
A Toxic Synergy between Aluminium and Amyloid Beta in Yeast



Abstract
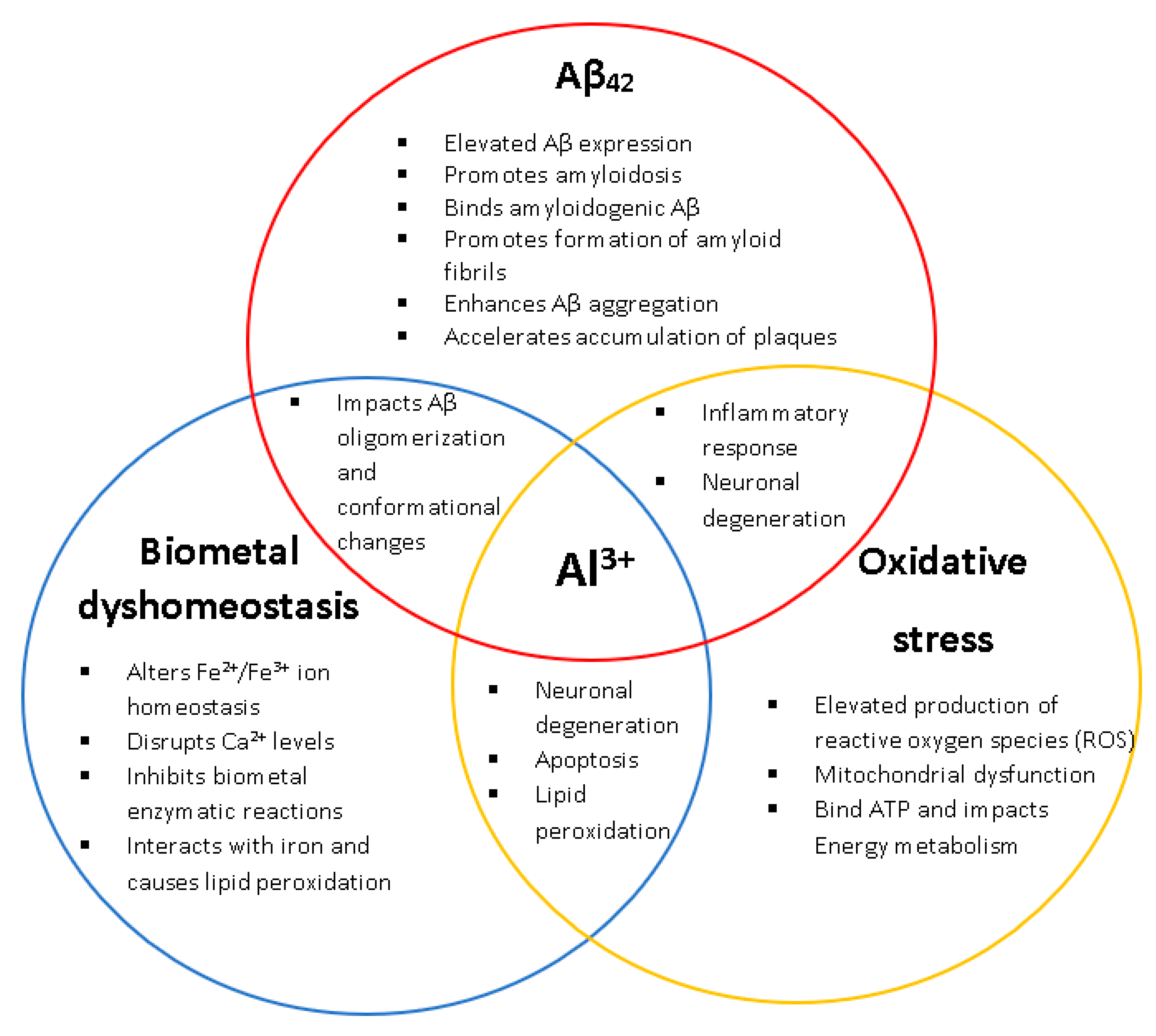
:1. Introduction
2. Results
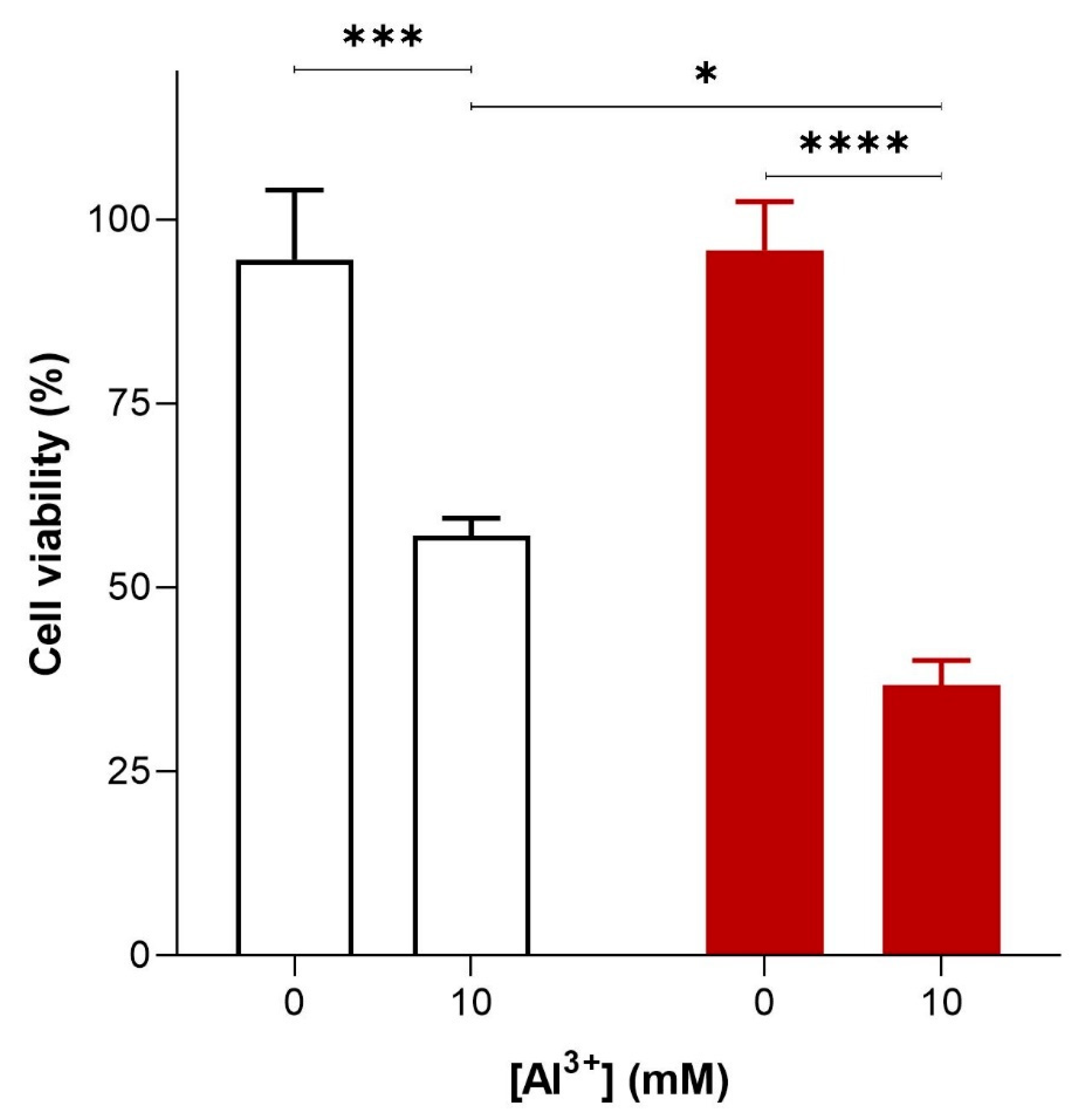
2.1. Aluminium Is Cytotoxic, Inhibits the Growth of Yeast Cells, and Its Toxicity is Exacerbated by the Presence of Aβ42
2.2. Fe3+ Increases Al3+ Toxicity, and Al3+ and Aβ42 Toxic Synergy
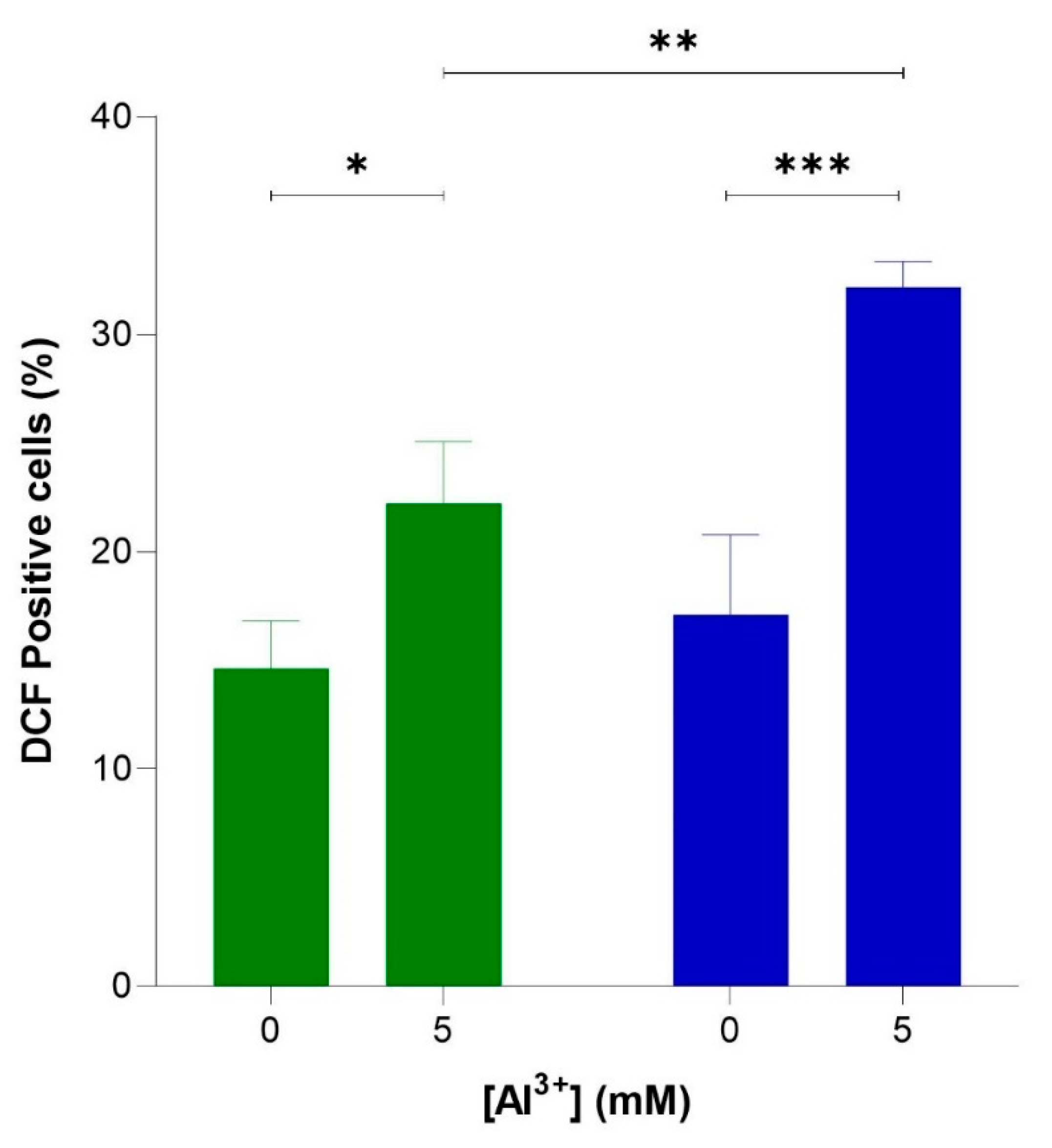
2.3. Aluminium Elevates ROS Levels in Yeast, Enhancing Oxidative Stress in Yeast Producing Aβ42
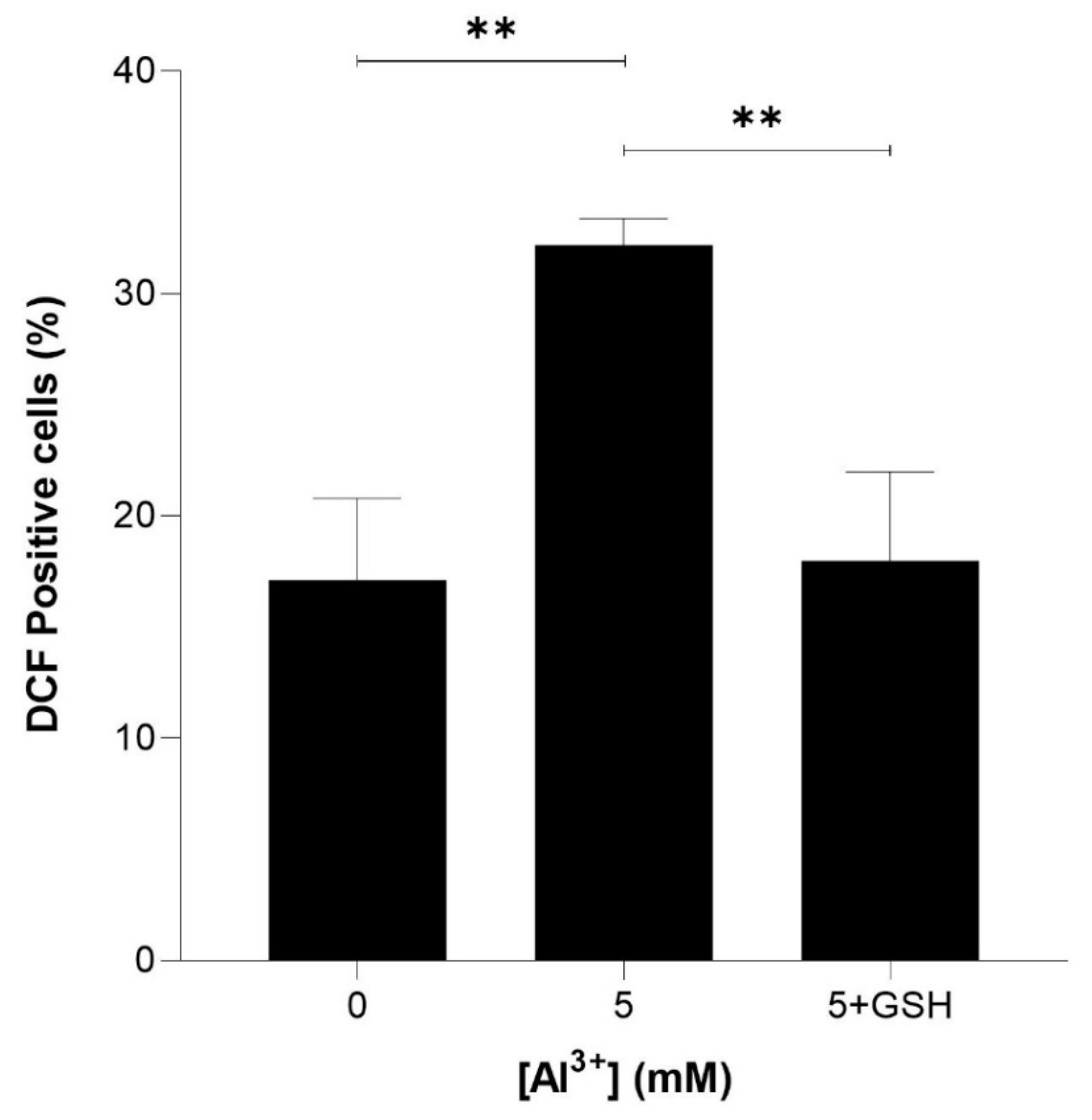
2.4. Glutathione Alleviates Al3+- and Aβ42-Enhanced Induction of ROS
3. Discussion
4. Materials and Methods
4.1. Yeast Strains, Plasmids, and Growth Media
4.2. Yeast Viability Measurements
4.3. Growth Inhibition Assays
4.4. Aluminium-Induced-ROS Detection in Yeast
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid beta |
| GFP | Green fluorescent protein |
| Al3+ | Aluminium ion |
| ROS | Reactive oxygen species |
| GFP–Aβ42 | Green fluorescent protein tagged with amyloid beta 42 |
| Fe3+ | Ferric ion |
| Fe2+ | Ferrous ion |
| H2DCFDA | 2’,7’-dichlorodihydrofluorescein diacetate |
| DCF | Dichlorofluorescein |
| GSH | Glutathione |
| YEPD | Yeast extract peptone dextrose |
| YEPE | Yeast extract peptone ethanol |
| YNB | Yeast nitrogen base |
| MAO | Monoamine oxidase |
| LPP | Low pH and low phosphate |
References
- Tomljenovic, L. Aluminum and Alzheimer’s Disease: After a century of controversy, is there a plausible link? J. Alzheimer’s Dis. 2011, 23, 567–598. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, M.; Kato-Negishi, M. Link between aluminum and the pathogenesis of Alzheimer’s Disease: The integration of the aluminum and amyloid cascade hypotheses. Int. J. Alzheimer’s 2011, 2011, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Rondeau, V.; Commenges, D.; Jacqmin-Gadda, H.; Dartigues, J-F. Relation between aluminum concentrations in drinking water and Alzheimer’s Disease: An 8-year follow-up study. Am. J. Epidemiol. 2000, 152, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Jiao, Q.; Xu, H.; Du, X.; Shi, L.; Jia, F.; Jiang, H. Biometal dyshomeostasis and toxic metal accumulations in the development of Alzheimer’s Disease. Front. Mol. Neurosci. 2017, 10, 339. [Google Scholar] [CrossRef]
- Yokel, R.A. Blood-brain barrier flux of aluminum, manganese, iron and other metals suspected to contribute to metal-induced neurodegeneration. J. Alzheimer’s Dis. 2006, 10, 223–253. [Google Scholar] [CrossRef]
- Exley, C. Aluminum should now be considered a primary etiological factor in Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2017, 1, 23–25. [Google Scholar] [CrossRef] [Green Version]
- Lupaescu, A.-V.; Humelnicu, I.; Petre, B.A.; Ciobanu, C.-I.; Drochioiu, G. Direct evidence for binding of aluminum to NAP anti-amyloid peptide and its analogs. Eur. J. Mass Spectrom. 2020, 26, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Mold, M.; Linhart, C.; Gómez-Ramírez, J.; Villegas-Lanau, A.; Exley, C. Aluminum and amyloid-β in familial Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, F.; Ni, Y.; Kokot, S. Effects of aluminum on amyloid-beta aggregation in the context of Alzheimer’s disease. Arab. J. Chem. 2019, 12, 2897–2904. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Sharma, P.; Srivastava, P.; Seth, A.; Tripathi, P.N.; Banerjee, A.G.; Shrivastava, S.K. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog. Neurobiol. 2019, 174, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Seynnaeve, D.; Vecchio, M.; Fruhmann, G.; Verelst, J.; Cools, M.; Beckers, J.; Mulvihill, D.P.; Winderickx, J.; Franssens, V. Recent insights on Alzheimer’s Disease originating from yeast models. Int. J. Mol. Sci. 2018, 19, 1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcdonald, J.B.; Dhakal, S.; Macreadie, I.G. Yeast contributions to Alzheimer’s Disease. J. Hum. Clin. Gen. 2020, 2, 1–19. [Google Scholar] [CrossRef]
- Dhakal, S.; Macreadie, I. Protein homeostasis networks and the use of yeast to guide interventions in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 8014. [Google Scholar] [CrossRef]
- Porzoor, A.; Macreadie, I. Yeast as a model for studies on Aβ aggregation toxicity in Alzheimer’s Disease, autophagic responses, and drug screening. In Systems Biology of Alzheimer’s Disease; Castrillo, J.I., Oliver, S.G., Eds.; Springer: New York, NY, USA, 2016; pp. 217–226. [Google Scholar]
- Dhakal, S.; Subhan, M.; Fraser, J.M.; Gardiner, K.; Macreadie, I. Simvastatin efficiently reduces levels of Alzheimer’s amyloid beta in yeast. Int. J. Mol. Sci. 2019, 20, 3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakal, S.; Macreadie, I. Tyramine and amyloid beta 42: A toxic synergy. Biomedicines 2020, 8, 145. [Google Scholar] [CrossRef]
- Crapper, D.R.; Krishnan, S.S.; Dalton, A.J. Brain aluminum distribution in Alzheimer’s Disease and experimental neurofibrillary degeneration. Science 1973, 180, 511–513. [Google Scholar] [CrossRef]
