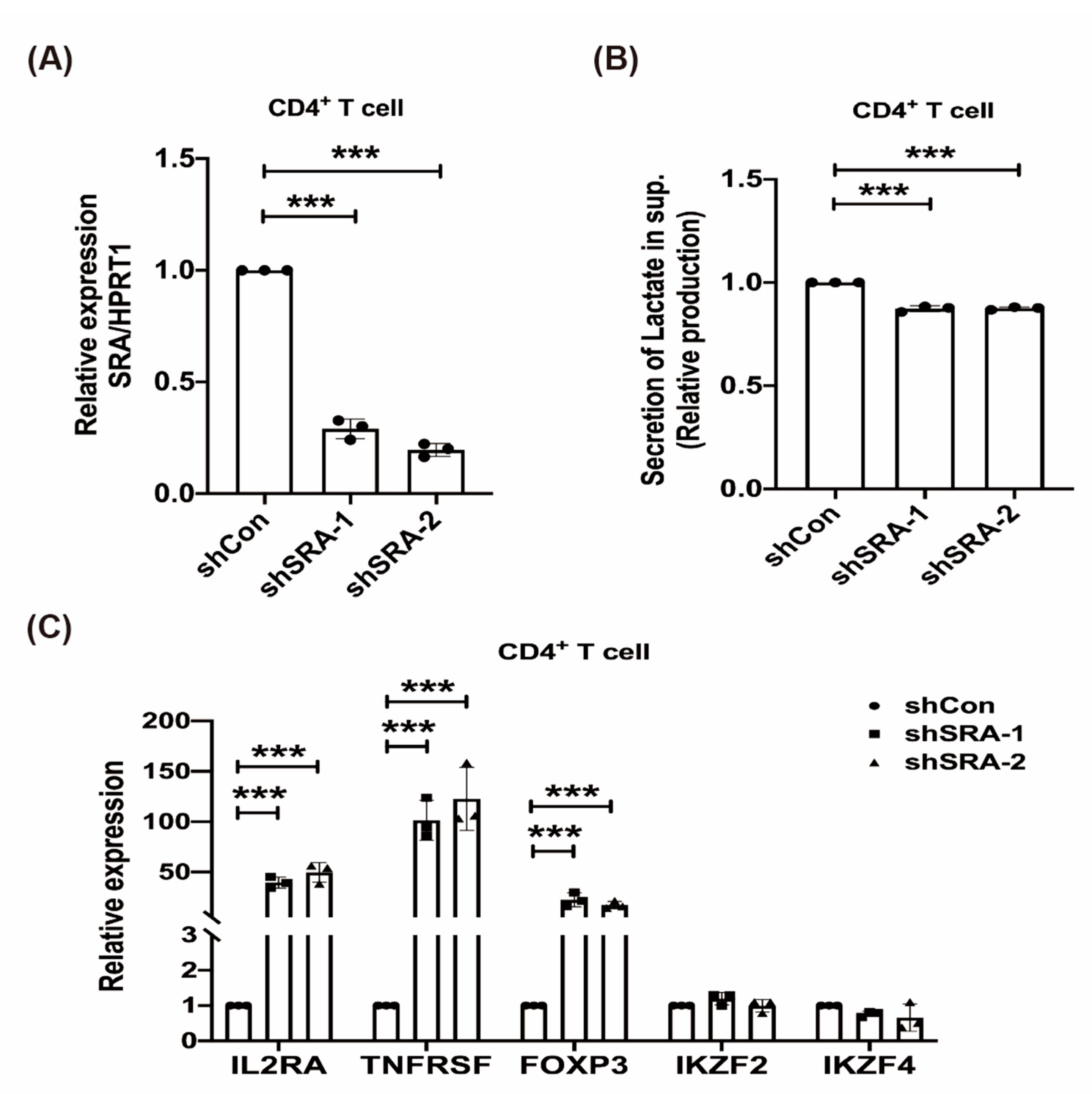
2.4. LncRNA SRA Suppresses CD4+ Treg Function
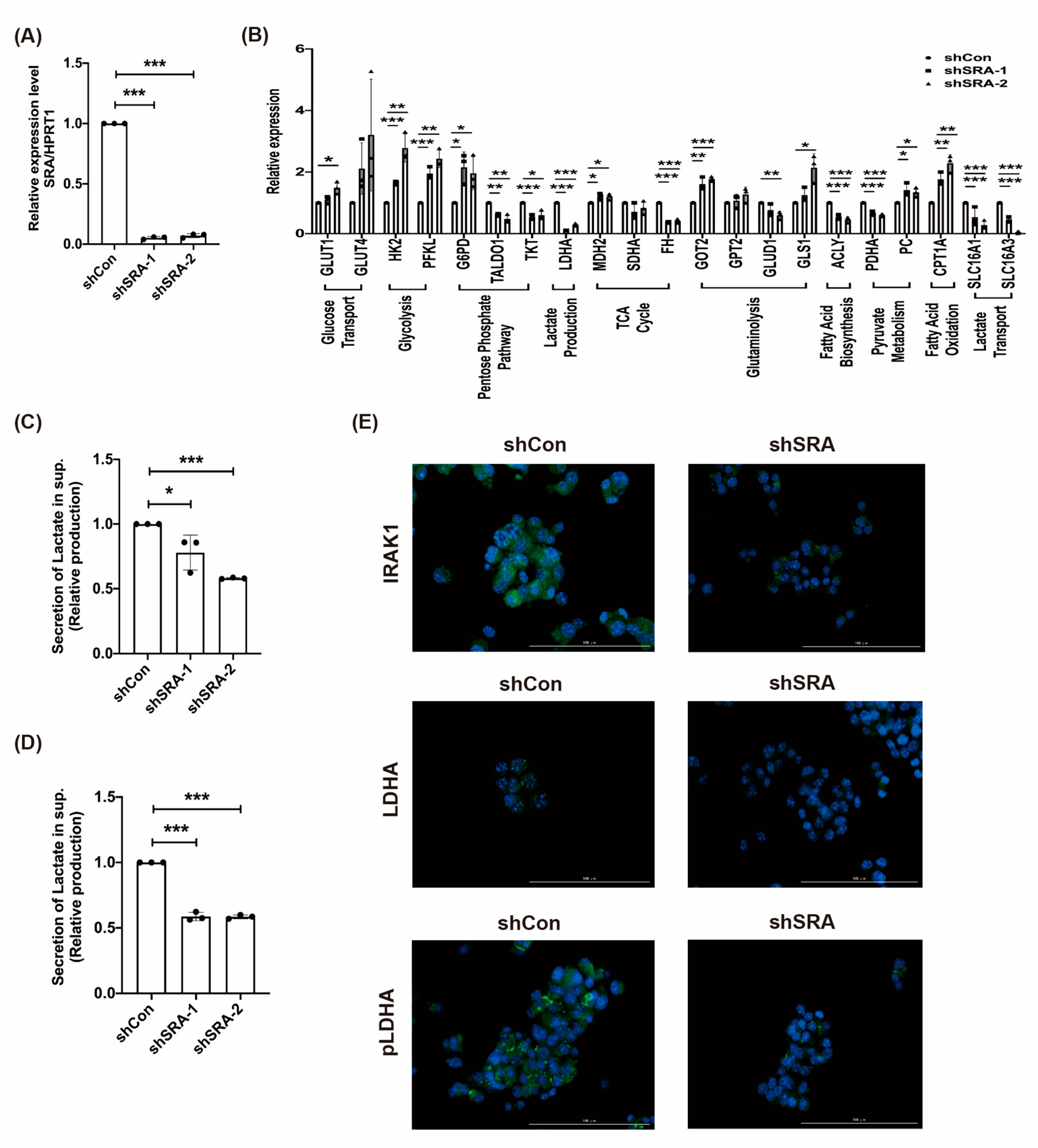
2.5. LncRNA SRA Induces MIN6 β-Cell Metabolic Reprogramming and the IRAK1/LDHA/pLDHA Signaling Pathway
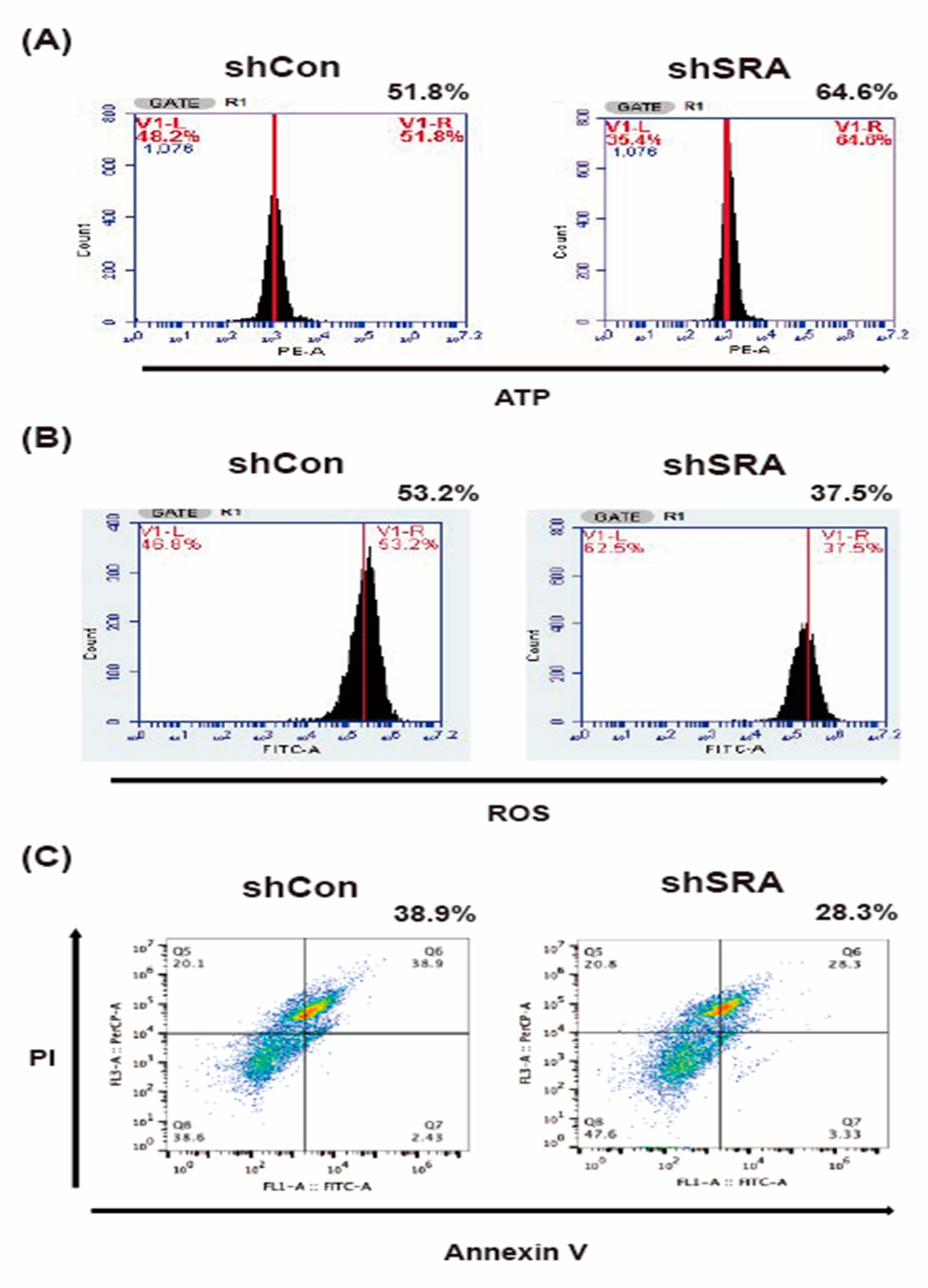
2.6. LncRNA SRA Silencing Induces ATP Production, Represses ROS Levels, and Inhibits MIN6 β-Cell Apoptosis
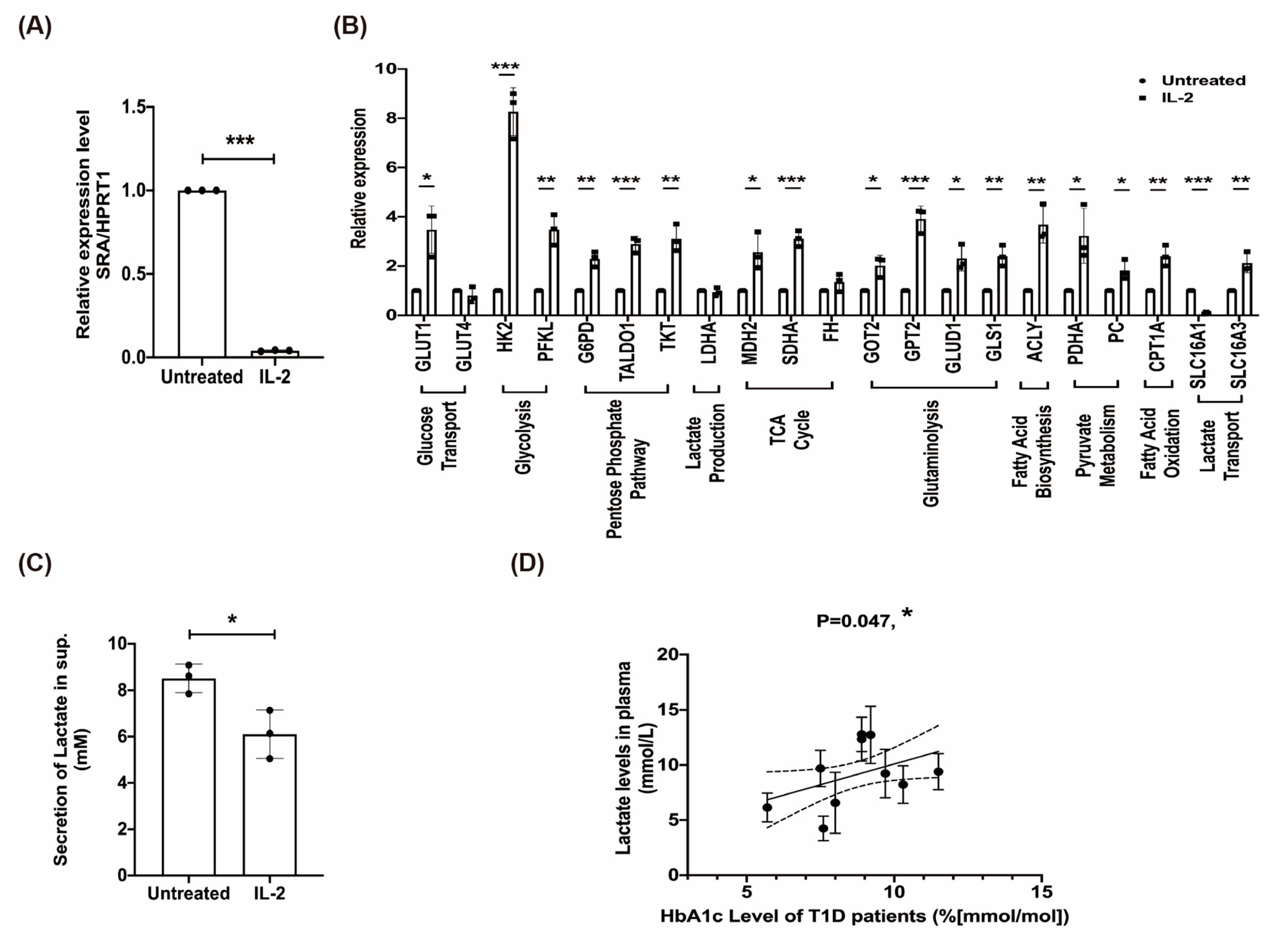
2.7. The Effect of rhIL-2 Treatment on the Expression of lncRNA SRA, Metabolic Pathways in MIN6 β-Cells, and Lactate Associated with Poor Clinical Outcome
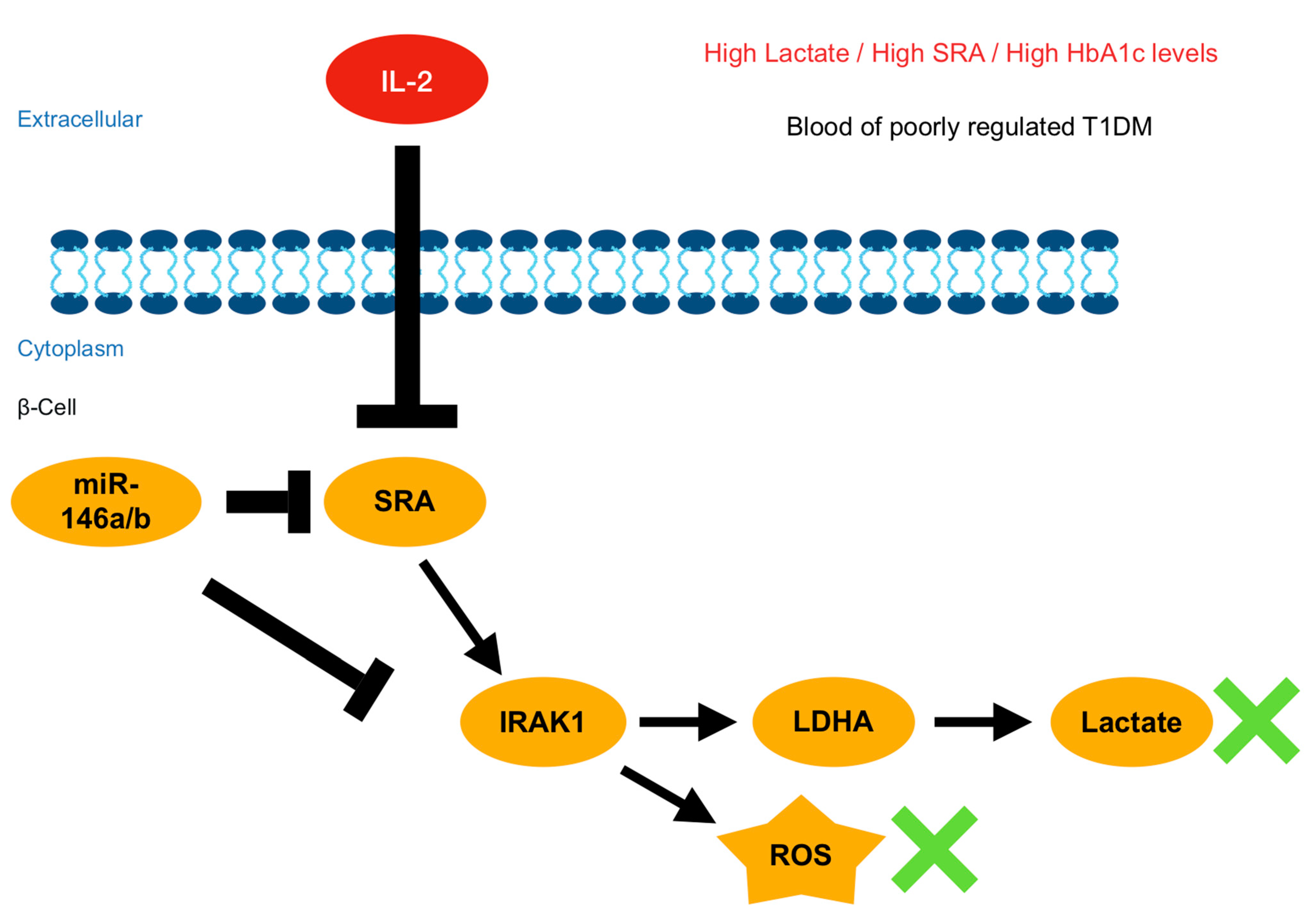
3. Discussion
4. Materials and Methods
4.1. Clinical Participants
4.2. Cell Culture
4.3. LncRNA Profiling
4.4. LncRNA and miRNA Expression Analysis
4.5. Vector Construction for Reporter Assays
4.6. miRNA Mimic Reporter Assay and Antagomir Transfection
4.7. shRNA Knockdown
4.8. Immunoblotting
4.9. Immunofluorescence
4.10. Analysis of Apoptosis, ROS, and ATP
4.11. Analysis of Lactate Secretion
4.12. IL-2 Treatment
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LncRNA | Long non-coding RNA |
| SRA | Steroid receptor RNA activator |
| T1D | Type 1 diabetes mellitus |
| IRAK1 | Interleukin-1 receptor-associated kinase 1 |
| PBMCs | Peripheral blood mononuclear cells |
| Tregs | Regulatory T cells |
| AKT | Protein kinase B |
| IL-2 | Interleukin-2 |
| HbA1c | Hemoglobin A1c |
| LDHA | Lactate dehydrogenase A |
| ROS | Reactive oxygen species |
References
- American Diabetes Association. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018, 41, S13–S27. [Google Scholar] [CrossRef] [Green Version]
