
The Role of mTOR Signaling as a Therapeutic Target in Cancer


Abstract
:1. Mechanistic/Mammalian Target of Rapamycin (mTOR)
2. Activation of mTOR
2.1. PI3K/AKT/mTOR Signalling Pathway
2.2. Regulation of mTORC1 by Other Factors
3. Deregulation of the PI3K/AKT/mTOR Pathway in Cancer
3.1. PI3K in Human Cancer
3.2. AKT in Cancers
3.3. Alterations of Receptor Tyrosine Kinases
3.4. PTEN
3.5. mTOR and Cancer Development
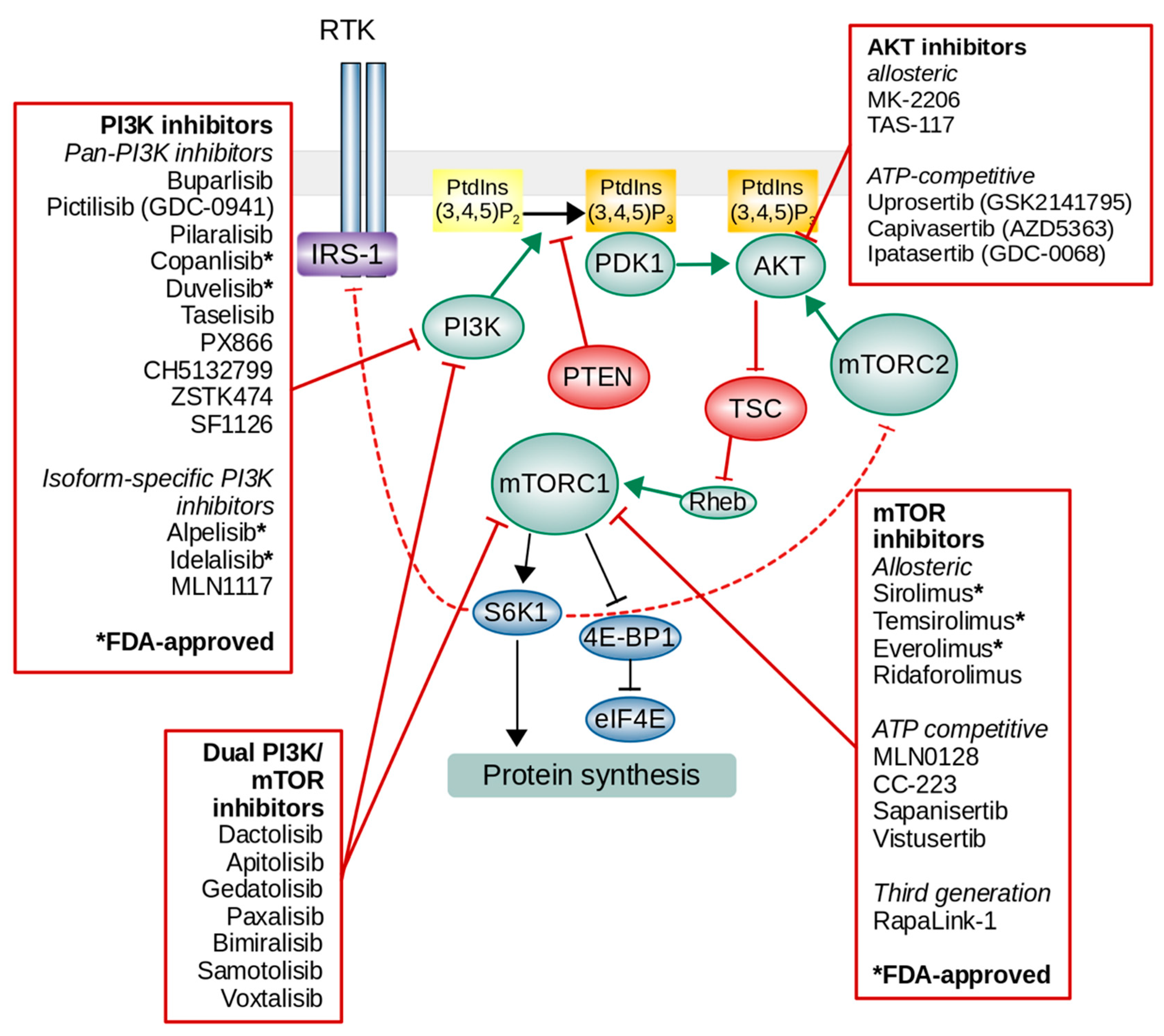
4. Targeting PI3K/AKT/mTOR
4.1. PI3K Inhibitors
4.2. AKT Inhibitors
4.3. mTOR Inhibitors
4.3.1. The First Generation: Allosteric mTOR Inhibitors
4.3.2. The Second Generation: ATP-Competitive mTOR Inhibitors
4.3.3. The Third Generation: RapaLink
4.4. Dual PI3K/mTOR Inhibitors
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AICAR | 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside |
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| BAD | BCL2-associated agonist of cell death |
| CTMP | carboxyl-terminal modulator protein |
| DEPTOR | DEP domain-containing mTOR-interacting protein |
| DNA-PK | DNA-dependent protein kinase |
| eIF4E | eukaryotic translation initiation factor 4E |
| ERK | extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| FKBP12 | FK506-binding protein 12 |
| FoxO | forkhead box O transcription factors |
| FRB | FKBP12-rapamycin-binding |
| GAP | GTPase activating protein |
| GβL | G protein β-subunit-like protein |
| GSK3 | glycogen synthase kinase 3 |
| IGF-1 | insulin-like growth factor-1 |
| INPP4B | inositol polyphosphate 4-phosphatase type II |
| IRS-1 | insulin receptor substrate 1 |
| MAP kinase | mitogen-activated protein kinase |
| mLST8 | mammalian lethal with SEC13 protein 8 |
| mSIN1 | stress-activated map kinase-interacting protein 1 |
| mTOR | mechanistic/mammalian target of rapamycin |
| PDCD4 | programmed cell death protein 4 |
| PDK1 | phosphoinositide-dependent kinase 1 |
| PI3K | phosphatidylinositol-3-kinase |
| PIKK | phosphatidylinositol 3-kinase-related kinase |
| PI(4,5)P2 | phosphatidylinositol 4,5 bisphosphate |
| PI(3,4,5)P3 | phosphatidylinositol 3,4,5 trisphosphate |
| PLD | phospholipase D |
| PRAS40 | proline-rich AKT1 substrate 1 |
| Protor | protein observed with RICTOR |
| PTEN | phosphatase and tensin homolog |
| RAPTOR | regulatory-associated protein of mTOR |
| Rheb | Ras homolog enriched in brain |
| RICTOR | rapamycin insensitive companion of mTOR |
| RSK | ribosomal S6 kinase |
| RTK | receptor tyrosine kinase |
| S6K1 | p70 S6 kinase, ribosomal protein S6 kinase beta-1 |
| SGK1 | serum and glucocorticoid-activated kinase 1 |
| SHIP | Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase |
| TBC1D7 | TBC1 domain family member 7 |
| TIF-1A | transcription initiation factor 1A |
| TSC | tuberous sclerosis complex |
| UBF | upstream binding factor |
References
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A Mammalian Protein Targeted by G1-Arresting Rapamycin-Receptor Complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A Mammalian Protein That Binds to FKBP12 in a Rapamycin-Dependent Fashion and Is Homologous to Yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a Protein Target of the FKBP12-Rapamycin Complex in Mammalian Cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR Complexes, Only One of Which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.P.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GβL, a Positive Regulator of the Rapamycin-Sensitive Pathway Required for the Nutrient-Sensitive Interaction between Raptor and MTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. MTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex That Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the MTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an MTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The Mammalian Target of Rapamycin (MTOR) Partner, Raptor, Binds the MTOR Substrates P70 S6 Kinase and 4E-BP1 through Their TOR Signaling (TOS) Motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [Green Version]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS Motif-Mediated Raptor Binding Regulates 4E-BP1 Multisite Phosphorylation and Function. Curr. Biol. CB 2003, 13, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Haar, E.V.; Lee, S.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin Signalling to MTOR Mediated by the Akt/PKB Substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Scaiola, A.; Mangia, F.; Imseng, S.; Boehringer, D.; Berneiser, K.; Shimobayashi, M.; Stuttfeld, E.; Hall, M.N.; Ban, N.; Maier, T. The 3.2-Å Resolution Structure of Human MTORC2. Sci. Adv. 2020, 6, eabc1251. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of MTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway That Regulates the Cytoskeleton. Curr. Biol. CB 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.-L. Identification of Sin1 as an Essential TORC2 Component Required for Complex Formation and Kinase Activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.M.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a Novel Rictor-Binding Component of MTOR Complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-H.; Sarbassov, D.D. The MTOR (Mammalian Target of Rapamycin) Kinase Maintains Integrity of MTOR Complex 2. J. Biol. Chem. 2011, 286, 40386–40394. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Sommer, E.M.; Sakamoto, K.; Wullschleger, S.; Alessi, D.R. Protor-1 Is Required for Efficient MTORC2-Mediated Activation of SGK1 in the Kidney. Biochem. J. 2011, 436, 169–179. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR Complex 2 Controls the Actin Cytoskeleton and Is Rapamycin Insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Bierer, B.E.; Mattila, P.S.; Standaert, R.F.; Herzenberg, L.A.; Burakoff, S.J.; Crabtree, G.; Schreiber, S.L. Two Distinct Signal Transmission Pathways in T Lymphocytes Are Inhibited by Complexes Formed between an Immunophilin and Either FK506 or Rapamycin. Proc. Natl. Acad. Sci. USA 1990, 87, 9231–9235. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-FKBP Specifically Blocks Growth-Dependent Activation of and Signaling by the 70 Kd S6 Protein Kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. MTOR Kinase Structure, Mechanism and Regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, M.; Tian, Y.; Li, J.; Qi, Y.; Zhao, D.; Wu, Z.; Huang, M.; Wong, C.C.L.; Wang, H.-W.; et al. Cryo-EM Structure of Human MTOR Complex 2. Cell Res. 2018, 28, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Stuttfeld, E.; Aylett, C.H.; Imseng, S.; Boehringer, D.; Scaiola, A.; Sauer, E.; Hall, M.N.; Maier, T.; Ban, N. Architecture of the Human MTORC2 Core Complex. eLife 2018, 7, e33101. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-Induced Insulin Resistance Is Mediated by MTORC2 Loss and Uncoupled from Longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits MTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Fruman, D.A.; Meyers, R.E.; Cantley, L.C. Phosphoinositide Kinases. Annu. Rev. Biochem. 1998, 67, 481–507. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Pawson, T.; Gish, G.D.; Nash, P. SH2 Domains, Interaction Modules and Cellular Wiring. Trends Cell Biol. 2001, 11, 504–511. [Google Scholar] [CrossRef]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. PTEN: A Tumour Suppressor That Functions as a Phospholipid Phosphatase. Trends Cell Biol. 1999, 9, 125–128. [Google Scholar] [CrossRef]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative Regulation of PKB/Akt-Dependent Cell Survival by the Tumor Suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Cantley, L.C. Regulation of MTORC1 by PI3K Signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.; Komander, D.; van Aalten, D.M.F.; Alessi, D.R. PDK1, the Master Regulator of AGC Kinase Signal Transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-Phosphoinositide-Dependent Protein Kinase Which Phosphorylates and Activates Protein Kinase Balpha. Curr. Biol. CB 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Stephens, L.; Anderson, K.; Stokoe, D.; Erdjument-Bromage, H.; Painter, G.F.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; McCormick, F.; Tempst, P.; et al. Protein Kinase B Kinases That Mediate Phosphatidylinositol 3,4,5-Trisphosphate-Dependent Activation of Protein Kinase B. Science 1998, 279, 710–714. [Google Scholar] [CrossRef]
- Hresko, R.C.; Mueckler, M. MTOR.RICTOR Is the Ser473 Kinase for Akt/Protein Kinase B in 3T3-L1 Adipocytes. J. Biol. Chem. 2005, 280, 40406–40416. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The Nuts and Bolts of AGC Protein Kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 Is Phosphorylated and Inhibited by Akt and Suppresses MTOR Signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of MTORC1 at the Lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt Regulates Growth by Directly Phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 Is a Third Subunit of the TSC1-TSC2 Complex Upstream of MTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [Green Version]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between Hamartin and Tuberin, the TSC1 and TSC2 Gene Products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase Is a Direct Target of TSC2 GAP Activity and Regulates MTOR Signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control MTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb Binds and Regulates the MTOR Kinase. Curr. Biol. CB 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets MTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C. PRAS40 Regulates MTORC1 Kinase Activity by Functioning as a Direct Inhibitor of Substrate Binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and Functional Inactivation of TSC2 by Erk Implications for Tuberous Sclerosis and Cancer Pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-Promoting Phorbol Esters and Activated Ras Inactivate the Tuberous Sclerosis Tumor Suppressor Complex via P90 Ribosomal S6 Kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Fu, W.; Hall, M.N. Regulation of MTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 Phosphorylation of the Translational Regulators P70 S6 Kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar]
- Brunn, G.J.; Hudson, C.C.; Sekulić, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C.; Abraham, R.T. Phosphorylation of the Translational Repressor PHAS-I by the Mammalian Target of Rapamycin. Science 1997, 277, 99–101. [Google Scholar] [CrossRef]
- Gingras, A.-C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 Phosphorylation: A Novel Two-Step Mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar]
- Hara, K.; Yonezawa, K.; Kozlowski, M.T.; Sugimoto, T.; Andrabi, K.; Weng, Q.P.; Kasuga, M.; Nishimoto, I.; Avruch, J. Regulation of EIF-4E BP1 Phosphorylation by MTOR. J. Biol. Chem. 1997, 272, 26457–26463. [Google Scholar] [CrossRef] [Green Version]
- Hannan, K.; Brandenburger, Y.; Jenkins, A.; Sharkey, K.; Cavanaugh, A.; Rothblum, L.; Moss, T.; Poortinga, G.; Mcarthur, G.; Pearson, R.; et al. MTOR-Dependent Regulation of Ribosomal Gene Transcription Requires S6K1 and Is Mediated by Phosphorylation of the Carboxy-Terminal Activation Domain of the Nucleolar Transcription Factor UBF†. Mol. Cell. Biol. 2004, 23, 8862–8877. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. MTOR-Dependent Activation of the Transcription Factor TIF-IA Links RRNA Synthesis to Nutrient Availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Michels, A.A.; Robitaille, A.M.; Buczynski-Ruchonnet, D.; Hodroj, W.; Reina, J.H.; Hall, M.N.; Hernandez, N. MTORC1 Directly Phosphorylates and Regulates Human MAF1. Mol. Cell. Biol. 2010, 30, 3749–3757. [Google Scholar] [CrossRef] [Green Version]
- Shor, B.; Wu, J.; Shakey, Q.; Toral-Barza, L.; Shi, C.; Follettie, M.; Yu, K. Requirement of the MTOR Kinase for the Regulation of Maf1 Phosphorylation and Control of RNA Polymerase III-Dependent Transcription in Cancer Cells. J. Biol. Chem. 2010, 285, 15380–15392. [Google Scholar] [CrossRef] [Green Version]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. MTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and BetaTRCP-Mediated Degradation of PDCD4 Promotes Protein Translation and Cell Growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 Tumor Suppressor Controls Insulin–PI3K Signaling via Regulation of IRS Proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Shah, O.J.; Wang, Z.; Hunter, T. Inappropriate Activation of the TSC/Rheb/MTOR/S6K Cassette Induces IRS1/2 Depletion, Insulin Resistance, and Cell Survival Deficiencies. Curr. Biol. 2004, 14, 1650–1656. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 Phosphorylation Impairs MTORC2 Complex Integrity and Inhibits Downstream Akt Signalling to Suppress Tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef]
- Bunney, T.D.; Katan, M. Phosphoinositide Signalling in Cancer: Beyond PI3K and PTEN. Nat. Rev. Cancer 2010, 10, 342–352. [Google Scholar] [CrossRef]
- Woscholski, R.; Finan, P.M.; Radley, E.; Totty, N.F.; Sterling, A.E.; Hsuan, J.J.; Waterfield, M.D.; Parker, P.J. Synaptojanin Is the Major Constitutively Active Phosphatidylinositol-3,4,5-Trisphosphate 5-Phosphatase in Rodent Brain. J. Biol. Chem. 1997, 272, 9625–9628. [Google Scholar] [CrossRef] [Green Version]
- Brognard, J.; Sierecki, E.; Gao, T.; Newton, A.C. PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms. Mol. Cell 2007, 25, 917–931. [Google Scholar] [CrossRef]
- Maira, S.M.; Galetic, I.; Brazil, D.P.; Kaech, S.; Ingley, E.; Thelen, M.; Hemmings, B.A. Carboxyl-Terminal Modulator Protein (CTMP), a Negative Regulator of PKB/Akt and v-Akt at the Plasma Membrane. Science 2001, 294, 374–380. [Google Scholar] [CrossRef]
- Foster, D.A. Regulation of MTOR by Phosphatidic Acid? Cancer Res. 2007, 67, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Bond, P. Regulation of MTORC1 by Growth Factors, Energy Status, Amino Acids and Mechanical Stimuli at a Glance. J. Int. Soc. Sports Nutr. 2016, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gulati, P.; Thomas, G. Nutrient Sensing in the MTOR/S6K1 Signalling Pathway. Biochem. Soc. Trans. 2007, 35, 236–238. [Google Scholar] [CrossRef]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino Acids Mediate MTOR/Raptor Signaling through Activation of Class 3 Phosphatidylinositol 3OH-Kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in Nutrient Response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to MTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK—A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-Activated/SNF1 Protein Kinases: Conserved Guardians of Cellular Energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic Acid-Mediated Mitogenic Activation of MTOR Signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef]
