
Central Role of Dendritic Cells in Pulmonary Arterial Hypertension in Human and Mice

 ,
,  , , ,
, , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
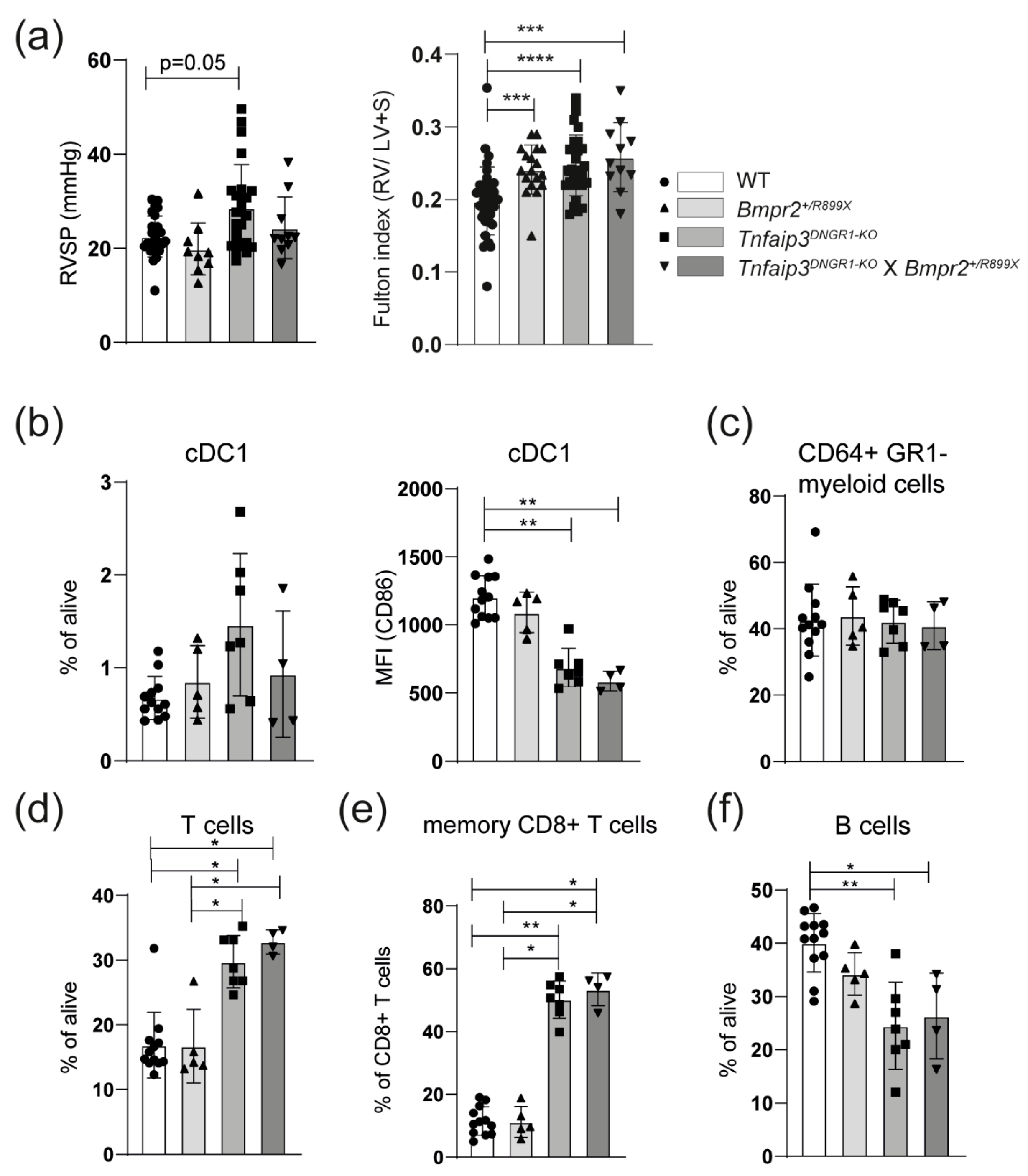
2.1. Increased Myocardial Infiltration of Dendritic Cells in the RV of Tnfaip3DNGR1-KO Mice
2.2. TLR4 Activation Leads to RV Hypertrophy but Not to Increased RV Pressures in Young Tnfaip3DNGR1-KO Mice
2.3. The PH Phenotype of Tnfaip3DNGR1-KO Mice Is Not Enhanced by Concomitant BMPR2 Mutation
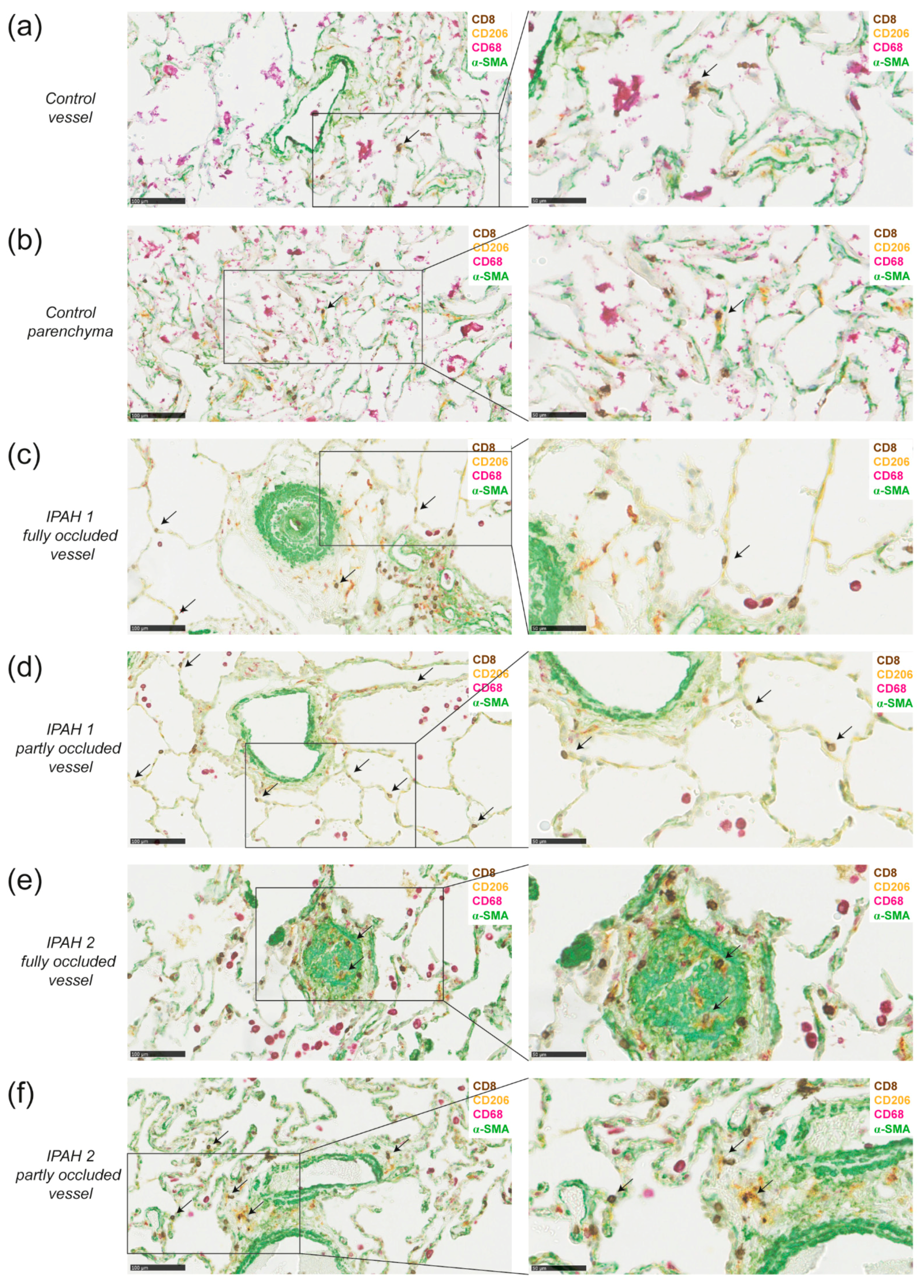
2.4. DCs and CD8+ T Cells Co-Localize around Blood Vessels in Lungs of IPAH Patients
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Exposure of TLR Ligand in Tnfaip3DNGR1-KO Mice
4.3. Right Heart Catheterization and Fulton Index
4.4. RNA Extraction Real-Time Quantitative RT-PCR
4.5. Histology of Lung and Heart Tissues
4.6. Flow Cytometry
4.7. Statistic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AM | Alveolar macrophages |
| BATF3 | Basic leucine zipper transcriptional factor ATF-like 3 |
| BMPR2 | Bone morphogenetic protein receptor type 2 |
| cDC1s | Type 1 conventional DCs |
| cDC2s | Type 2 conventional DCs |
| CTD-PAH | Connective tissue disease PAH |
| CXCL8 | X-X-C-motif chemokine ligand 8 |
| DCs | Dendritic cells |
| EvG | Elastic von Gieson |
| H&E | Hematoxylin and eosin |
| HPAH | Heritable PAH |
| i.p. | Intraperitoneal |
| i.t. | Intratracheally |
| IL | Interleukin |
| IM | Interstitial macrophages |
| IPAH | Idiopathic PAH |
| LPS | Lipopolysaccharide |
| LV | Left ventricle |
| Mo-DC | Monocyte-derived-DC |
| NaCl | Sodium chloride |
| NF-kB | Nuclear factor-kappa B |
| PAH | Pulmonary arterial hypertension |
| PAP | Pulmonary arterial pressure |
| PASMCs | Pulmonary artery smooth muscle cells |
| PH | Pulmonary hypertension |
| pDC | Plasmacytoid DC |
| PFA | paraformaldehyde |
| Poly I:C | Polyinosinic-polycytidylic acif high molecular weight |
| RV | Right ventricle |
| RVSP | Right ventricular systolic pressure |
| S | Septum |
| TGFβ | Transforming growth factor-β |
| TLOs | Tertiary lymphoid organs |
| TLR | Toll like receptor |
| Tnfaip3 | TNFα-induced protein-3 |
| WHO | World health organization |
| WT | Wild type |
| α-SMA | Alpha smooth muscle actin |
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Southgate, L.; Machado, R.D.; Graf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2020, 17, 85–95. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [Green Version]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [Green Version]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and Beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2499. [Google Scholar] [CrossRef] [Green Version]