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017. [Google Scholar]
- Kühtreiber, W.M.; Tran, L.; Kim, T.; Dybala, M.; Nguyen, B.; Plager, S.; Huang, D.; Janes, S.; Defusco, A.; Baum, D.; et al. Long-term reduction in hyperglycemia in advanced type 1 diabetes: The value of induced aerobic glycolysis with BCG vaccinations. NPJ Vaccines 2018, 3, 23. [Google Scholar] [CrossRef]
- Jones, A.R., IV; Coleman, E.L.; Husni, N.R.; Deeney, J.T.; Raval, F.; Steenkamp, D.; Dooms, H.; Nikolajczyk, B.S.; Corkey, B.E. Type 1 diabetes alters lipid handling and metabolism in human fibroblasts and peripheral blood mononuclear cells. PLoS ONE 2017, 12, e0188474. [Google Scholar] [CrossRef] [Green Version]
- Hull, C.M.; Peakman, M.; Tree, T.I.M. Regulatory T cell dysfunction in type 1 diabetes: What’s broken and how can we fix it? Diabetologia 2017, 10, 1839–1850. [Google Scholar] [CrossRef] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Todd, J.A.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Downes, K.; Plagnol, V.; Bailey, R.; Nejentsev, S.; Field, S.F.; Payne, F.; et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007, 39, 857–864. [Google Scholar] [CrossRef]
- Casellas, A.; Mallol, C.; Salavert, A.; Jimenez, V.; Garcia, M.; Agudo, J.; Obach, M.; Haurigot, V.; Vila, L.; Molas, M.; et al. Insulin-like Growth Factor 2 Overexpression Induces beta-Cell Dysfunction and Increases Beta-cell Susceptibility to Damage. J. Biol Chem 2015, 290, 16772–16785. [Google Scholar] [CrossRef] [Green Version]