- Saito, M.; Kanfer, J. Phosphatidohydrolase Activity in a Solubilized Preparation from Rat Brain Particulate Fraction. Arch. Biochem. Biophys. 1975, 169, 318–323. [Google Scholar] [CrossRef]
- Singer, W.D.; Brown, H.A. Sternweis, P.C. regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu. Rev. Biochem. 1997, 66, 475–509. [Google Scholar] [CrossRef]
- Ballou, L.M.; Jiang, Y.-P.; Du, G.; Frohman, M.A.; Lin, R.Z. Ca(2+)- and Phospholipase D-Dependent and -Independent Pathways Activate MTOR Signaling. FEBS Lett. 2003, 550, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Park, I.-H.; Wu, A.-L.; Du, G.; Huang, P.; Frohman, M.A.; Walker, S.J.; Brown, H.A.; Chen, J. PLD1 Regulates MTOR Signaling and Mediates Cdc42 Activation of S6K1. Curr. Biol. CB 2003, 13, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Kam, Y.; Exton, J.H. Role of Phospholipase D1 in the Regulation of MTOR Activity by Lysophosphatidic Acid. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 311–319. [Google Scholar] [CrossRef]
- du Rusquec, P.; Blonz, C.; Frenel, J.S.; Campone, M. Targeting the PI3K/Akt/MTOR Pathway in Estrogen-Receptor Positive HER2 Negative Advanced Breast Cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920940939. [Google Scholar] [CrossRef]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/MTOR Signaling Pathway as a Therapeutic Target for Ovarian Cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef]
- Dobashi, Y.; Watanabe, Y.; Miwa, C.; Suzuki, S.; Koyama, S. Mammalian Target of Rapamycin: A Central Node of Complex Signaling Cascades. Int. J. Clin. Exp. Pathol. 2011, 4, 476–495. [Google Scholar]
- Fedele, C.G.; Ooms, L.M.; Ho, M.; Vieusseux, J.; O’Toole, S.A.; Millar, E.K.; Lopez-Knowles, E.; Sriratana, A.; Gurung, R.; Baglietto, L.; et al. Inositol Polyphosphate 4-Phosphatase II Regulates PI3K/Akt Signaling and Is Lost in Human Basal-like Breast Cancers. Proc. Natl. Acad. Sci. USA 2010, 107, 22231–22236. [Google Scholar] [CrossRef] [Green Version]
- Saal, L.H.; Gruvberger-Saal, S.K.; Persson, C.; Lövgren, K.; Jumppanen, M.; Staaf, J.; Jönsson, G.; Pires, M.M.; Maurer, M.; Holm, K.; et al. Recurrent Gross Mutations of the PTEN Tumor Suppressor Gene in Breast Cancers with Deficient DSB Repair. Nat. Genet. 2008, 40, 102–107. [Google Scholar] [CrossRef]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An Integrative Genomic and Proteomic Analysis of PIK3CA, PTEN, and AKT Mutations in Breast Cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [Green Version]
- Toker, A.; Cantley, L.C. Signalling through the Lipid Products of Phosphoinositide-3-OH Kinase. Nature 1997, 387, 673–676. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH Kinase as a Direct Target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The Evolution of Phosphatidylinositol 3-Kinases as Regulators of Growth and Metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Vanhaesebroeck, B. PI3K in Lymphocyte Development, Differentiation and Activation. Nat. Rev. Immunol. 2003, 3, 317–330. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA Oncogene in Human Cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Lee, H.W.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. PIK3CA Gene Is Frequently Mutated in Breast Carcinomas and Hepatocellular Carcinomas. Oncogene 2005, 24, 1477–1480. [Google Scholar] [CrossRef] [Green Version]
- Levine, D.A.; Bogomolniy, F.; Yee, C.J.; Lash, A.; Barakat, R.R.; Borgen, P.I.; Boyd, J. Frequent Mutation of the PIK3CA Gene in Ovarian and Breast Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 2875–2878. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.Y.; Wei, S.J.; Lin, Y.C.; Lung, J.C.; Chang, T.C.; Whang-Peng, J.; Liu, J.M.; Yang, D.M.; Yang, W.K.; Shen, C.Y. PIK3CA as an Oncogene in Cervical Cancer. Oncogene 2000, 19, 2739–2744. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [Green Version]
- Vadas, O.; Burke, J.E.; Zhang, X.; Berndt, A.; Williams, R.L. Structural Basis for Activation and Inhibition of Class I Phosphoinositide 3-Kinases. Sci. Signal. 2011, 4, re2. [Google Scholar] [CrossRef]
- Hao, Y.; Wang, C.; Cao, B.; Hirsch, B.M.; Song, J.; Markowitz, S.D.; Ewing, R.M.; Sedwick, D.; Liu, L.; Zheng, W.; et al. Gain of Interaction with IRS1 by P110α-Helical Domain Mutants Is Crucial for Their Oncogenic Functions. Cancer Cell 2013, 23, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Pang, H.; Flinn, R.; Patsialou, A.; Wyckoff, J.; Roussos, E.T.; Wu, H.; Pozzuto, M.; Goswami, S.; Condeelis, J.S.; Bresnick, A.R.; et al. Differential Enhancement of Breast Cancer Cell Motility and Metastasis by Helical and Kinase Domain Mutations of Class IA Phosphoinositide 3-Kinase. Cancer Res. 2009, 69, 8868–8876. [Google Scholar] [CrossRef] [Green Version]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, Regulation and Function of PKB/AKT—a Major Therapeutic Target. Biochim. Biophys. Acta BBA—Proteins Proteom. 2004, 1697, 3–16. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Felicioni, L.; Buttitta, F.; Lamba, S.; Cardone, L.; Rodolfo, M.; Scarpa, A.; Leenstra, S.; Frattini, M.; Barbareschi, M.; et al. AKT1(E17K) in Human Solid Tumours. Oncogene 2008, 27, 5648–5650. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Huang, L.; Dong, Y.; Tao, C.; Zhang, R.; Shao, H.; Shen, H. Effect of AKT1 (p. E17K) Hotspot Mutation on Malignant Tumorigenesis and Prognosis. Front. Cell Dev. Biol. 2020, 8, 996. [Google Scholar] [CrossRef]
- Landgraf, K.E.; Pilling, C.; Falke, J.J. Molecular Mechanism of an Oncogenic Mutation That Alters Membrane Targeting: Glu17Lys Modifies the PIP Lipid Specificity of the AKT1 PH Domain. Biochemistry 2008, 47, 12260–12269. [Google Scholar] [CrossRef] [Green Version]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A Transforming Mutation in the Pleckstrin Homology Domain of AKT1 in Cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef]
