
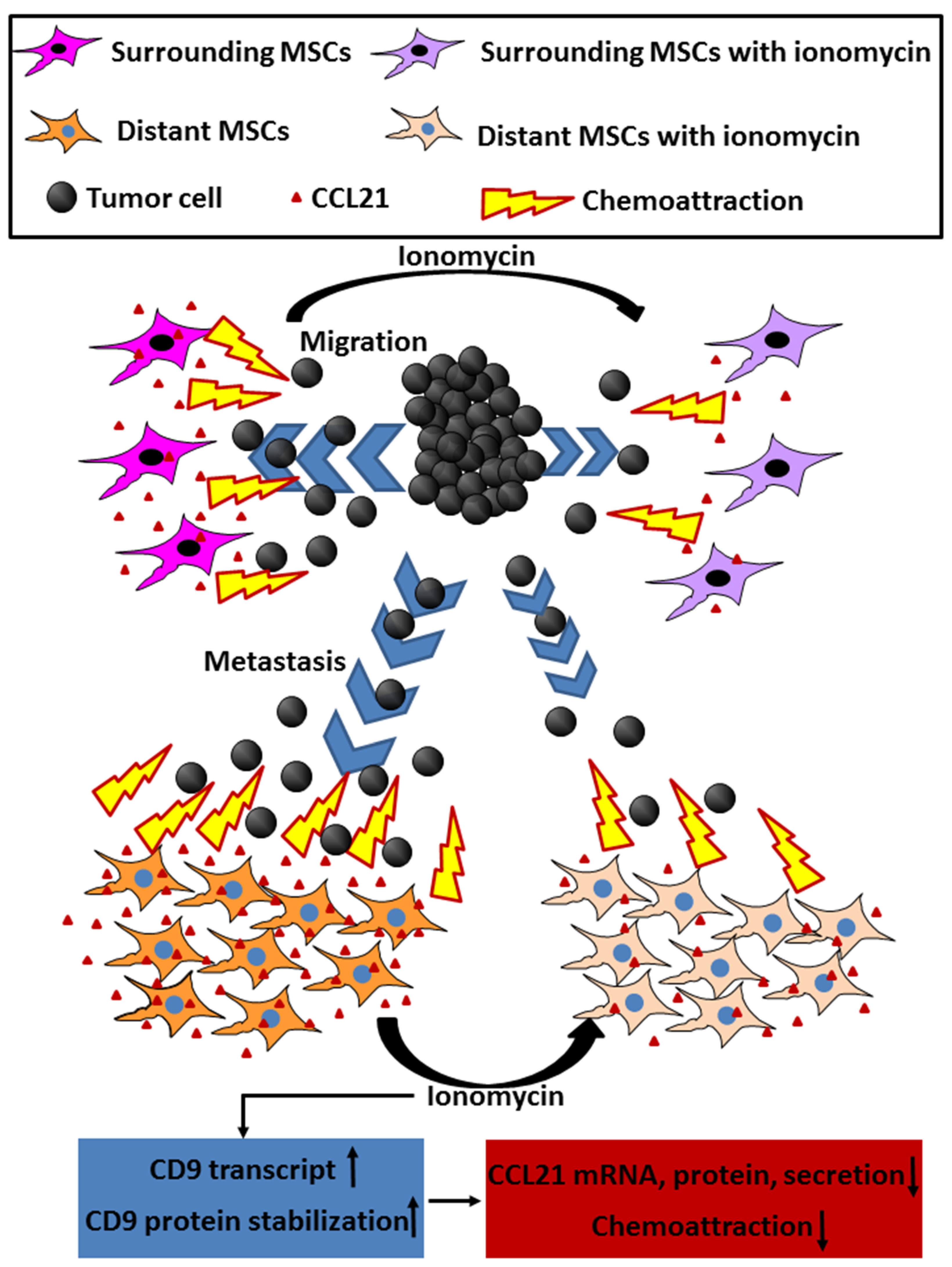
CD9 Upregulation-Decreased CCL21 Secretion in Mesenchymal Stem Cells Reduces Cancer Cell Migration


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
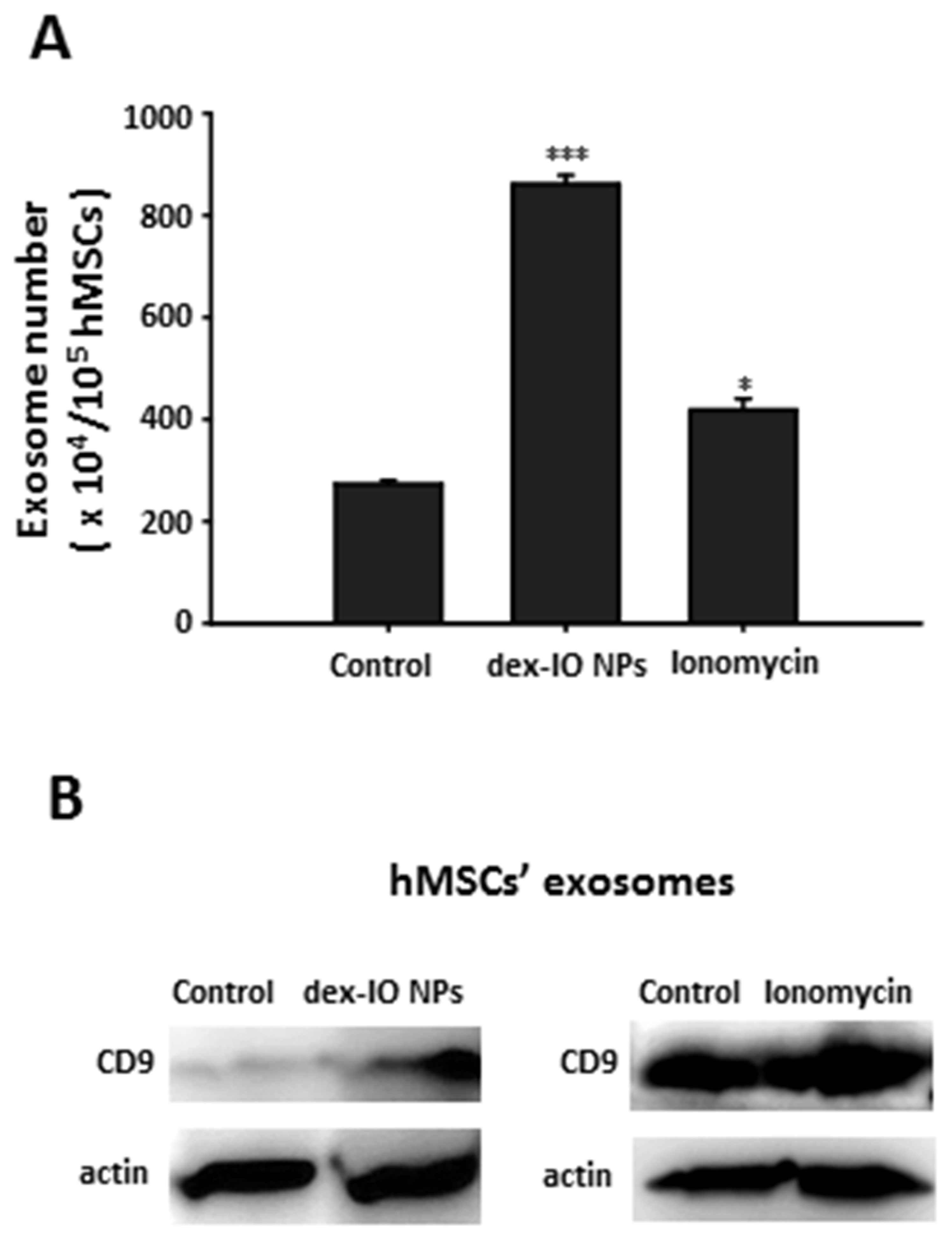
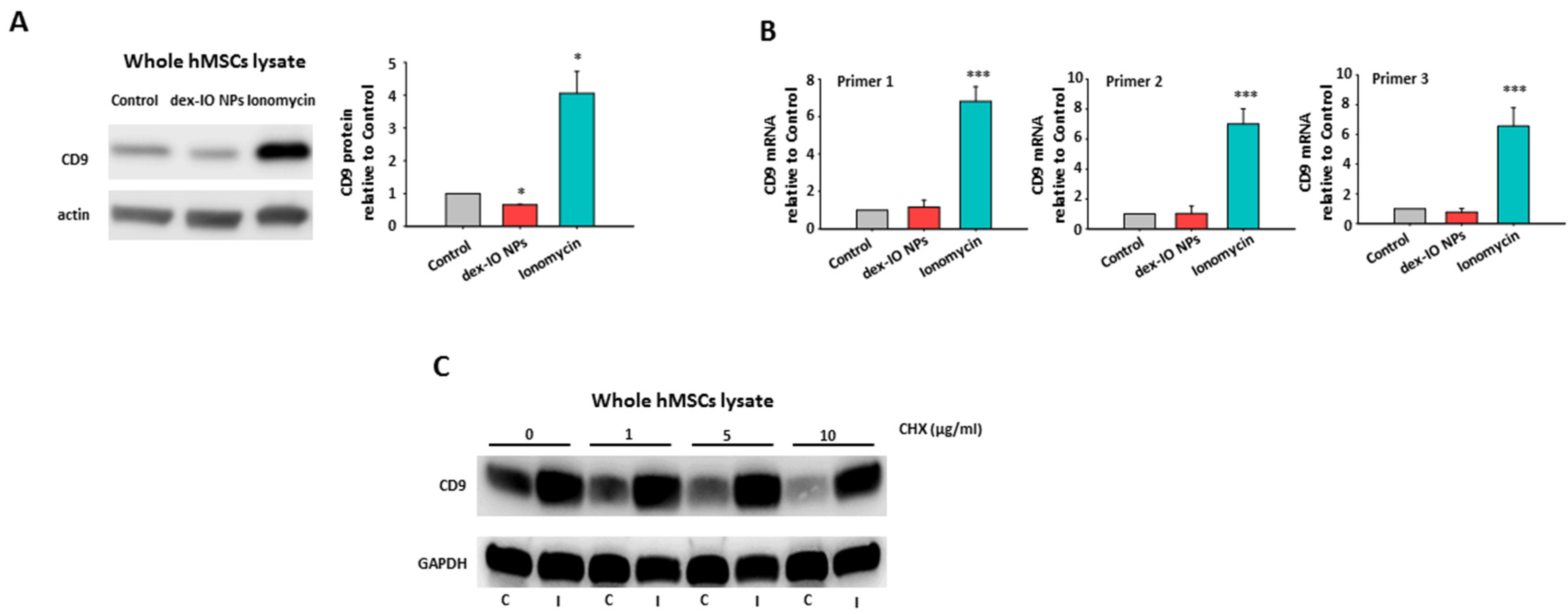
2.1. Dex-IO NPs and Ionomycin Stimulated hMSCs’ Exocytosis and Exosomal CD9 Expression
2.2. Expression of CD9 Inhibited B16F10 Cell Migration in Wound Healing Assay
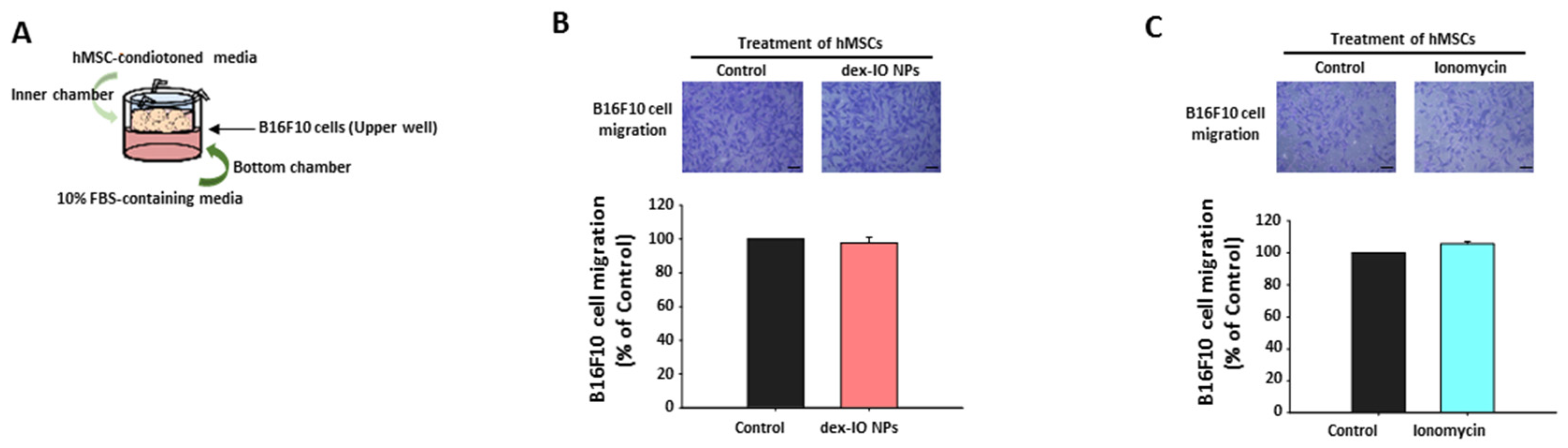
2.3. The Effect of hMSC-Conditioned Media in the Upper Chamber on B16F10 Cell Migration
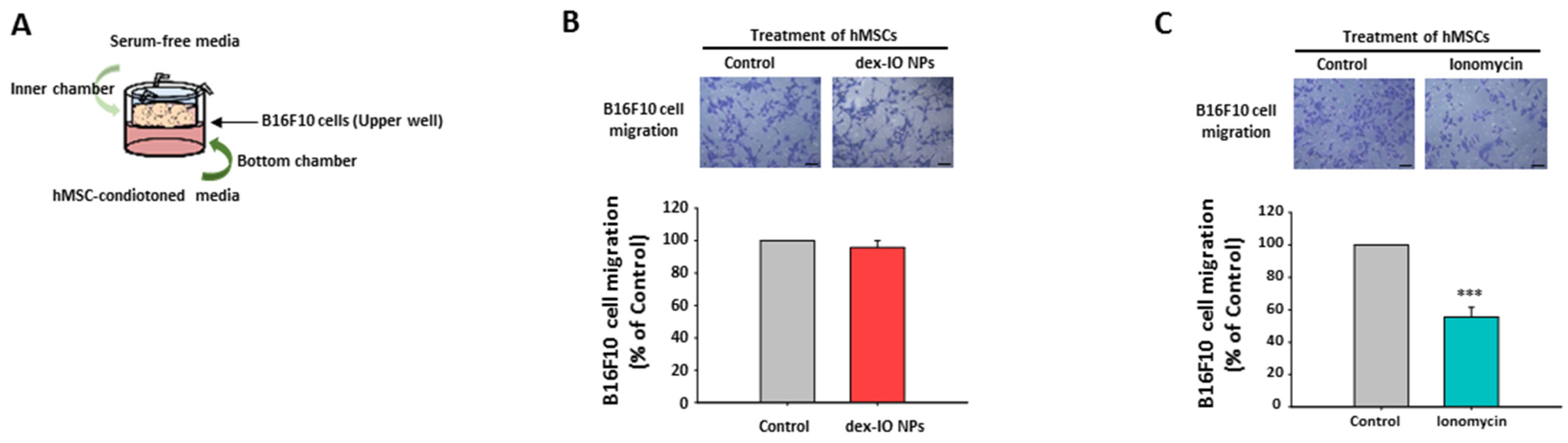
2.4. The Effect of hMSC-Conditioned Media in the Bottom Chamber on B16F10 Cell Migration
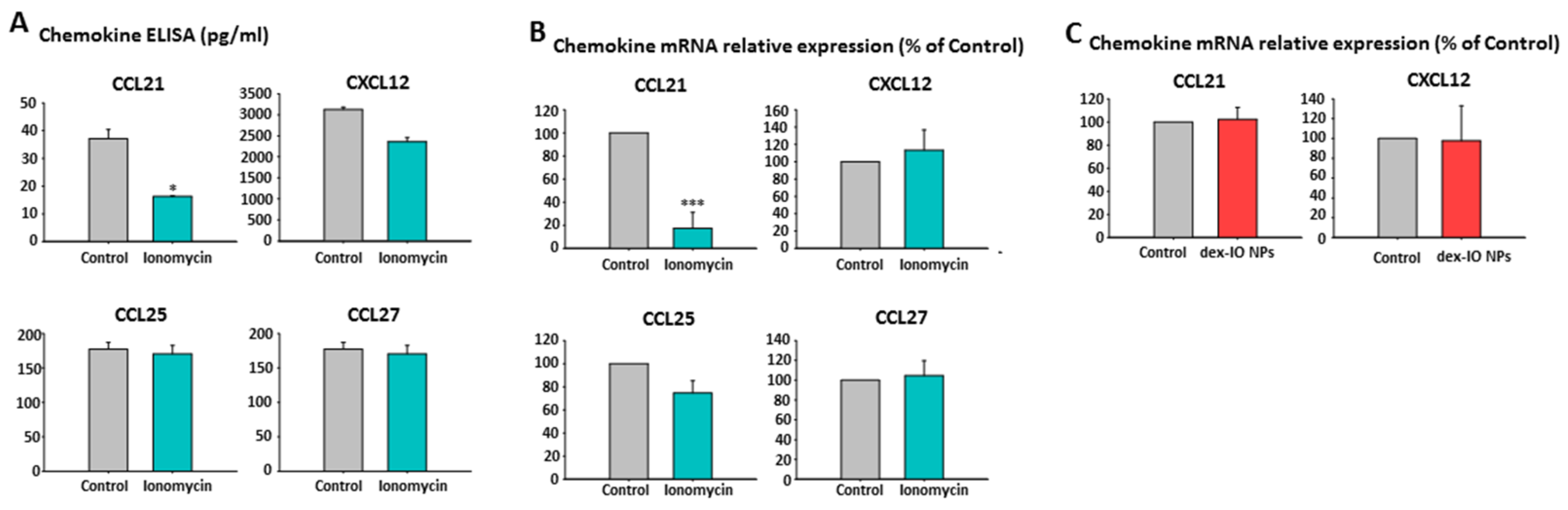
2.5. Ionomycin-Incubated hMSC-Conditioned Media Reduced B16F10 Cell Migration by Chemokine CCL21 Decrease
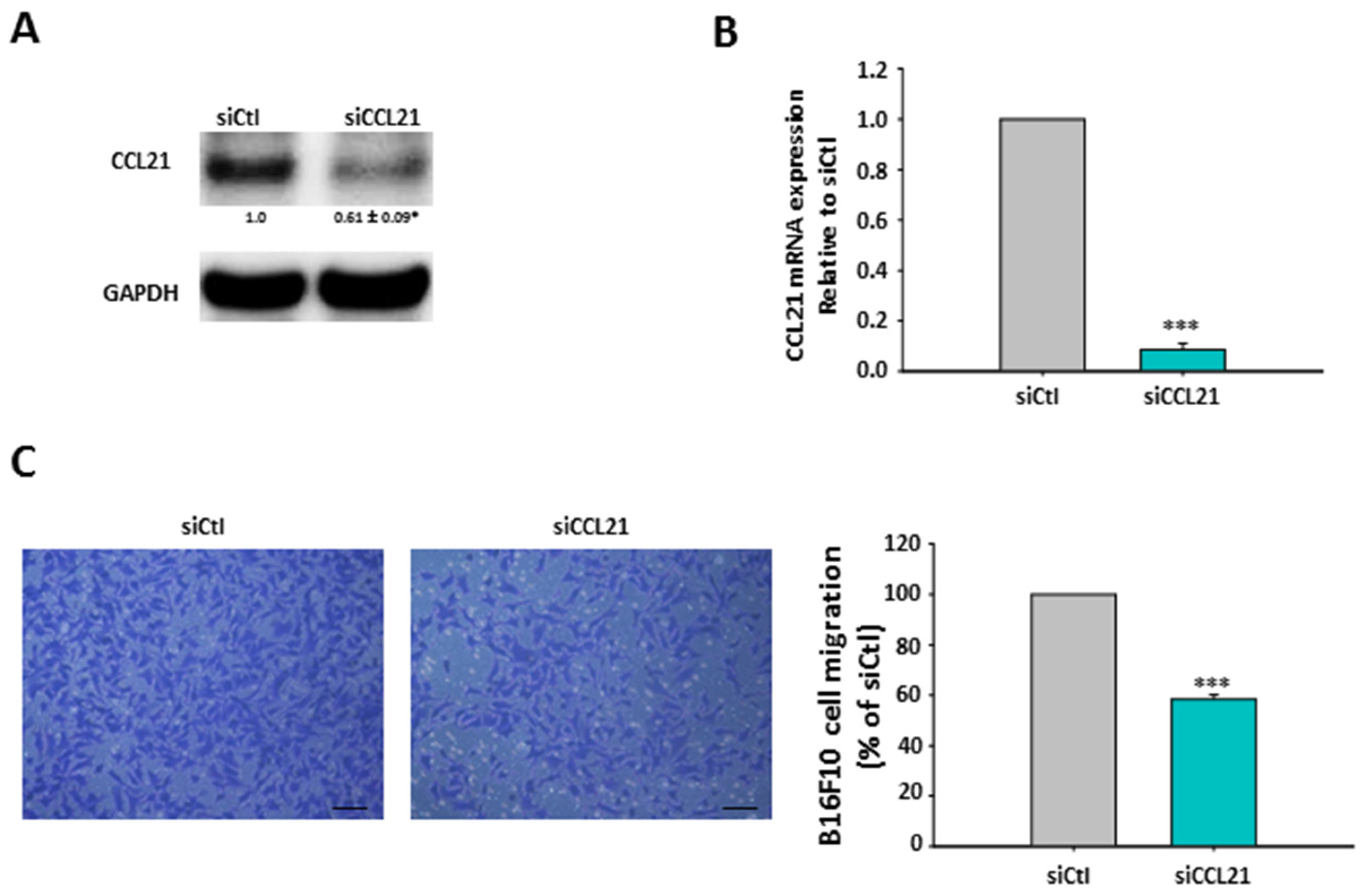
2.6. Silencing of hMSCs’ CCL21 Reduced B16F10 Cell Migration
2.7. The Mechanism for the Opposite Effects of dex-IO NPs and Ionomycin on hMSCs’ CD9 Expression
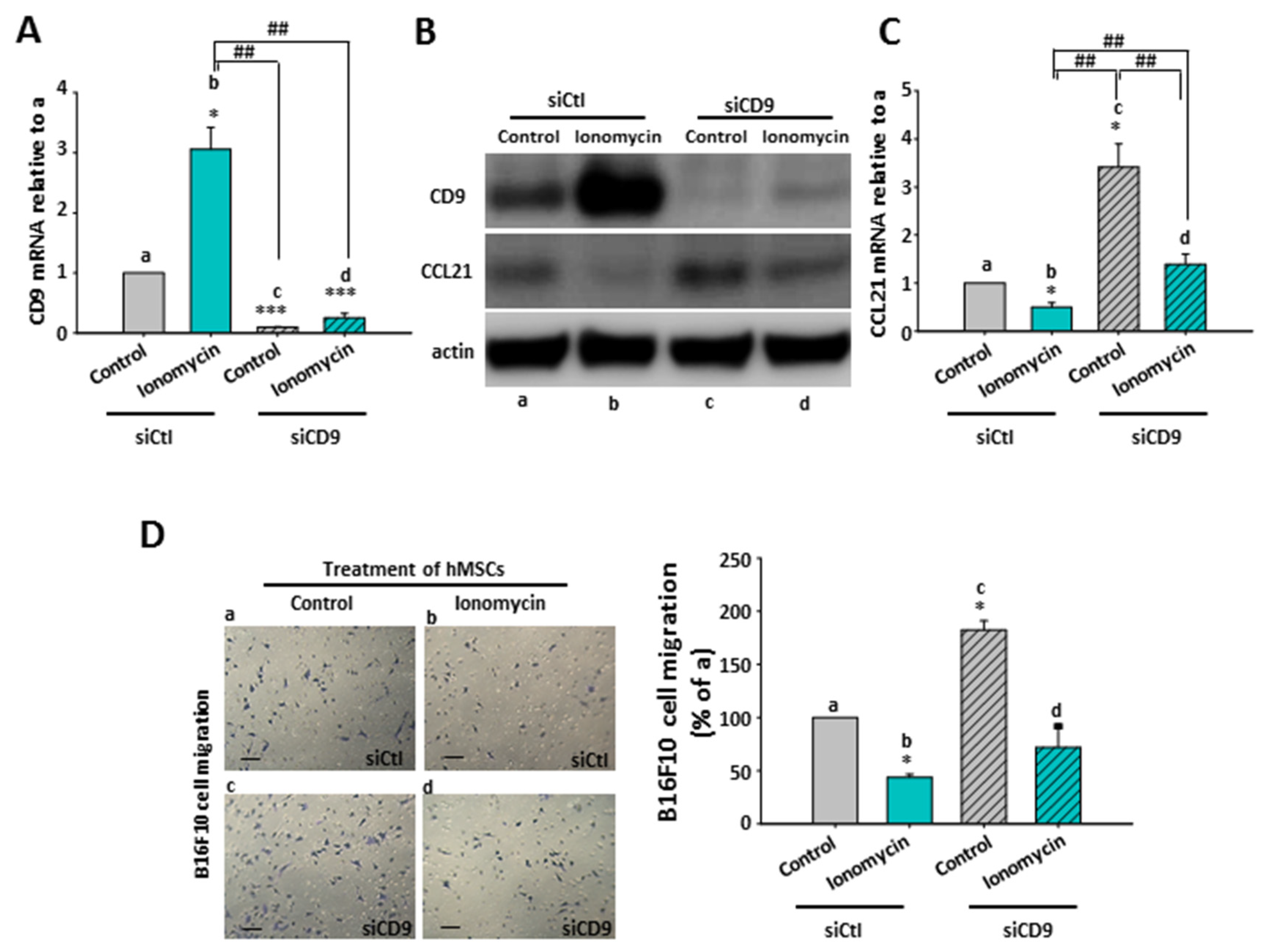
2.8. The Inhibitory Role of Ionomycin-Upregulated Cellular CD9 in hMSCs’ CCL21 Expression and B16F10 Cell Migration
2.9. The Effect of CD9-Regulated hMSCs’ CCL21 Expression and Secretion in B16F10 Cell Migration