- Arnes, L.; Akerman, I.; Balderes, D.A.; Ferrer, J.; Sussel, L. Betalinc1 encodes a long noncoding RNA that regulates islet beta-cell formation and function. Genes Dev. 2016, 30, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Wallace, C.; Smyth, D.J.; Maisuria-Armer, M.; Walker, N.M.; Todd, J.A.; Clayton, D.G. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 2010, 42, 68–71. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wang, N.; Yin, D.; Wang, L.; Jin, F.; Zhu, Y.; Yuan, Q.; De, W. Downregulation of Long Noncoding RNA Meg3 Affects Insulin Synthesis and Secretion in Mouse Pancreatic Beta Cells. J. Cell Physiol 2016, 231, 852–862. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, F.; Zhou, J.; Yan, L.; Jurczak, M.J.; Lee, H.Y.; Yang, L.; Mueller, M.; Zhou, X.B.; Dandolo, L.; et al. The H19/let-7 double-negative feedback loop contributes to glucose metabolism in muscle cells. Nucleic Acids Res. 2014, 42, 13799–13811. [Google Scholar] [CrossRef]
- Liu, S.; Sheng, L.; Miao, H.; Saunders, T.L.; MacDougald, O.A.; Koenig, R.J.; Xu, B. SRA gene knockout protects against diet-induced obesity and improves glucose tolerance. J. Biol. Chem. 2014, 289, 13000–13009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanz, R.B.; McKenna, N.J.; Onate, S.A.; Albrecht, U.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 1999, 97, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Gerin, I.; Miao, H.; Vu-Phan, D.; Johnson, C.N.; Xu, R.; Chen, X.W.; Cawthorn, W.P.; MacDougald, O.A.; Koenig, R.J. Multiple roles for the non-coding RNA SRA in regulation of adipogenesis and insulin sensitivity. PLoS ONE 2010, 5, e14199. [Google Scholar] [CrossRef] [Green Version]