- Mancini, M.L.; Lien, E.C.; Toker, A. Oncogenic AKT1(E17K) Mutation Induces Mammary Hyperplasia but Prevents HER2-Driven Tumorigenesis. Oncotarget 2016, 7, 17301–17313. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-Y.; Di, M.-Y.; Yuan, J.-Q.; Shen, W.-X.; Zheng, D.-Y.; Chen, J.-Z.; Mao, C.; Tang, J.-L. The Prognostic Value of Phosphorylated Akt in Breast Cancer: A Systematic Review. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.-X.; Zhang, K.; Qiu, X.-S.; Zhou, M.; Li, W.-M. The Prognostic Value of Phosphorylated AKT Expression in Non-Small Cell Lung Cancer: A Meta-Analysis. PLoS ONE 2013, 8, e81451. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, J.; Zhai, X.; Fu, Z.; Tang, Z.; Liu, L.; Chen, M.; Zhu, Y. Prognostic Value of Phospho-Akt in Patients with Non-Small Cell Lung Carcinoma: A Meta-Analysis. Int. J. Cancer 2014, 135, 1417–1424. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Zhang, C.; Han, W.; Gao, X.-J.; Ma, J.; Hu, Y.-W.; Gu, X.; Ding, H.-Z.; Zhu, L.-X.; Liu, Q. P-Akt as a Potential Poor Prognostic Factor for Gastric Cancer: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 59878–59888. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Xu, L.; Tang, H.; Yang, Q.; Yi, X.; Fang, Y.; Zhu, Y.; Wang, Z. The Role of the PTEN/PI3K/Akt Pathway on Prognosis in Epithelial Ovarian Cancer: A Meta-Analysis. Oncologist 2014, 19, 528–535. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective Use of PI3K and MEK Inhibitors to Treat Mutant Kras G12D and PIK3CA H1047R Murine Lung Cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, P.J.A.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.L.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-Kinase Hyperactivation Results in Lapatinib Resistance That Is Reversed by the MTOR/Phosphatidylinositol 3-Kinase Inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [Google Scholar] [CrossRef] [Green Version]
- McKay, J.A.; Murray, L.J.; Curran, S.; Ross, V.G.; Clark, C.; Murray, G.I.; Cassidy, J.; McLeod, H.L. Evaluation of the Epidermal Growth Factor Receptor (EGFR) in Colorectal Tumours and Lymph Node Metastases. Eur. J. Cancer Oxf. Engl. 2002, 38, 2258–2264. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal Growth Factor Receptor in Glioma: Signal Transduction, Neuropathology, Imaging, and Radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Pines, G. The ERBB Network: At Last, Cancer Therapy Meets Systems Biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Engelman, J.A. ERBB Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [Green Version]
- Castellanos, E.; Feld, E.; Horn, L. Driven by Mutations: The Predictive Value of Mutation Subtype in EGFR-Mutated Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 612–623. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, R.; Gerald, W.L.; Li, A.R.; Pan, Q.; Lal, P.; Ladanyi, M.; Chen, B. EGFR Gene Amplification in Breast Cancer: Correlation with Epidermal Growth Factor Receptor MRNA and Protein Expression and HER-2 Status and Absence of EGFR-Activating Mutations. Mod. Pathol. 2005, 18, 1027–1033. [Google Scholar] [CrossRef]
- Hsu, J.L.; Hung, M.-C. The Role of HER2, EGFR, and Other Receptor Tyrosine Kinases in Breast Cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Worby, C.A.; Dixon, J.E. PTEN. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal Structure of the PTEN Tumor Suppressor: Implications for Its Phosphoinositide Phosphatase Activity and Membrane Association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Sansal, I.; Sellers, W.R. The Biology and Clinical Relevance of the PTEN Tumor Suppressor Pathway. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 2954–2963. [Google Scholar] [CrossRef]
- Tamguney, T.; Stokoe, D. New Insights into PTEN. J. Cell Sci. 2007, 120, 4071–4079. [Google Scholar] [CrossRef] [Green Version]
- Cristofano, A.D.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten Is Essential for Embryonic Development and Tumour Suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Furnari, F.B.; Lin, H.; Huang, H.-J.S.; Cavenee, W.K. Growth Suppression of Glioma Cells by PTEN Requires a Functional Phosphatase Catalytic Domain. Proc. Natl. Acad. Sci. USA 1997, 94, 12479–12484. [Google Scholar] [CrossRef] [Green Version]
- Weng, L.-P.; Smith, W.M.; Dahia, P.L.M.; Ziebold, U.; Gil, E.; Lees, J.A.; Eng, C. PTEN Suppresses Breast Cancer Cell Growth by Phosphatase Activity-Dependent G1 Arrest Followed by Cell Death. Cancer Res. 1999, 59, 5808–5814. [Google Scholar]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; McGowan, E.M.; Nassif, N.T. PTEN/PTENP1: ‘Regulating the Regulator of RTK-Dependent PI3K/Akt Signalling’, New Targets for Cancer Therapy. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the Human PTEN Gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Soria, J.-C.; Lee, H.-Y.; Lee, J.I.; Wang, L.; Issa, J.-P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN Expression in Non-Small Cell Lung Cancer Could Be Related to Promoter Methylation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 1178–1184. [Google Scholar]
- Marsit, C.J.; Zheng, S.; Aldape, K.; Hinds, P.W.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. PTEN Expression in Non-Small-Cell Lung Cancer: Evaluating Its Relation to Tumor Characteristics, Allelic Loss, and Epigenetic Alteration. Hum. Pathol. 2005, 36, 768–776. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G. MicroRNA-21 (MiR-21) Represses Tumor Suppressor PTEN and Promotes Growth and Invasion in Non-Small Cell Lung Cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef]
- Murugan, A.K. MTOR: Role in Cancer, Metastasis and Drug Resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef]
- Sato, T.; Nakashima, A.; Guo, L.; Coffman, K.; Tamanoi, F. Single Amino-Acid Changes That Confer Constitutive Activation of MTOR Are Discovered in Human Cancer. Oncogene 2010, 29, 2746–2752. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Park, G.; Huuhtanen, J.; Lundgren, S.; Khajuria, R.K.; Hurtado, A.M.; Muñoz-Calleja, C.; Cardeñoso, L.; Gómez-García de Soria, V.; Chen-Liang, T.H.; et al. Somatic MTOR Mutation in Clonally Expanded T Lymphocytes Associated with Chronic Graft versus Host Disease. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A Diverse Array of Cancer-Associated MTOR Mutations Are Hyperactivating and Can Predict Rapamycin Sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming MTOR Resistance Mutations with a New-Generation MTOR Inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, Z.; Huang, J.; Cheng, S.; Du, H.; Che, G.; Peng, Y. Clinicopathological and Prognostic Significance of MTOR and Phosphorylated MTOR Expression in Patients with Esophageal Squamous Cell Carcinoma: A Systematic Review and Meta-Analysis. BMC Cancer 2016, 16, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Du, Z.; Zhu, Y.; Song, Y.; Pang, L.; Chen, Z. The Expression and Prognostic Impact of the PI3K/AKT/MTOR Signaling Pathway in Advanced Esophageal Squamous Cell Carcinoma. Technol. Cancer Res. Treat. 2018, 17, 1533033818758772. [Google Scholar] [CrossRef]
- Zhao, W.; Qiu, Y.; Kong, D. Class I Phosphatidylinositol 3-Kinase Inhibitors for Cancer Therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Workman, P.; Clarke, P.A.; Raynaud, F.I.; Montfort, R.L.M. van Drugging the PI3 Kinome: From Chemical Tools to Drugs in the Clinic. Cancer Res. 2010, 70, 2146–2157. [Google Scholar] [CrossRef] [Green Version]