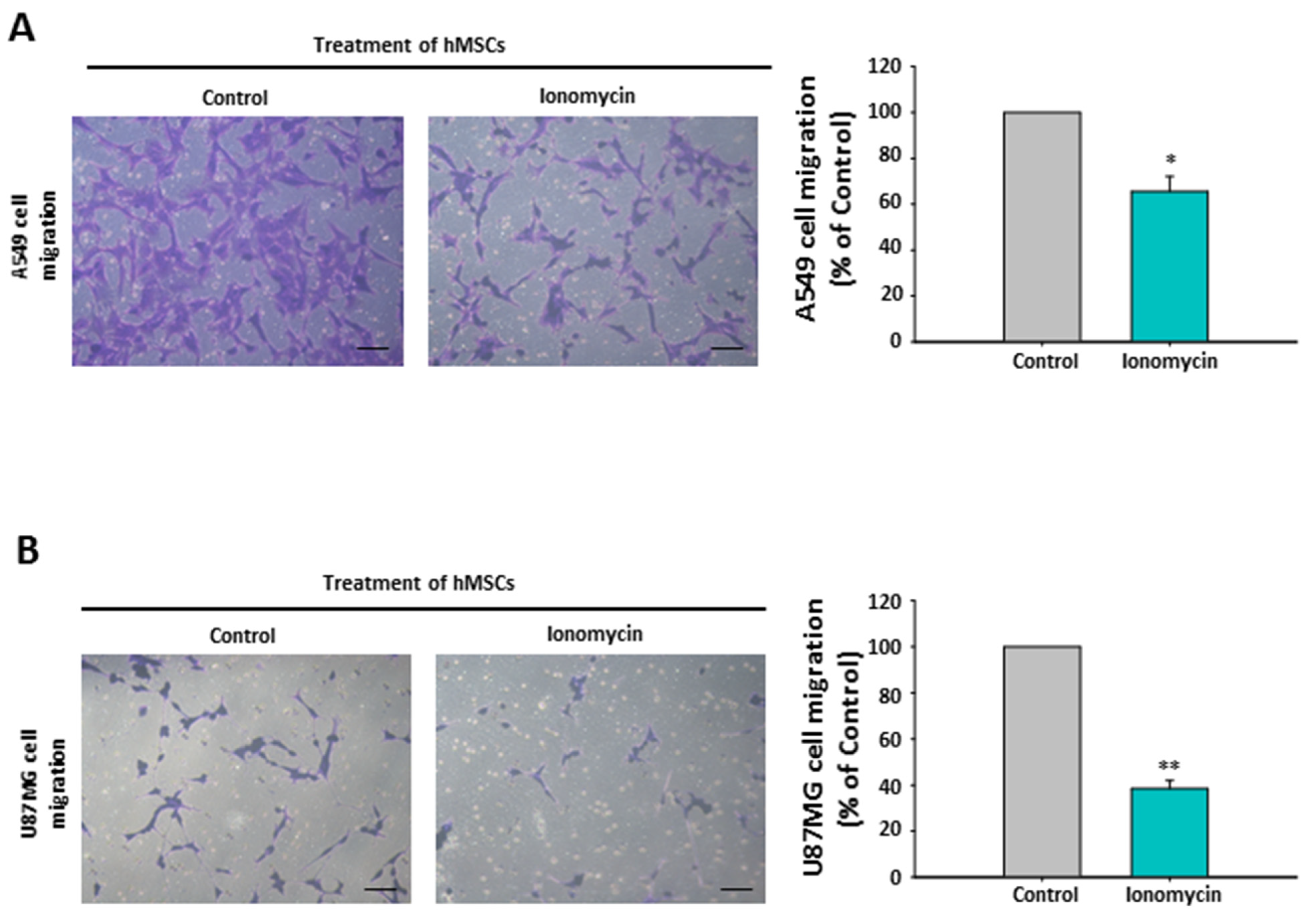
2.10. The Chemoattraction Reduction of Ionomycin-Incubated hMSC-Conditioned Media in Other Cancer Cell Lines
3. Materials and Methods
3.1. Cell Culture
3.2. Preparation and Characterization of dex-IO NPs and Ionomycin Treatment
3.3. Exosome Extraction and Identification
3.4. Transwell Migration Assay
3.5. Chemokine Protein Quantification
3.6. CD9 and Chemokine mRNA Determination
3.7. CD9 Silencing
3.8. Western Blot Analysis
3.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| hMSCs/MSCs | (Human) Mesenchymal stem cells |
| EVs | Extracellular vesicles |
| miRNAs/miR | Micro RNAs |
| CAF | Cancer-associated fibroblstas |
| IO | Iron oxide |
| NPs | Nanoparticles |
| EVs | Extracellular vesicles |
| miRNAs/miR | Micro RNAs |
| CAF | Cancer-associated fibroblstas |
| IO | Iron oxide |
| NPs | Nanoparticles |
| MRI | Magnetic resonance imaging |
| EGF/EGFR | Epidermal growth factor (receptor) |
| CCL | C-C motif chemokine ligand |
| CCR | C-C motif chemokine receptor |
| CXCL | C-X-C motif chemokine ligand |
| CXCR | C-X-C motif chemokine receptor |
| mRNA | Messenger RNA |
| CHX | Cycloheximide |
| siRNA | Small interfering RNA |
References