- Elinoff, J.M.; Mazer, A.J.; Cai, R.; Lu, M.; Graninger, G.; Harper, B.; Ferreyra, G.A.; Sun, J.; Solomon, M.A.; Danner, R.L. Meta-analysis of blood genome-wide expression profiling studies in pulmonary arterial hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L98–L111. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Zhuang, W.; Wang, T.; Lian, G.; Luo, L.; Ye, C.; Wang, H.; Xie, L. Transcriptomic analysis identifies Toll-like and Nod-like pathways and necroptosis in pulmonary arterial hypertension. J. Cell. Mol. Med. 2020, 24, 11409–11421. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Xie, H.; Zhang, Y. Identification of Differentially Expressed Genes Associated with Idiopathic Pulmonary Arterial Hypertension by Integrated Bioinformatics Approaches. J. Comput. Biol. 2021. [Google Scholar] [CrossRef]
- Chesne, J.; Danger, R.; Botturi, K.; Reynaud-Gaubert, M.; Mussot, S.; Stern, M.; Danner-Boucher, I.; Mornex, J.F.; Pison, C.; Dromer, C.; et al. Systematic analysis of blood cell transcriptome in end-stage chronic respiratory diseases. PLoS ONE 2014, 9, e109291. [Google Scholar] [CrossRef]
- George, P.M.; Badiger, R.; Shao, D.; Edwards, M.R.; Wort, S.J.; Paul-Clark, M.J.; Mitchell, J.A. Viral Toll Like Receptor activation of pulmonary vascular smooth muscle cells results in endothelin-1 generation; relevance to pathogenesis of pulmonary arterial hypertension. Biochem. Biophys. Res. Commun. 2012, 426, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Cool, C.D.; Voelkel, N.F.; Bull, T. Viral infection and pulmonary hypertension: Is there an association? Expert Rev. Respir. Med. 2011, 5, 207–216. [Google Scholar] [CrossRef]
- Kubrycht, J.; Novotna, J. Sequence-based prediction of linear autoepitopes involved in pathogenesis of IPAH and the corresponding organism sources of molecular mimicry. Int. J. Bioinform. Res. Appl. 2014, 10, 587–612. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Sweatt, A.J.; Hedlin, H.K.; Balasubramanian, V.; Hsi, A.; Blum, L.K.; Robinson, W.H.; Haddad, F.; Hickey, P.M.; Condliffe, R.; Lawrie, A.; et al. Discovery of Distinct Immune Phenotypes Using Machine Learning in Pulmonary Arterial Hypertension. Circ. Res. 2019, 124, 904–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perros, F.; Dorfmuller, P.; Montani, D.; Hammad, H.; Waelput, W.; Girerd, B.; Raymond, N.; Mercier, O.; Mussot, S.; Cohen-Kaminsky, S.; et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 311–321. [Google Scholar] [CrossRef] [PubMed]