- Yaguchi, S.; Fukui, Y.; Koshimizu, I.; Yoshimi, H.; Matsuno, T.; Gouda, H.; Hirono, S.; Yamazaki, K.; Yamori, T. Antitumor Activity of ZSTK474, a New Phosphatidylinositol 3-Kinase Inhibitor. JNCI J. Natl. Cancer Inst. 2006, 98, 545–556. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K Pathway in Cancer: Are We Making Headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Janku, F. Phosphoinositide 3-Kinase (PI3K) Pathway Inhibitors in Solid Tumors: From Laboratory to Patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Huw, L.-Y.; O’Brien, C.; Pandita, A.; Mohan, S.; Spoerke, J.M.; Lu, S.; Wang, Y.; Hampton, G.M.; Wilson, T.R.; Lackner, M.R. Acquired PIK3CA Amplification Causes Resistance to Selective Phosphoinositide 3-Kinase Inhibitors in Breast Cancer. Oncogenesis 2013, 2, e83. [Google Scholar] [CrossRef] [Green Version]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent Loss of PTEN Leads to Clinical Resistance to a PI(3)Kα Inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.-I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.A.; Brandhuber, B.J. Crystal Structure of Human AKT1 with an Allosteric Inhibitor Reveals a New Mode of Kinase Inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the Potential of AKT Inhibitors as Anti-Cancer Treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Huck, B.R.; Mochalkin, I. Recent Progress towards Clinically Relevant ATP-Competitive Akt Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2838–2848. [Google Scholar] [CrossRef]
- Lu, S.; Li, S.; Zhang, J. Harnessing Allostery: A Novel Approach to Drug Discovery. Med. Res. Rev. 2014, 34, 1242–1285. [Google Scholar] [CrossRef]
- Menon, S.; Manning, B.D. Common Corruption of the MTOR Signaling Network in Human Tumors. Oncogene 2008, 27, S43–S51. [Google Scholar] [CrossRef] [Green Version]
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a New Antifungal Antibiotic. I. Taxonomy of the Producing Streptomycete and Isolation of the Active Principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef]
- Calne, R.Y.; Collier, D.S.; Lim, S.; Pollard, S.G.; Samaan, A.; White, D.J.; Thiru, S. Rapamycin for Immunosuppression in Organ Allografting. Lancet Lond. Engl. 1989, 2, 227. [Google Scholar] [CrossRef]
- Thomson, A.W.; Woo, J. Immunosuppressive Properties of FK-506 and Rapamycin. Lancet Lond. Engl. 1989, 2, 443–444. [Google Scholar] [CrossRef]
- Douros, J.; Suffness, M. New Antitumor Substances of Natural Origin. Cancer Treat. Rev. 1981, 8, 63–87. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Faivre, S.; Serova, M.; Raymond, E. MTORC1 Inhibitors: Is Temsirolimus in Renal Cancer Telling Us How They Really Work? Br. J. Cancer 2008, 99, 1197–1203. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Frias, M.A.; Chatterjee, A.; Yellen, P.; Foster, D.A. The Enigma of Rapamycin Dosage. Mol. Cancer Ther. 2016, 15, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Choo, A.Y.; Yoon, S.-O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin Differentially Inhibits S6Ks and 4E-BP1 to Mediate Cell-Type-Specific Repression of MRNA Translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Zhang, X.; Lu, T.; Zhang, X.; Wang, X.; Meng, B.; Zhang, H.; Wang, P.; Vose, J.M.; Chan, W.C.; et al. Inhibition of 4EBP Phosphorylation Mediates the Cytotoxic Effect of Mechanistic Target of Rapamycin Kinase Inhibitors in Aggressive B-Cell Lymphomas. Haematologica 2017, 102, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Costa, M.; Zollo, O.; Davis, C.; Feldman, M.E.; Testa, J.R.; Meyuhas, O.; Shokat, K.M.; Ruggero, D. Genetic Dissection of the Oncogenic MTOR Pathway Reveals Druggable Addiction to Translational Control via 4EBP-EIF4E. Cancer Cell 2010, 17, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Mallya, S.; Fitch, B.A.; Lee, J.S.; So, L.; Janes, M.R.; Fruman, D.A. Resistance to MTOR Kinase Inhibitors in Lymphoma Cells Lacking 4EBP1. PLoS ONE 2014, 9, e88865. [Google Scholar] [CrossRef]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. MTORC2 Promotes Type I Insulin-like Growth Factor Receptor and Insulin Receptor Activation through the Tyrosine Kinase Activity of MTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The Translational Landscape of MTOR Signalling Steers Cancer Initiation and Metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Rojo, F.; Calvo, E.; Burris, H.; Judson, I.; Hazell, K.; Martinelli, E.; Ramon y Cajal, S.; Jones, S.; Vidal, L.; et al. Dose- and Schedule-Dependent Inhibition of the Mammalian Target of Rapamycin Pathway with Everolimus: A Phase I Tumor Pharmacodynamic Study in Patients with Advanced Solid Tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 1603–1610. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-Carboxamide-1-β-4-Ribofuranoside (AICAR) Enhances the Efficacy of Rapamycin in Human Cancer Cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frias, M.A.; Foster, D.A. Reciprocal Regulation of AMP-Activated Protein Kinase and Phospholipase D. J. Biol. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.-Q.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to MTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [Green Version]
- Oricchio, E.; Katanayeva, N.; Donaldson, M.C.; Sungalee, S.; Pasion, J.P.; Béguelin, W.; Battistello, E.; Sanghvi, V.R.; Jiang, M.; Jiang, Y.; et al. Genetic and Epigenetic Inactivation of SESTRIN1 Controls MTORC1 and Response to EZH2 Inhibition in Follicular Lymphoma. Sci. Transl. Med. 2017, 9, eaak9969. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Toscano, A.; Nickel, A.-C.; Li, G.; Kamp, M.A.; Muhammad, S.; Leprivier, G.; Fritsche, E.; Barker, R.A.; Sabel, M.; Steiger, H.-J.; et al. Rapalink-1 Targets Glioblastoma Stem Cells and Acts Synergistically with Tumor Treating Fields to Reduce Resistance against Temozolomide. Cancers 2020, 12, 3859. [Google Scholar] [CrossRef]
- La Manna, F.; De Menna, M.; Patel, N.; Karkampouna, S.; De Filippo, M.; Klima, I.; Kloen, P.; Beimers, L.; Thalmann, G.N.; Pelger, R.C.M.; et al. Dual-MTOR Inhibitor Rapalink-1 Reduces Prostate Cancer Patient-Derived Xenograft Growth and Alters Tumor Heterogeneity. Front. Oncol. 2020, 10, 1012. [Google Scholar] [CrossRef]
- Kuroshima, K.; Yoshino, H.; Okamura, S.; Tsuruda, M.; Osako, Y.; Sakaguchi, T.; Sugita, S.; Tatarano, S.; Nakagawa, M.; Enokida, H. Potential New Therapy of Rapalink-1, a New Generation Mammalian Target of Rapamycin Inhibitor, against Sunitinib-resistant Renal Cell Carcinoma. Cancer Sci. 2020, 111, 1607–1618. [Google Scholar] [CrossRef]
- Gazi, M.; Moharram, S.A.; Marhäll, A.; Kazi, J.U. The Dual Specificity PI3K/MTOR Inhibitor PKI-587 Displays Efficacy against T-Cell Acute Lymphoblastic Leukemia (T-ALL). Cancer Lett. 2017, 392, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.P.; Reynolds, C.P.; Kang, M.H. Modulation of Glucocorticoid Resistance in Pediatric T-Cell Acute Lymphoblastic Leukemia by Increasing BIM Expression with the PI3K/MTOR Inhibitor BEZ235. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Ewald, F.; Grabinski, N.; Grottke, A.; Windhorst, S.; Nörz, D.; Carstensen, L.; Staufer, K.; Hofmann, B.T.; Diehl, F.; David, K.; et al. Combined Targeting of AKT and MTOR Using MK-2206 and RAD001 Is Synergistic in the Treatment of Cholangiocarcinoma. Int. J. Cancer 2013, 133, 2065–2076. [Google Scholar] [CrossRef]
- Grabinski, N.; Ewald, F.; Hofmann, B.T.; Staufer, K.; Schumacher, U.; Nashan, B.; Jücker, M. Combined Targeting of AKT and MTOR Synergistically Inhibits Proliferation of Hepatocellular Carcinoma Cells. Mol. Cancer 2012, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Formisano, L.; Napolitano, F.; Rosa, R.; D’Amato, V.; Servetto, A.; Marciano, R.; De Placido, P.; Bianco, C.; Bianco, R. Mechanisms of Resistance to MTOR Inhibitors. Crit. Rev. Oncol. Hematol. 2020, 147, 102886. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Molecular Basis for Class Side Effects Associated with PI3K/AKT/MTOR Pathway Inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 1/883 (0.11%) | 1/267 (0.37%) | 1/267 (0.37%) | 1/79 (1.27%) | - |