- Perl, D.; Brody, A. Alzheimer’s disease: X-ray spectrometric evidence of aluminum accumulation in neurofibrillary tangle-bearing neurons. Science 1980, 208, 297–299. [Google Scholar] [CrossRef]
- Walton, J.R. Aluminum in hippocampal neurons from humans with Alzheimer’s disease. Neurotoxicology 2006, 27, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, S.; Kakimi, S.; Ohsaki, A.; Ishikawa, A. Demonstration of aluminum in amyloid fibers in the cores of senile plaques in the brains of patients with Alzheimer’s disease. J. Inorg. Biochem. 2009, 103, 1579–1584. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yamamoto, D.; Rokugawa, K.; Yoshimura, K.; Imura, Y.; Yoshimura, E.; Suzuki, M. Decreased aluminium tolerance in the growth of Saccharomyces cerevisiae with SSO2 gene disruption. Biometals 2018, 31, 203–215. [Google Scholar] [CrossRef]
- Tun, N.M.; O’Doherty, P.J.; Chen, Z.-H.; Wu, X.-Y.; Bailey, T.D.; Kersaitis, C.; Wu, M.J. Identification of aluminium transport-related genes via genome-wide phenotypic screening of Saccharomyces cerevisiae. Metallomics 2014, 6, 1558. [Google Scholar] [CrossRef]
- Praticò, D.; Uryu, K.; Sung, S.; Tang, S.; Trojanowski, J.Q.; Lee, V.M.-Y. Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J. 2002, 16, 1138–1140. [Google Scholar] [CrossRef]
- Zheng, K.; Pan, J.-W.; Ye, L.; Fu, Y.; Peng, H.-Z.; Wan, B.-Y.; Gu, Q.; Bian, H.-W.; Han, N.; Wang, J.-H.; et al. Programmed cell death-involved aluminum toxicity in yeast alleviated by antiapoptotic members with decreased calcium signals. Plant Physiol. 2007, 143, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Loo, D.T.; Copani, A.; Pike, C.J.; Whittemore, E.R.; Walencewicz, A.J.; Cotman, C.W. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc. Natl. Acad. Sci. USA 1993, 90, 7951–7955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maya, S.; Prakash, T.; Madhu, K.D.; Goli, D. Multifaceted effects of aluminium in neurodegenerative diseases: A review. Biomed. Pharmacother. 2016, 83, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Fernández, M.B.; Soldado, A.B.; Sanz-Medel, A.; Vega, J.-A.; Novelli, A.; Fernández-Sánchez, M.T. Aluminum-induced degeneration of astrocytes occurs via apoptosis and results in neuronal death. Brain Res. 1999, 835, 125–136. [Google Scholar] [CrossRef]
- Drago, D.; Cavaliere, A.; Mascetra, N.; Ciavardelli, D.; Di Ilio, C.; Zatta, P.; Sensi, S.L. Aluminum modulates effects of β Amyloid 1–42 on neuronal calcium homeostasis and mitochondria functioning and is altered in a triple transgenic mouse model of Alzheimer’s Disease. Rejuvenation Res. 2008, 11, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Ricchelli, F.; Drago, D.; Filippi, B.; Tognon, G.; Zatta, P. Aluminum-triggered structural modifications and aggregation of β-amyloids. Cell. Mol. Life Sci. 2005, 62, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Chen, S.C.; Lin, Y.F.; Lee, Y.C.; Huang, M.Y.; Chen, K.C.; Wu, H.-Y.; Lin, P.C.; Gozes, I.; Tian, Y.C. Reduction of aluminum ion neurotoxicity through a small peptide application—NAP treatment of Alzheimer’s disease. J. Food Drug Anal. 2019, 27, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-J.; Hu, Y.-Y.; Yang, Z.-J.; Jiang, L.-P.; Shi, S.-L.; Li, Y.