- Lovis, P.; Roggli, E.; Laybutt, D.R.; Gattesco, S.; Yang, J.Y.; Widmann, C.; Abderrahmani, A.; Regazzi, R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 2008, 57, 2728–2736. [Google Scholar] [CrossRef] [Green Version]
- Cobb, B.S.; Hertweck, A.; Smith, J.; O’Connor, E.; Graf, D.; Cook, T.; Smale, S.T.; Sakaguchi, S.; Livesey, F.J.; Fisher, A.G.; et al. A role for Dicer in immune regulation. J. Exp. Med. 2006, 203, 2519–2527. [Google Scholar] [CrossRef]
- Nakasa, T.; Miyaki, S.; Okubo, A.; Hashimoto, M.; Nishida, K.; Ochi, M.; Asahara, H. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2008, 58, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Luo, X.; Cui, H.; Ni, X.; Yuan, M.; Guo, Y.; Huang, X.; Zhou, H.; de Vries, N.; Tak, P.P.; et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009, 60, 1065–1075. [Google Scholar] [CrossRef]
- Legnini, I.; Morlando, M.; Mangiavacchi, A.; Fatica, A.; Bozzoni, I. A feedforward regulatory loop between HuR and the long noncoding RNA linc-MD1 controls early phases of myogenesis. Mol. Cell. 2014, 53, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Trotta, E.; Bessette, P.H.; Silveria, S.L.; Ely, L.K.; Jude, K.M.; Le, D.T.; Holst, C.R.; Coyle, A.; Potempa, M.; Lanier, L.L.; et al. A Human anti-IL-2 Antibody That Potentiates Regulatory T Cells by a Structure-Based Mechanism. Nat. Med. 2018, 7, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.J.; Kim, S.P.; Zhang, D.; Sun, H.; Cao, Q.; Lu, X.; Ying, Z.; Li, L.; Henry, R.R.; Ciaraldi, T.P.; et al. Deletion of interleukin 1 receptor-associated kinase 1 (Irak1) improves glucose tolerance primarily by increasing insulin sensitivity in skeletal muscle. J. Biol. Chem. 2017, 292, 12339–12350. [Google Scholar] [CrossRef] [Green Version]