| Autonomic ganglia | 7/1605 (0.44%) | - | - | - | - |
| Biliary tract | 180/2501 (7.2%) | - | - | - | - |
| Bone | 44/1277 (3.45%) | - | - | - | - |
| Breast | 6559/22748 (28.83%) | 27/1492 (1.81%) | - | 97/1104 (8.79%) | 8/1104 (0.72%) |
| – ductal carcinoma | 1335/5021 (26.59%) | 0/315 (0%) | 0/315 (0%) | - | - |
| – ER-positive carcinoma | 619/1592 (38.88%) | - | - | - | - |
| – HER-positive carcinoma | 325/1455 (22.34%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 136/932 (14.59%) | - | - | - | - |
| – ER-PR-positive carcinoma | 211/656 (32.16%) | - | - | - | - |
| Central nervous system | 422/5741 (7.35%) | 13/1035 (1.26%) | - | 38/679 (5.45%) | - |
| Cervix | 347/2530 (13.72%) | 61/299 (20.4%) | - | 105/307 (34.2%) | 7/307 (2.28%) |
| Endometrium | 1277/4662 (27.39%) | 24/586 (4.1%) | - | 85/602 (14.12%) | - |
| – endometrioid carcinoma | 698/1964 (35.54%) | 19/530 (3.58%) | - | 71/545 (13.03%) | - |
| Eye | 1/372 (0.27%) | - | 1/80 (1.25%) | - | - |
| Gastrointestinal tract | 42/958 (4.38%) | - | - | - | - |
| Hematopoietic and Lymphoid | 110/8604 (1.28%) | 1/661 (0.15%) | - | 12/221 (5.43%) | 3/221 (1.36%) |
| Kidney | 68/3166 (2.15%) | 3/995 (0.3%) | 1/995 (0.1%) | 14/600 (2.33%) | 4/600 (0.67%) |
| Large intestine | 3234/22,967 (14.08%) | 1/718 (0.14%) | - | 25/610 (4.1%) | 4/610 (0.66%) |
| Liver | 177/3513 (5.04%) | 2/663 (0.3%) | - | 21/373 (5.63%) | 2/373 (0.54%) |
| Lung | 805/16,563 (4.86%) | 187/1006 (18.59%) | - | 329/1019 (32.29%) | 8/1019 (0.79%) |
| – adenocarcinoma | 295/7542 (3.91%) | 6/375 (1.6%) | - | 34/378 (8.99%) | 5/378 (1.32%) |
| – non-small cell carcinoma | 125/3728 (3.35%) | - | - | - | - |
| – squamous cell carcinoma | 230/2832 (8.12%) | 181/500 (36.2%) | - | 279/502 (55.58%) | - |
| Meninges | 100/478 (20.92%) | - | - | - | - |
| Esophagus | 314/3866 (8.12%) | 21/510 (4.12%) | - | 45/125 (36%) | 1/125 (0.8%) |
| Ovary | 531/4615 (11.51%) | 58/684 (8.48%) | - | 131/266 (29.25%) | - |
| – clear cell carcinoma | 254/763 (33.29%) | - | - | - | - |
| – endometrioid carcinoma | 79/327 (24.16%) | - | - | - | - |
| – serous carcinoma | 52/1634 (3.18%) | 58/568 (10.21%) | - | 131/266 (49.25%) | - |
| Pancreas | 122/3893 (3.13%) | 1/898 (0.11%) | - | 8/179 (4.47%) | 2/179 (1.12%) |
| Penis | 38/233 (16.31%) | - | - | - | - |
| Peritoneum | 7/188 (3.72%) | - | - | - | - |
| Pituitary | 12/467 (2.57%) | - | - | - | - |
| Pleura | 7/639 (1.1%) | 1/87 (1.15%) | 1/87 (1.15%) | - | - |
| Prostate | 296/4124 (7.18%) | 5/949 (0.53%) | - | 15/498 (3.01%) | - |
| Salivary gland | 89/707 (12.59%) | - | - | - | - |
| Skin | 378/4887 (7.73%) | 1/587 (0.17%) | 1/587 (0.17%) | 24/473 (5.07%) | |
| Small intestine | 24/434 (5.53%) | - | - | - | - |
| Soft tissue | 288/3718 (7.75%) | - | - | 12/263 (4.56%) | - |
| Stomach | 484/4584 (10.56%) | 13/472 2.75%) | - | 23/285 (8.07%) | - |
| Testis | 10/628 (1.59%) | 2/149 (1.34%) | - | - | - |
| Thymus | 8/442 (1.81%) | - | - | - | - |
| Thyroid | 210/5263 (3.99%) | 1/490 (0.2%) | - | 17/513 (3.31%) | 3/513 (0.58%) |
| Upper aerodigestive tract | 507/4898 (10.35%) | 75/520 (14.42%) | - | 142/522 (27.2%) | 3/522 (0.57%) |
| Urinary tract | 552/2732 (20.2%) | 11/399 (2.76%) | - | 39/408 (9.56%) | 3/408 (0.74%) |
| Vulva | 27/142 (19.01%) | - | - | - | - |
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 0/790 (0%) | - | - | 9/79 (11.39%) | - |
| Autonomic ganglia | 0/1442 (0%) | - | - | - | - |
| Biliary tract | 20/1898 (1.05%) | - | - | - | - |
| Bone | 16/890 (1.8%) | - | - | - | - |
| Breast | 490/12,455 (3.93%) | 5/1492 (0.34%) | 2/1492 (0.13%) | 69/1104 (6.25%) | 3/1104 (0.27%) |
| – ductal carcinoma | 186/3743 (4.97%) | - | - | - | - |
| – ER-positive carcinoma | 71/1475 (4.81%) | - | - | - | - |
| – HER-positive carcinoma | 1/301 (0.33%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 9/553 (1.63%) | - | - | - | - |
| – ER-PR-positive carcinoma | 22/364 (6.04%) | - | - | - | - |
| Central nervous system | 15/4711 (0.32%) | 4/1035 (0.39%) | 1/1035 (0.1%) | 22/697 (3.16%) | 43/697 (6.17%) |
| Cervix | 11/901 (1.22%) | 1/299 (0.33%) | - | 15/307 (4.89%) | 7/307 (2.28%) |
| Endometrium | 58/1575 (3.68%) | 2/586 (0.34%) | - | 49/602 (8.14%) | 6/602 (1%) |
| – endometrioid carcinoma | 44/954 (4.61%) | 2/530 (0.38%) | - | 39/545 (7.16%) | 6/545 (1.1%) |
| Eye | 1/370 (0.27%) | - | - | - | - |
| Gastrointestinal tract | 0/1 (0%) | - | - | - | - |
| Hematopoietic and lymphoid | 33/8040 (0.41%) | - | 1/661 (0.15%) | 7/221 (3.17%) | 2/221 (0.9%) |
| Kidney | 10/3008 (0.33%) | - | 4/995 (0.4%) | 26/600 (4.33%) | 27/600 (4.5%) |
| Large intestine | 147/8066 (1.82%) | - | - | 18/610 (2.95%) | 44/610 (7.21%) |
| Liver | 31/2822 (1.1%) | 1/663 (0.15%) | 1/663 (0.15%) | 24/373 (6.43%) | 24/373 (6.43%) |
| Lung | 96/11467 (0.84%) | 10/1006 (0.99%) | 2/1006 (0.2%) | 89/1019 (8.73%) | 46/1019 (4.51%) |
| – adenocarcinoma | 41/6799 (0.6%) | 4/375 (1.07%) | - | 44/378 (11.64%) | 12/378 (3.17%) |
| – non-small cell carcinoma | 4/644 (0.62%) | - | - | - | - |
| – squamous cell carcinoma | 18/2138 (0.84%) | 6/500 (1.2%) | - | 37/502 (7.37%) | 26/502 (5.18%) |
| Meninges | 269/1766 (15.23%) | - | - | - | - |
| Esophagus | 14/2601 (0.54%) | 1/510 (0.2%) | - | 9/125 (7.2%) | - |
| Ovary | 32/2012 (1.59%) | 8/684 (1.17%) | - | 33/266 (12.41%) | 6/266 (2.26%) |
| Pancreas | 29/3155 (0.92%) | - | - | 10/179 (5.59%) | 8/179 (4.47%) |
| Penis | 1/101 (0.99%) | - | - | - | - |
| Peritoneum | 3/201 (1.49%) | - | - | - | - |
| Pituitary | 0/88 (0%) | - | - | - | - |
| Pleura | 1/511 (0.2%) | - | - | - | - |
| Prostate | 161/4202 (3.83%) | - | - | 22/498 (4.42%) | 5/498 (1%) |
| Salivary gland | 7/535 (1.31%) | - | - | - | - |
| Skin | 128/3730 (3.43%) | - | - | 21/473 (4.44%) | - |
| Small intestine | 2/315 (0.63%) | - | - | - | - |
| Soft tissue | 35/2336 (1.5%) | 1/264 (0.38%) | - | 28/263 (10.65%) | 2/263 (0.76%) |
| Stomach | 42/3628 (1.16%) | - | - | 15/285 (5.26%) | 10/285 (3.51%) |
| Testis | 2/602 (0.33%) | 1/149 (0.67%) | - | - | - |
| Thymus | 1/379 (0.26%) | - | - | - | - |
| Thyroid | 39/3657 (1.07%) | - | - | 24/513 (4.68%) | 1/513 (0.19%) |
| Upper aerodigestive tract | 27/2621 (1.03%) | 2/520 (0.38%) | - | 63/522 (12.07%) | 23/522 (4.41%) |
| Urinary tract | 47/1749 (2.69%) | - | 2/399 (0.5%) | 21/408 (5.15%) | 12/408 (2.94%) |
| Vulva | 0/64 (0%) | - | - | - | - |
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 14/1112 (1.26%) | - | 1/267 (0.37%) | 4/79 (5.06%) | - |
| Breast | 304/10,708 (2.84%) | 14/1492 (0.94%) | 2/1492 (0.13%) | 65/1104 (5.89%) | - |
| – ductal carcinoma | 157/3346 (4.69%) | - | - | - | - |
| – ER-positive carcinoma | 8/466 (1.72%) | - | - | - | - |
| – HER-positive carcinoma | 33/609 (5.42%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 19/1182 (1.61%) | - | - | - | - |
| – ER-PR-positive carcinoma | 2/312 (0.64%) | - | - | - | - |
| Central nervous system | 581/6021 (9.65%) | 280/1035 (27.05%) | - | 142/697 (20.37%) | - |