- Jiang, Y.; Jahagirdar, B.N.; Reinhardt, R.L.; Schwartz, R.E.; Keene, C.D.; Ortiz-Gonzalez, X.R.; Reyes, M.; Lenvik, T.; Lund, T.; Blackstad, M.; et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002, 418, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.R.; Soria, B.; Capilla-Gonzalez, V. Therapeutic potential of mesenchymal stem cells for cancer therapy. Front. Bioeng. Biotechnol. 2020, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I.; Dennis, J.E. Mesenchymal stem cells as tropic mediators. J. Cell. Biochem. 2006, 98, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, S.; Hashemi, S.M.; Ghanbarian, H.; Salehi, M.; Mohammadi-Yeganeh, S. Effect of mesenchymal stem cells-derived exosomes on tumor microenvironment: Tumor progression versus tumor suppression. J. Cell. Physiol. 2019, 234, 3394–3409. [Google Scholar] [CrossRef]
- Dai, L.J.; Moniri, M.R.; Zeng, Z.R.; Zhou, J.X.; Rayat, J.; Warnock, G.L. Potential implications of mesenchymal stem cells in cancer therapy. Cancer Lett. 2011, 305, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Klopp, A.H.; Gupta, A.; Spaeth, E.; Andreeff, M.; Marini, F., 3rd. Concise review: Dissecting a discrepancy in the literature: Do mesenchymal stem cells support or suppress tumor growth? Stem Cells 2011, 29, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Vakhshiteh, F.; Atyibi, F.; Ostad, S.Y. Mesenchymal stem cell exosomes: A two-edged sword in cancer therapy. Int. J. Nanomed. 