-R.; Guo, M.Y.; Wu, H.L.; Wan, Y.Y. Amyloid β-42 induces neuronal apoptosis by targeting mitochondria. Mol. Med. Rep. 2017, 16, 4521–4528. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Misschops, M.M.M.; Agarwal, N.R.; Ji, B.; Shanmugavel, K.P.; Petranovic, D. Interplay of energetics and ER stress exacerbates Alzheimer’s amyloid-β (Aβ) toxicity in yeast. Front. Mol. Neurosci. 2017, 10, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayton, S.; Lei, P.; Bush, A.I. Biometals and their therapeutic implications in Alzheimer’s Disease. Neurotherapeutics 2015, 12, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oteiza, P.I. A mechanism for the stimulatory effect of aluminum on iron-induced lipid peroxidation. Arch. Biochem. Biophys. 1994, 308, 374–379. [Google Scholar] [CrossRef]
- Kaneko, N.; Sugioka, T.; Sakurai, H. Aluminum compounds enhance lipid peroxidation in liposomes: Insight into cellular damage caused by oxidative stress. J. Inorg. Biochem. 2007, 101, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Metal-catalyzed disruption of membrane protein and lipid signaling in the pathogenesis of neurodegenerative disorders. Ann. N. Y. Acad. Sci. 2004, 1012, 37–50. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Zhao, Y.; Pogue, A.I.; Tarr, M.A.; Kruck, T.P.A.; Percy, M.E.; Cui, J.G.; Lukiw, W.J. Synergistic effects of iron and aluminum on stress-related gene expression in primary human neural cells. J. Alzheimer’s Dis. 2005, 8, 117–127. [Google Scholar] [CrossRef]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in neurodegeneration—Cause or consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Dobson, J.P.; Exley, C. Redox cycling of iron by Aβ42. Free Radic. Biol. Med. 2006, 40, 557–569. [Google Scholar] [CrossRef]