| Cervix | 15/1038 (1.45%) | 6/299 (2.01%) | - | 27/307 (8.79%) | - |
| Endometrium | 91/1432 (6.35%) | 3/586 (0.51%) | - | 41/602 (6.81%) | - |
| – endometrioid carcinoma | 51/819 (6.23%) | 3/530 (0.57%) | - | 41/545 (7.52%) | - |
| Hematopoietic and lymphoid | 193/8438 (2.29%) | 1/661 (0.15%) | 1/661 (0.15%) | 18/221 (8.14%) | - |
| Kidney | 56/3554 (1.58%) | 2/995 (0.2%) | 3/995 (0.3%) | 20/600 (3.33%) | - |
| Large intestine | 345/9946 (3.47%) | 13/718 (1.81%) | 1/718 (0.14%) | 61/610 (10%) | 5/610 (0.82%) |
| Liver | 225/3015 (7.46%) | 2/663 (0.3%) | - | 20/373 (5.36%) | - |
| Lung | 26,499/99,694 (26.58%) | 53/1006 (5.27%) | 1/1006 (0.1%) | 148/1019 (14.52%) | - |
| – adenocarcinoma | 14,832/48,449 (30.61%) | 14/375 (3.73%) | - | 58/378 (15.34%) | - |
| – non-small cell carcinoma | 9016/35,920 (25.1%) | - | - | - | - |
| – squamous cell carcinoma | 413/5824 (7.09%) | 32/500 (6.4%) | 1/500 (0.2%) | 65/502 (12.95%) | - |
| Meninges | 150/394 (38.07%) | - | - | - | - |
| Esophagus | 134/3500 (3.83%) | 15/510 (2.94%) | 1/510 (0.2%) | 26/125 (20.08%) | - |
| Ovary | 89/2481 (3.59%) | 2/684 (0.29%) | 1/684 (0.15%) | 26/266 (9.77%) | - |
| Pancreas | 136/3681 (3.69%) | - | 1/898 (0.11%) | 4/179 (2.23%) | - |
| Prostate | 282/3949 (7.14%) | 1/949 (0.11%) | 1/949 (0.11%) | 23/498 (4.62%) | - |
| Skin | 242/3858 (6.27%) | 4/587 (0.68%) | - | 10/473 (2.11%) | - |
| Soft tissue | 66/2966 (2.23%) | 3/264 (1.14%) | 2/264 (0.76%) | 30/263 (11.41%) | - |
| Stomach | 146/3335 (4.38%) | 19/472 (4.03%) | 2/472 (0.42%) | 33/285 (11.58%) | 1/285 (0.35%) |
| Thyroid | 33/3623 (0.91%) | - | - | 27/513 (5.26%) | - |
| Upper aerodigestive tract | 153/4981 (3.07%) | 44/520 (8.46%) | 1/520 (0.19%) | 78/522 (14.94%) | - |
| Urinary tract | 51/1649 (3.09%) | 19/399 (4.76%) | 1/399 (0.25%) | 42/408 (10.29%) | - |
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 1/816 (0.12%) | - | - | 6/79 (7.59%) | 2/79 (2.53%) |
| Autonomic ganglia | 3/1404 (0.21%) | - | - | - | - |
| Biliary tract | 92/1922 (4.81%) | - | - | - | - |
| Bone | 11/889 (1.24%) | - | - | - | - |
| Breast | 567/10,325 (5.49%) | 1/1492 (0.07%) | 31/1492 (2.08%) | 21/1104 (1.9%) | 47/1104 (4.26%) |
| – ductal carcinoma | 214/2832 (7.56%) | - | - | - | - |
| – ER-positive carcinoma | 94/1477 (6.36%) | - | - | - | - |
| – HER-positive carcinoma | 11/275 (4%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 21/469 (4.48%) | - | - | - | - |
| – ER-PR-positive carcinoma | 12/403 (2.98%) | - | - | - | - |
| Central nervous system | 1040/7783 (13.36%) | - | 46/1035 (4.44%) | 19/697 (2.73%) | 146/679 (20.95%) |
| Cervix | 74/1598 (4.63%) | - | 12/299 (4.01%) | 13/307 (4.23%) | 21/307 (6.84%) |
| Endometrium | 1565/3944 (39.68%) | 1/586 (0.17%) | 19/586 (3.24%) | 50/602 (8.31) | 37/602 (6.15%) |
| – endometrioid carcinoma | 1170/2100 (55.71%) | 1/530 (0.19%) | 17/530 (3.21%) | 34/545 (6.24%) | 27/545 (4.95%) |
| Eye | 15/401 (3.74%) | - | - | - | - |
| Hematopoietic and lymphoid | 388/14,559 (2.67%) | - | - | 9/221 (4.07%) | 1/221 (0.45%) |
| Kidney | 132/3451 (3.82%) | - | 4/995 (0.4%) | 19/600 (3.17%) | 33/600 (5.5%) |
| Large intestine | 476/8913 (5.34%) | - | 11/718 (1.53%) | 14/610 (2.3%) | 46/610 (7.54%) |
| Liver | 184/2979 (6.18%) | 1/663 (0.15%) | 10/663 (1.51%) | 10/373 (2.68%) | 13/373 (3.49%) |
| Lung | 319/9054 (3.52%) | 1/1006 (0.1%) | 21/1006 (2.09%) | 28/1019 (2.75%) | 83/1019 (8.15%) |
| – adenocarcinoma | 94/4362 (2.15%) | - | 2/375 (0.53%) | 13/378 (3.44%) | 23/378 (6.08%) |
| – non-small cell carcinoma | 22/1521 (1.45%) | - | - | - | - |
| – squamous cell carcinoma | 113/1554 (7.27%) | 1/500 (0.2%) | 18/500 (3.6%) | 12/502 (2.39%) | 57/502 (11.35%) |
| Meninges | 30/513 (5.85%) | - | - | - | - |
| Esophagus | 63/2117 (2.98%) | - | 4/510 (0.78%) | 9/125 (7.2%) | 10/125 (8%) |
| Ovary | 141/2831 (4.98%) | 2/684 (0.29%) | 13/684 (1.9%) | 7/266 (2.63%) | 47/266 (17.67%) |
| Pancreas | 103/3192 (3.23%) | 1/898 (0.11%) | 1/898 (0.11%) | 11/179 (6.15%) | 4/179 (2.23%) |
| Penis | 2/74 (2.7%) | - | - | - | - |
| Peritoneum | 1/225 (0.44%) | - | - | - | - |
| Pituitary | 0/89 (0%) | - | - | - | - |
| Pleura | 2/530 (0.38%) | - | 1/87 (1.15%) | - | - |
| Prostate | 393/4517 (8.7%) | 1/949 (0.11%) | 71/949 (7.48%) | 16/498 (3.21%) | 91/498 (18.27%) |
| Salivary gland | 22/623 (3.53%) | - | - | - | - |
| Skin | 309/3679 (8.4%) | - | 28/587 (4.77%) | 17/473 (3.59%) | 30/473 (6.34%) |
| Small intestine | 2/326 (0.61%) | - | - | - | - |
| Soft tissue | 59/2514 (2.35%) | - | 14/264 (5.3%) | 5/263 (1.9%) | 15/263 (5.7%) |
| Stomach | 130/2980 (4.36%) | - | 15/472 (3.18%) | 7/285 (2.46%) | 26/285 (9.12%) |
| Testis | 7/639 (1.1%) | - | 1/149 (0.67%) | - | - |
| Thymus | 0/372 (0%) | - | - | - | - |
| Thyroid | 110/3936 (2.79%) | - | 1/490 (0.2%) | 25/513 (4.87%) | 9/513 (1.75%) |
| Upper aerodigestive tract | 126/3441 (3.66%) | - | 12/520 (2.31%) | 17/522 (3.26%) | 16/522 (3.07%) |
| Urinary tract | 45/1614 (2.79%) | 1/399 (0.25%) | 8/399 (2.01%) | 7/408 (1.72%) | 20/408 (4.9%) |
| Vulva | 12/157 (7.64%) | - | - | - | - |
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 0/658 (0%) | - | 4/267 (1.5%) | 1/79 (1.27%) | - |
| Autonomic ganglia | 0/1225 (0%) | - | - | - | - |
| Biliary tract | 51/1388 (3.67%) | - | - | - | - |
| Bone | 3/725 (0.41%) | - | - | - | - |
| Breast | 258/7666 (3.37%) | 1/1492 (0.07%) | 2/1492 (0.13%) | 39/1104 (3.53%) | 32/1104 (2.9%) |
| – ductal carcinoma | 120/2483 (4.83%) | - | - | - | - |
| – ER-positive carcinoma | 29/1363 (2.13%) | - | - | - | - |
| – HER-positive carcinoma | 8/205 (3.9%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 5/372 (1.34%) | - | - | - | - |
| ER-PR-positive carcinoma | 6/308 (1.95%) | - | - | - | - |
| Central nervous system | 56/3499 (1.6%) | 1/1035 (0.1%) | - | 21/697 (3.01%) | 91/697 (13.06%) |
| Cervix | 21/388 (5.41%) | - | - | 18/307 (5.86%) | 5/307 (1.63%) |
| Endometrium | 103/988 (10.43%) | - | - | 27/602 (4.49%) | 9/602 (1.5%) |
| – endometrioid carcinoma | 82/649 (12.63%) | - | - | 23/545 (4.22%) | - |
| Eye | 3/179 (1.68%) | - | - | - | - |
| Hematopoietic and lymphoid | 118/6030 (1.96%) | - | - | 2/221 (0.9%) | 5/221 (2.26%) |
| Kidney | 165/2998 (5.5%) | - | - | 20/600 (3.33%) | 12/600 (2%) |
| Large intestine | 344/4526 (7.6%) | - | 1/718 (0.14%) | 16/610 (2.62%) | 35/610 (5.74%) |
| Liver | 175/2381 (7.35%) | - | - | 5/373 (1.34%) | - |
| Lung | 228/4930 (4.62%) | 3/1006 (0.3%) | 2/1006 (0.2%) | 44/1019 (4.32%) | 17/1019 (1.67%) |
| – adenocarcinoma | 126/2724 (4.63%) | 1/375 (0.27%) | 1/375 (0.27%) | 23/378 (6.08%) | 12/378 (3.17%) |
| – non-small cell carcinoma | 4/124 (3.23%) | - | - | - | - |
| – squamous cell carcinoma | 44/1004 (4.38%) | 1/500 (0.2%) | 1/500 (0.2%) | 11/502 (2.19%) | 5/502 (1%) |
| Meninges | 57/306 (18.63%) | - | - | - | - |
| Esophagus | 79/1673 (4.72%) | 1/510 (0.2%) | - | 5/125 (4%) | - |
| Ovary | 63/1396 (4.51%) | 1/684 (0.15%) | - | 26/266 (9.77%) | 11/266 (4.14%) |
| Pancreas | 83/2505 (3.31%) | - | - | 6/179 (3.35%) | 10/179 (5.59%) |
| Penis | 2/17 (11.76%) | - | - | - | - |
| Peritoneum | 1/43 (2.33%) | - | - | - | - |
| Pituitary | 0/89 (0%) | - | - | - | - |
| Pleura | 0/470 (0%) | - | - | - | - |