2019, 14, 2847–2859. [Google Scholar] [CrossRef] [Green Version]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Shimbo, K.; Miyaki, S.; Ishitobi, H.; Kato, Y.; Kubo, T.; Shimose, S.; Ochi, M. Exosome-formed synthetic microRNA-143 is transferred to osteosarcoma cells and inhibits their migration. Biochem. Biophys. Res. Commun. 2014, 445, 381–387. [Google Scholar] [CrossRef]
- Maacha, S.; Sidahmed, H.; Jacob, S.; Gentilcore, G.; Calzone, R.; Grivel, J.-C.; Cugno, C. Paracrine mechanisms of mesenchymal stromal cells in angiogenesis. Stem Cells Int. 2020, 2020, 4356359. [Google Scholar] [CrossRef]
- Park, W.S.; Ahn, S.Y.; Sung, S.I.; Ahn, J.-Y.; Chang, Y.S. Strategies to enhance paracrine potency of transplanted mesenchymal stem cells in intractable neonatal disorders. Pediatr. Res. 2018, 83, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrazzo, P.; Crupi, A.N.; Alviano, F.; Teodori, L.; Bonsi, L. Exploring the roles of MSCs in infections: Focus on bacterial diseases. J. Mol. Med. 2019, 97, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Wang, S.; Zhao, R.C. Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol. Cell. Biochem. 