- Alkanani, A.K.; Rewers, M.; Dong, F.; Waugh, K.; Gottlieb, P.A.; Zipris, D. Dysregulated Toll-like receptor-induced interleukin-1beta and interleukin-6 responses in subjects at risk for the development of type 1 diabetes. Diabetes 2012, 61, 2525–2533. [Google Scholar] [CrossRef] [Green Version]
- Bevan, M.J. Helping the CD8(+) T-cell response. Nat. Rev. Immunol. 2004, 4, 595–602. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.S.; von Herrath, M. CD4 T cell differentiation in type 1 diabetes. Clin. Exp. Immunol. 2016, 183, 16–29. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.K.; Choi, Y.A.; Park, W.; Lee, T.; Ryu, S.H.; Kim, S.Y.; Kim, J.R.; Kim, J.H.; Baek, S.H. Phosphatidic acid regulates systemic inflammatory responses by modulating the Akt-mammalian target of rapamycin-p70 S6 kinase 1 pathway. J. Biol. Chem. 2003, 278, 45117–45127. [Google Scholar] [CrossRef] [Green Version]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [Green Version]
- Parisi, C.; Arisi, I.; D’Ambrosi, N.; Storti, A.E.; Brandi, R.; D’Onofrio, M.; Volonté, C. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013, 12, e959. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.L.; Wang, J.; Chan, C.H.; Lee, S.W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009, 325, 1134–1138. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Chen, K.