- Ott, S.; Dziadulewicz, N.; Crowther, D.C. Iron is a specific cofactor for distinct oxidation- and aggregation-dependent A toxicity mechanisms in a Drosophila model. Dis. Model Mech. 2015, 8, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.L.; Fan, Y.G.; Yang, Z.S.; Wang, Z.-Y.; Guo, C. Iron and Alzheimer’s Disease: From pathogenesis to therapeutic implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef] [Green Version]
- Coyle, J.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Petranovic, D. Amyloid-β peptide-induced cytotoxicity and mitochondrial dysfunction in yeast. FEMS Yeast. Res. 2015, 15, fov061. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, U.; Mancon, M.; Battaglia, V.; Ceccon, S.; Cardellini, P.; Toninello, A. Influence of reactive oxygen species production by monoamine oxidase activity on aluminum-induced mitochondrial permeability transition. Cell. Mol. Life Sci. 2004, 61, 2664–2671. [Google Scholar] [CrossRef]
- Nehru, B.; Bhalla, P. Reversal of an aluminium induced alteration in redox status in different regions of rat brain by administration of centrophenoxine. Mol. Cell. Biochem. 2006, 290, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Khan, M.F.; Jan, S.U.; Ullah, N. Effect of aluminium metal on glutathione (GSH) level in plasma and cytosolic fraction of human blood. Pak. J. Pharm. Sci. 2011, 24, 13–18. [Google Scholar]
- Rao, J.K.S.; Rao, G.V. Effect of aluminium (Al) on brain mitochondrial monoamine oxidase-A (MAO-A) activity? An in vitro kinetic study. Mol. Cell. Biochem. 1994, 137, 57–60. [Google Scholar] [PubMed]