2013, 383, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, C.; Shi, H.; Zhang, B.; Zhang, L.; Zhang, X.; Wang, S.; Wu, X.; Yang, T.; Huang, F.; et al. Deregulated microRNAs in gastric cancer tissue-derived mesenchymal stem cells: Novel biomarkers and a mechanism for gastric cancer. Br. J. Cancer 2014, 110, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Finniss, S.; Cazacu, S.; Bucris, E.; Ziv-Av, A.; Xiang, C.; Bobbitt, K.; Rempel, S.A.; Hasselbach, L.; Mikkelsen, T.; et al. Mesenchymal stem cells deliver synthetic microRNA mimics to glioma cells and glioma stem cells and inhibit their cell migration and self-renewal. Oncotarget 2013, 4, 346–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zöller, M. Tetraspanins: Push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer 2009, 9, 40–55. [Google Scholar] [CrossRef]
- Powner, D.; Kopp, P.M.; Monkley, S.J.; Critchley, D.R.; Berditchevski, F. Tetraspanin CD9 in cell migration. Biochem. Soc. Trans. 2011, 39, 563–567. [Google Scholar] [CrossRef]
- Malla, R.R.; Pandrangi, S.; Kumari, S.; Gavara, M.M.; Badana, A.K. Exosomal tetraspanins as regulators of cancer progression and metastasis and novel diagnostic markers. Asia Pac. J. Clin. Oncol. 