-B.; Tsai, C.; Tsai, F.; Huang, C.; Tang, C.; Yang, J.; Hsu, Y.; Peng, S.; Chung, J. Casticin inhibits human prostate cancer DU 145 cell migration and invasion via Ras/Akt/NF-κB signaling pathways. J. Food Biochem. 2019, 43, e12902. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Patterson, S.J.; Levings, M.K. The Role of the PI3K Signaling Pathway in CD4(+) T Cell Differentiation and Function. Front. Immunol. 2012, 3, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, M.C.G.J.; Ham, J.C.; Wisse, E.; Misra, S.; Landewe, S.; Rosenthal, M.; Patel, D.; Oliver, N.; Bilo, H.J.G.; Murphy, E. Elevated Lactate Levels in Patients With Poorly Regulated Type 1 Diabetes and Glycogenic Hepatopathy: A New Feature of Mauriac Syndrome. Diabetes Care. 2015, 2, e11–e12. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Dong, Y.; Atefi, M.; Liu, Y.; Elshimali, Y.; Vadgama, J.V. Lactate, a Neglected Factor for Diabetes and Cancer Interaction. Mediat. Inflamm. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinberg-Bleyer, Y.; Baeyens, A.; You, S.; Elhage, R.; Fourcade, G.; Gregoire, S.; Cagnard, N.; Carpentier, W.; Tang, Q.; Bluestone, J.; et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J. Exp. Med. 2010, 207, 1871–1878. [Google Scholar] [CrossRef]
- Hartemann, A.; Bensimon, G.; Payan, C.A.; Jacqueminet, S.; Bourron, O.; Nicolas, N.; Fonfrede, M.; Rosenzwajg, M.; Bernard, C.; Klatzmann, D. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013, 1, 295–305. [Google Scholar] [CrossRef]
- McMurray, R.W.; Ndebele, K.; Hardy, K.J.; Jenkins, J.K. 17-beta-estradiol suppresses IL-2 and IL-2 receptor. Cytokine 2001, 6, 324–333. [Google Scholar] [CrossRef]