- Zatta, P.; Zambenedetti, P.; Milanese, M. Activation of monoamine oxidase type-B by aluminum in rat brain homogenate. NeuroReport 1999, 10, 3645–3648. [Google Scholar] [CrossRef] [PubMed]
- Sherman, F. Getting started with yeast. Methods Enzymol. 2002, 350, 3–41. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transformant Yeast Strain | 3 Days | ||||||
|---|---|---|---|---|---|---|---|
| 0 Al | 0.4 Al | 0.8 Al | 1.6 Al | 3.2 Al | 4.8 Al | 6.4 Al | |
| BY4743 [p416GPD.GFP] | +++ | +++ | +++ | +++ | ++ | ++ | + |
| BY4743 [p416GPD.GFP.Aβ] | ++ | ++ | ++ | + | + | + | − |
| 7 Days | |||||||
| 0 Al | 0.4 Al | 0.8 Al | 1.6 Al | 3.2 Al | 4.8 Al | 6.4 Al | |
| BY4743 [p416GPD.GFP] | +++ | +++ | +++ | +++ | +++ | ++ | + |
| BY4743 [p416GPD.GFP.Aβ] | +++ | +++ | +++ | ++ | ++ | + | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mcdonald, J.B.; Dhakal, S.; Macreadie, I. A Toxic Synergy between Aluminium and Amyloid Beta in Yeast. Int. J. Mol. Sci. 2021, 22, 1835. https://doi.org/10.3390/ijms22041835
Mcdonald JB, Dhakal S, Macreadie I. A Toxic Synergy between Aluminium and Amyloid Beta in Yeast. International Journal of Molecular Sciences. 2021; 22(4):1835. https://doi.org/10.3390/ijms22041835
Chicago/Turabian StyleMcdonald, Jamieson B., Sudip Dhakal, and Ian Macreadie. 2021. "A Toxic Synergy between Aluminium and Amyloid Beta in Yeast" International Journal of Molecular Sciences 22, no. 4: 1835. https://doi.org/10.3390/ijms22041835
APA StyleMcdonald, J. B., Dhakal, S., & Macreadie, I. (2021). A Toxic Synergy between Aluminium and Amyloid Beta in Yeast. International Journal of Molecular Sciences, 22(4), 1835. https://doi.org/10.3390/ijms22041835