- GeurtsvanKessel, C.H.; Willart, M.A.; Bergen, I.M.; van Rijt, L.S.; Muskens, F.; Elewaut, D.; Osterhaus, A.D.; Hendriks, R.; Rimmelzwaan, G.F.; Lambrecht, B.N. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J. Exp. Med. 2009, 206, 2339–2349. [Google Scholar] [CrossRef]
- Halle, S.; Dujardin, H.C.; Bakocevic, N.; Fleige, H.; Danzer, H.; Willenzon, S.; Suezer, Y.; Hammerling, G.; Garbi, N.; Sutter, G.; et al. Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J. Exp. Med. 2009, 206, 2593–2601. [Google Scholar] [CrossRef] [PubMed]
- van Uden, D.; Boomars, K.; Kool, M. Dendritic Cell Subsets and Effector Function in Idiopathic and Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Front. Immunol. 2019, 10, 11. [Google Scholar] [CrossRef]
- Perros, F.; Dorfmuller, P.; Souza, R.; Durand-Gasselin, I.; Mussot, S.; Mazmanian, M.; Herve, P.; Emilie, D.; Simonneau, G.; Humbert, M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 462–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorfmuller, P.; Chaumais, M.C.; Giannakouli, M.; Durand-Gasselin, I.; Raymond, N.; Fadel, E.; Mercier, O.; Charlotte, F.; Montani, D.; Simonneau, G.; et al. Increased oxidative stress and severe arterial remodeling induced by permanent high-flow challenge in experimental pulmonary hypertension. Respir. Res. 2011, 12, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hautefort, A.; Girerd, B.; Montani, D.; Cohen-Kaminsky, S.; Price, L.; Lambrecht, B.N.; Humbert, M.; Perros, F. T-helper 17 cell polarization in pulmonary arterial hypertension. Chest 2015, 147, 1610–1620. [Google Scholar] [CrossRef]
- Marsh, L.M.; Jandl, K.; Grunig, G.; Foris, V.; Bashir, M.; Ghanim, B.; Klepetko, W.; Olschewski, H.; Olschewski, A.; Kwapiszewska, G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Yan, H.; Zhu, W.; Cui, Y.; Chen, J.; Wang, X.; Li, S.; Zhu, J. Impairment of monocyte-derived dendritic cells in idiopathic pulmonary arterial hypertension. J. Clin. Immunol. 2009, 29, 705–713. [Google Scholar] [CrossRef]
- Koudstaal, T.; van Hulst, J.A.C.; Das, T.; Neys, S.F.H.; Merkus, D.; Bergen, I.M.; de Raaf, M.A.; Bogaard, H.J.; Boon, L.; van Loo, G.; et al. DNGR1-Cre-mediated Deletion of Tnfaip3/A20 in Conventional Dendritic Cells Induces Pulmonary Hypertension in Mice. Am. J. Respir. Cell Mol. Biol. 2020, 63, 665–680. [Google Scholar] [CrossRef]