- Wu, M.; Chen, W.; Zhang, S.; Huang, S.; Zhang, A.; Zhang, Y.; Jia, Z. Rotenone protects against β-cell apoptosis and attenuates type 1 diabetes mellitus. Apoptosis 2019, 24, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T.; Weiss, A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat. Rev. Immunol. 2004, 4, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Peng, S.-F.; Huang, Y.; Tsai, C.; Tsai, F.; Huang, C.; Tang, C.; Yang, J.; Hsu, Y.; Yin, M.; et al. Combinational treatment of all-trans retinoic acid (ATRA) and bisdemethoxycurcumin (BDMC)-induced apoptosis in liver cancer Hep3B cells. J. Food Biochem. 2020, 44, e13122. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | First Onset | Recent Onset (Enrolled) |
|---|---|---|
| Gender, male/female = 10/15 | ||
| Age, mean (SD) | 9.5 (4.8) | 18.4 (5.2) |
| Fasting glucose level, mg/dL (SD) | 345.2 (132.6) | 186.0 (89.3) |
| Hemoglobin A1c (HbA1c) level, % (SD) | 12.7 (2.5) | 9.0 (1.8) |
| C-peptide, ng/mL (SD) | 0.3 (0.2) | 0.4 (0.2) |
| Hemoglobin, g/dL (SD) | 14.2 (1.7) | 14.2 (1.6) |
| Hematocrit, % (SD) | 40.4 (3.7) | 40.6 (4.4) |
| Red blood cells, 106 cells/L (SD) | 5.3 (0.6) | 5.4 (0.8) |
| White blood cells, 103 cells/mm3 (SD) | 14.4 (8.2) | 14.1 (7.6) |
| Platelets, 103 cells/mm3 (SD) | 328.1 (106.2) | 327.1 (99.8) |
| Neutrophils, % (SD) | 68.9 (19.5) | 64.5 (21.2) |
| Lymphocytes, % (SD) | 25.9 (17.6) | 28.9 (20.7) |
| Monocytes, % (SD) | 5.6 (2.7) | 6.4 (3.0) |
| Total cholesterol, mg/dL (SD) | 198.1 (45.2) | 190.1 (37.6) |