| Prostate | 217/3317 (6.54%) | - | 1/949 (0.11%) | 14/498 (2.81%) | - |
| Salivary gland | 7/410 (1.71%) | - | - | - | - |
| Skin | 236/2248 (10.5%) | 2/587 (0.34%) | - | 39/473 (8.25%) | 24/473 (5.07%) |
| Small intestine | 5/252 (1.98%) | - | - | - | - |
| Soft tissue | 18/1489 (1.21%) | - | - | - | - |
| Stomach | 124/1912 (6.49%) | 2/472 (0.42%) | - | 18/285 (6.32%) | 4/285 (1.4%) |
| Testis | 9/662 (1.36%) | - | - | - | - |
| Thymus | 0/173 (0%) | - | - | - | - |
| Thyroid | 42/2131 (1.97%) | - | - | 20/513 (3.9%) | 7/513 (1.36%) |
| Upper aerodigestive tract | 44/1836 (2.4%) | - | - | 26/522 (4.98%) | 5/522 (0.96%) |
| Urinary tract | 72/1209 (5.96%) | - | - | 34/408 (8.33%) | 2/408 (0.49%) |
| Vulva | 1/30 (3.33%) | - | - | - | - |
| Inhibitor | Target | Condition or Disease | ClinicalTrials.gov Identifier |
|---|---|---|---|
| Ipatasertib and Paclitaxel | AKT and microtubules | Locally advanced or metastatic TNBC; locally advanced or metastatic HR+/HER2– breast adenocarcinoma | NCT03337724 Study start date: January 2018 Estimated study completion date: December 2021 |
| Ipatasertib and Abiraterone | AKT and CYP17 | Metastatic castrate-resistant prostate cancer | NCT03072238 Study start date: June 2017 Estimated study completion date: November 2023 |
| Ipatasertib, Palbociclib, and Fulvestrant | AKT, CDK4/6, and ER | HR+ and HER2– locally advanced unresectable or metastatic breast cancer | NCT04060862 Study start date: November 2019 Estimated study completion date: January 2026 |
| Ipatasertib, Paclitaxel, and Atezolizumab | AKT, microtubules, and PD-L1 | Locally advanced or metastatic TNBC | NCT04177108 Study start date: November 2019 Estimated study completion date: October 2025 |
| Ipatasertib and Fulvestrant | AKT and ER | Advanced HER2– and ER+ breast cancer | NCT04650581 Estimated study start date: December 2020 Estimated study completion date: December 2026 |
| Capivasertib and Paclitaxel | AKT and microtubules | Locally advanced or metastatic TNBC | NCT03997123 Study start date: June 2019 Estimated study completion date: January 2023 |
| Capivasertib and Abiraterone | AKT and CYP17 | Hormone-sensitive prostate cancer | NCT04493853 Study start date: July 2020 Estimated study completion date: November 2025 |
| Capivasertib and Fulvestrant | AKT and ER | Locally advanced (inoperable) or metastatic HR+/HER2– breast cancer | NCT04305496 Study start date: April 2020 Estimated study completion date: July 2024 |
| Inhibitor (Trade Name) | Target | Indications | Approval Date |
|---|---|---|---|
| Sirolimus (Rapamune) | mTORC1 | Treatment of patients with lymphangioleiomyomatosis | August 2000 |
| Temsirolimus (Torisel) | mTORC1 | Treatment of advanced renal cell carcinoma | May 2007 |
| Everolimus (Afinitor) | mTORC1 |
| March 2009 August 2012 February 2016 |
| Inhibitor | Target | Phase | Condition or Disease | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| Allosteric | ||||
| Ridaforolimus | mTORC1 | 3 | Metastatic soft-tissue sarcomas and metastatic bone sarcomas | NCT00538239 Study start date: October 2007 Study completion date: December 2012 |
| mTORC1 | 2 | Advanced sarcoma | NCT00093080 Study start date: October 2004 Study completion date: November 2008 | |
| mTORC1 | 2 | Relapsed or refractory hematologic malignancies | NCT00086125 Study start date: June 2004 Study completion date: June 2006 | |
| mTORC1 | 2 | Endometrial cancer | NCT00122343 Study start date: August 2005 Study completion date: January 2008 | |
| mTORC1 | 2 | Breast cancer | NCT00736970 Study start date: July 2008 Study completion date: May 2011 | |
| ATP-Competitive | ||||
| MLN0128 | mTORC1/2 | 2 | Metastatic castration-resistant prostate cancer | NCT02091531 Study start date: March 2014 Study completion date: October 2018 |
| mTORC1/2 | 2 | Metastatic anaplastic thyroid cancer | NCT02244463 Study start date: July 2015 Estimated study completion date: January 2022 | |
| CC-223 | mTORC1/2 | 1/2 | Multiple Myeloma Diffuse large B-cell lymphoma Glioblastoma multiforme Hepatocellular carcinoma non-small cell lung cancer Neuroendocrine tumors of non-pancreatic origin Hormone receptor-positive breast cancer | NCT01177397 Study start date: July 2010 Study completion date: December 2016 |
| Sapanisertib (TAK-228 or MLN0128) | mTORC1/2 | 2 | Estrogen receptor-positive breast cancer | NCT02988986 Study start date: April 2017 Study completion date: March 2019 |
| mTORC1/2 | 2 | Soft-tissue sarcoma | NCT02987959 Study start date: February 2017 Estimated study completion date: September 2020 | |
| mTORC1/2 | 2 | Locally advanced or metastatic bladder cancer | NCT03047213 Study start date: December 2016 Estimated study completion date: June 2021 | |
| Vistusertib (AZD2014) | mTORC1/2 | 2 | Progressive or symptomatic meningioma | NCT02831257 Study start date: August 2016 Study completion date: October 2020 |
| mTORC1/2 | 2 | Estrogen receptor positive breast cancer | NCT02216786 Study start date: January 2014 Estimated study completion date: July 2020 | |
| mTORC1/2 | 2 | Meningioma | NCT03071874 Study start date: October 2017 Estimated study completion date: July 2024 | |
| Dual PI3K/mTOR | ||||
| Dactolisib (BEZ235) | PI3K/mTOR | 2 | Pancreatic neuroendocrine tumors (pNET) | NCT01658436 Study start date: November 2012 Study completion date: July 2015 |
| Samotolisib (LY3023414) | PI3K/mTOR | 2 | Metastatic castration resistant prostate cancer | NCT02407054 Study start date: April 2015 Study completion date: April 2020 |
| Bimiralisib (PQR309) | PI3K/mTOR | 2 | Primary central nervous system lymphoma | NCT02669511 Study start date: November 2015 Study completion date: January 2018 |
| PI3K/mTOR | 2 | Relapsed or refractory lymphoma | NCT02249429 Study start date: May 2015 Study completion date: September 2018 | |
| Gedatolisib(PKI587) and Talazoparib | PI3K/mTORandPARP | 2 | Triple-Negative Breast Cancer | NCT03911973 Study start date: April 2019 Estimated study completion date: May 2022 |
| Apitolisib (GDC-0980) | PI3K/mTOR | 2 | Prostate Cancer | NCT01485861 Study start date: January 2019 Estimated study completion date: April 2021 |
| Voxtalisib (SAR245409, XL765) | PI3K/mTOR | 2 | Ovarian Cancer | NCT01936363 Study start date: September 2013 Study completion date: November 2017 |
| PI3K/mTOR | 2 | Lymphoma | NCT01403636 Study start date: October 2011 Study completion date: September 2014 | |
| Paxalisib (GDC-0084) | PI3K/mTOR | 2 | Glioblastoma | NCT03522298 Study start date: May 2018 Estimated study completion date: December 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. https://doi.org/10.3390/ijms22041743
Popova NV, Jücker M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. International Journal of Molecular Sciences. 2021; 22(4):1743. https://doi.org/10.3390/ijms22041743
Chicago/Turabian StylePopova, Nadezhda V., and Manfred Jücker. 2021. "The Role of mTOR Signaling as a Therapeutic Target in Cancer" International Journal of Molecular Sciences 22, no. 4: 1743. https://doi.org/10.3390/ijms22041743
APA StylePopova, N. V., & Jücker, M. (2021). The Role of mTOR Signaling as a Therapeutic Target in Cancer. International Journal of Molecular Sciences, 22(4), 1743. https://doi.org/10.3390/ijms22041743