- Koudstaal, T.; Boomars, K.A.; Kool, M. Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective. J. Clin. Med. 2020, 9, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatel, C.; Radermecker, C.; Fievez, L.; Paulissen, G.; Chakarov, S.; Fernandes, C.; Olivier, S.; Toussaint, M.; Pirottin, D.; Xiao, X.; et al. Exposure to Bacterial CpG DNA Protects from Airway Allergic Inflammation by Expanding Regulatory Lung Interstitial Macrophages. Immunity 2017, 46, 457–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frump, A.; Prewitt, A.; de Caestecker, M.P. BMPR2 mutations and endothelial dysfunction in pulmonary arterial hypertension (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045894018765840. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Graf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.Q.; Abbate, A.; Bogaard, H.J. Role of cardiac inflammation in right ventricular failure. Cardiovasc. Res. 2017, 113, 1441–1452. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, M.J.; Mouchaers, K.T.; Niessen, H.M.; Hadi, A.M.; Kupreishvili, K.; Boonstra, A.; Voskuyl, A.E.; Belien, J.A.; Smit, E.F.; Dijkmans, B.C.; et al. Characteristics of interstitial fibrosis and inflammatory cell infiltration in right ventricles of systemic sclerosis-associated pulmonary arterial hypertension. Int. J. Rheumatol. 2010, 2010, 604615. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Zaman, R.; Hamidzada, H.; Epelman, S. Exploring cardiac macrophage heterogeneity in the healthy and diseased myocardium. Curr. Opin. Immunol. 2020, 68, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Siamwala, J.H.; Zhao, A.; Barthel, H.; Pagano, F.S.; Gilbert, R.J.; Rounds, S. Adaptive and innate immune mechanisms in cardiac fibrosis complicating pulmonary arterial hypertension. Physiol. Rep. 2020, 8, e14532. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Vande Walle, L.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Zhivaki, D.; Borriello, F.; Chow, O.A.; Doran, B.; Fleming, I.; Theisen, D.J.; Pallis, P.; Shalek, A.K.; Sokol, C.L.; Zanoni, I.; et al. Inflammasomes within Hyperactive Murine Dendritic Cells Stimulate Long-Lived T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2020, 33, 108381. [Google Scholar] [CrossRef]
- Abdi, K.; Singh, N.J.; Matzinger, P. Lipopolysaccharide-activated dendritic cells: "exhausted" or alert and waiting? J. Immunol. 2012, 188, 5981–5989. [Google Scholar] [CrossRef] [PubMed]