2018, 14, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Yashiro, M.; Okuno, T.; Kitayama, K.; Masuda, G.; Hirakawa, K.; Ohire, M. CD9-positive exosomes from cancer-associated fibroblasts stimulate the migration ability of scirrhous-type gastric cancer cells. Br. J. Cancer 2018, 118, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Chung, T.-H.; Hsiao, J.-K.; Hsu, S.-C.; Yao, M.; Wang, S.-W.; Kuo, M.-K.; Yang, C.-S.; Huang, D.-M. Iron oxide nanoparticle-induced epidermal growth factor receptor expression in human stem cells for tumor therapy. ACS Nano 2011, 5, 9807–9816. [Google Scholar] [CrossRef]
- Chung, T.-H.; Hsiao, J.-K.; Yao, M.; Hsu, S.-C.; Liu, H.-M.; Huang, D.-M. Ferucarbotran, a carboxydextran-coated superparamagnetic iron oxide nanoparticle, induces endosomal recycling contributing to cellular and exosomal EGFR overexpression for cancer therapy. RSC Adv. 2015, 5, 88932–88939. [Google Scholar] [CrossRef]
- Chung, T.-H.; Hsieh, C.-C.; Hsiao, J.-K.; Yao, M.; Hsu, S.-C.; Huang, D.-M. Dextran-coated iron oxide nanoparticles turn protumor mesenchymal stem cells (MSCs) into antitumor MSCs. RSC Adv. 2016, 6, 45553–45561. [Google Scholar] [CrossRef]
- Hwangbo, H.; Choi, E.O.; Kim, M.Y.; Kwon, D.H.; Ji, S.Y.; Lee, H.; Hong, S.H.; Kim, G.Y.; Hwang, H.J.; Hong, S.H.; et al. Suppression of tumor growth and metastasis by ethanol extract of