| High-density lipoprotein, mg/dL (SD) | 60.2 (18.3) | 62.3 (19.0) |
| Low-density lipoprotein, mg/dL (SD) | 119.2 (37.6) | 104.2 (29.1) |
| Triglyceride, mg/dL (SD) | 73. (68.5) | 105.8 (133.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-N.; Chiang, S.-L.; Lin, Y.-J.; Liu, S.-C.; Li, Y.-H.; Liao, Y.-C.; Lee, M.-R.; Su, P.-H.; Tsai, F.-J.; Hung, H.-C.; et al. Long, Noncoding RNA SRA Induces Apoptosis of β-Cells by Promoting the IRAK1/LDHA/Lactate Pathway. Int. J. Mol. Sci. 2021, 22, 1720. https://doi.org/10.3390/ijms22041720
Huang Y-N, Chiang S-L, Lin Y-J, Liu S-C, Li Y-H, Liao Y-C, Lee M-R, Su P-H, Tsai F-J, Hung H-C, et al. Long, Noncoding RNA SRA Induces Apoptosis of β-Cells by Promoting the IRAK1/LDHA/Lactate Pathway. International Journal of Molecular Sciences. 2021; 22(4):1720. https://doi.org/10.3390/ijms22041720
Chicago/Turabian StyleHuang, Yu-Nan, Shang-Lun Chiang, Yu-Jung Lin, Su-Ching Liu, Yen-Hsien Li, Yu-Chen Liao, Maw-Rong Lee, Pen-Hua Su, Fuu-Jen Tsai, Hui-Chih Hung, and et al. 2021. "Long, Noncoding RNA SRA Induces Apoptosis of β-Cells by Promoting the IRAK1/LDHA/Lactate Pathway" International Journal of Molecular Sciences 22, no. 4: 1720. https://doi.org/10.3390/ijms22041720
APA StyleHuang, Y. -N., Chiang, S. -L., Lin, Y. -J., Liu, S. -C., Li, Y. -H., Liao, Y. -C., Lee, M. -R., Su, P. -H., Tsai, F. -J., Hung, H. -C., & Wang, C. -H. (2021). Long, Noncoding RNA SRA Induces Apoptosis of β-Cells by Promoting the IRAK1/LDHA/Lactate Pathway. International Journal of Molecular Sciences, 22(4), 1720. https://doi.org/10.3390/ijms22041720