- Nogueira-Ferreira, R.; Vitorino, R.; Ferreira, R.; Henriques-Coelho, T. Exploring the monocrotaline animal model for the study of pulmonary arterial hypertension: A network approach. Pulm. Pharmacol. Ther. 2015, 35, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Schraml, B.U.; van Blijswijk, J.; Zelenay, S.; Whitney, P.G.; Filby, A.; Acton, S.E.; Rogers, N.C.; Moncaut, N.; Carvajal, J.J.; Reis e Sousa, C. Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell 2013, 154, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Vroman, H.; Bergen, I.M.; van Hulst, J.A.C.; van Nimwegen, M.; van Uden, D.; Schuijs, M.J.; Pillai, S.Y.; van Loo, G.; Hammad, H.; Lambrecht, B.N.; et al. TNF-alpha-induced protein 3 levels in lung dendritic cells instruct TH2 or TH17 cell differentiation in eosinophilic or neutrophilic asthma. J. Allergy Clin. Immunol. 2018, 141, 1620–1633.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Uden, D.; Koudstaal, T.; van Hulst, J.A.C.; Bergen, I.M.; Gootjes, C.; Morrell, N.W.; van Loo, G.; von der Thüsen, J.H.; van den Bosch, T.P.P.; Ghigna, M.-R.; et al. Central Role of Dendritic Cells in Pulmonary Arterial Hypertension in Human and Mice. Int. J. Mol. Sci. 2021, 22, 1756. https://doi.org/10.3390/ijms22041756
van Uden D, Koudstaal T, van Hulst JAC, Bergen IM, Gootjes C, Morrell NW, van Loo G, von der Thüsen JH, van den Bosch TPP, Ghigna M-R, et al. Central Role of Dendritic Cells in Pulmonary Arterial Hypertension in Human and Mice. International Journal of Molecular Sciences. 2021; 22(4):1756. https://doi.org/10.3390/ijms22041756
Chicago/Turabian Stylevan Uden, Denise, Thomas Koudstaal, Jennifer A. C. van Hulst, Ingrid M. Bergen, Chelsea Gootjes, Nicholas W. Morrell, Geert van Loo, Jan H. von der Thüsen, Thierry P. P. van den Bosch, Maria-Rosa Ghigna, and et al. 2021. "Central Role of Dendritic Cells in Pulmonary Arterial Hypertension in Human and Mice" International Journal of Molecular Sciences 22, no. 4: 1756. https://doi.org/10.3390/ijms22041756
APA Stylevan Uden, D., Koudstaal, T., van Hulst, J. A. C., Bergen, I. M., Gootjes, C., Morrell, N. W., van Loo, G., von der Thüsen, J. H., van den Bosch, T. P. P., Ghigna, M.-R., Perros, F., Montani, D., Kool, M., Boomars, K. A., & Hendriks, R. W. (2021). Central Role of Dendritic Cells in Pulmonary Arterial Hypertension in Human and Mice. International Journal of Molecular Sciences, 22(4), 1756. https://doi.org/10.3390/ijms22041756