Angelica dahurica Radix in murine melanoma B16F10 cells. Biosci. Trends 2020, 14, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Xiong, H.; Duan, L.; Li, Q.; Li, X.; Zhou, Y. MiR-1246 promotes metastasis and invasion of A549 cells by targeting GSK-3β‒mediated Wnt/β-catenin pathway. Cancer Res. Treat. 2019, 51, 1420–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwon, D.H.; Lee, W.Y.; Shin, N.; Kim, S.I.; Jeong, K.; Lee, W.H.; Kim, D.W.; Hong, J.; Lee, S.Y. BMAL1 suppresses proliferation, migration, and invasion of U87MG cells by downregulating cyclin B1, phospho-AKT, and metalloproteinase-9. Int. J. Mol. Sci. 2020, 21, 2352. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.D.; Holmberg, C.; Balabanova, S.; Borysova, L.; Burdyga, T.; Beynon, R.; Dockray, G.J.; Varro, A. Mesenchymal stem cells exhibit regulated exocytosis in response to chemerin and IGF. PLoS ONE 2015, 10, e0141331. [Google Scholar] [CrossRef]
- Jacquelot, N.; Duong, C.P.M.; Belz, G.T.; Zitvogel, L. Targeting chemokines and chemokine receptors in melanoma and other cancers. Front. Immunol. 2018, 9, 2480. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Zoller, M. Exosome target cell selection and the importance of exosomal tetraspanins: A hypothesis. Biochem. Soc. Trans. 2011, 11, 559–562. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.; Yue, S.; Stadel, D.; Zoller, M. Toward tailored exosomes: The exosomal tetraspanin web contributes to target cell selection. Int. J. Biochem. Cell Biol. 2012, 44, 1574–1584. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, C.-C.; Hsu, S.-C.; Yao, M.; Huang, D.-M. CD9 Upregulation-Decreased CCL21 Secretion in Mesenchymal Stem Cells Reduces Cancer Cell Migration. Int. J. Mol. Sci. 2021, 22, 1738. https://doi.org/10.3390/ijms22041738
Hsieh C-C, Hsu S-C, Yao M, Huang D-M. CD9 Upregulation-Decreased CCL21 Secretion in Mesenchymal Stem Cells Reduces Cancer Cell Migration. International Journal of Molecular Sciences. 2021; 22(4):1738. https://doi.org/10.3390/ijms22041738
Chicago/Turabian StyleHsieh, Chia-Chu, Szu-Chun Hsu, Ming Yao, and Dong-Ming Huang. 2021. "CD9 Upregulation-Decreased CCL21 Secretion in Mesenchymal Stem Cells Reduces Cancer Cell Migration" International Journal of Molecular Sciences 22, no. 4: 1738. https://doi.org/10.3390/ijms22041738
APA StyleHsieh, C.-C., Hsu, S.-C., Yao, M., & Huang, D.-M. (2021). CD9 Upregulation-Decreased CCL21 Secretion in Mesenchymal Stem Cells Reduces Cancer Cell Migration. International Journal of Molecular Sciences, 22(4), 1738. https://doi.org/10.3390/ijms22041738