The Neurovascular Unit Dysfunction in Alzheimer’s Disease

 ,
,  , , ,
, , , 

Abstract
:1. Introduction
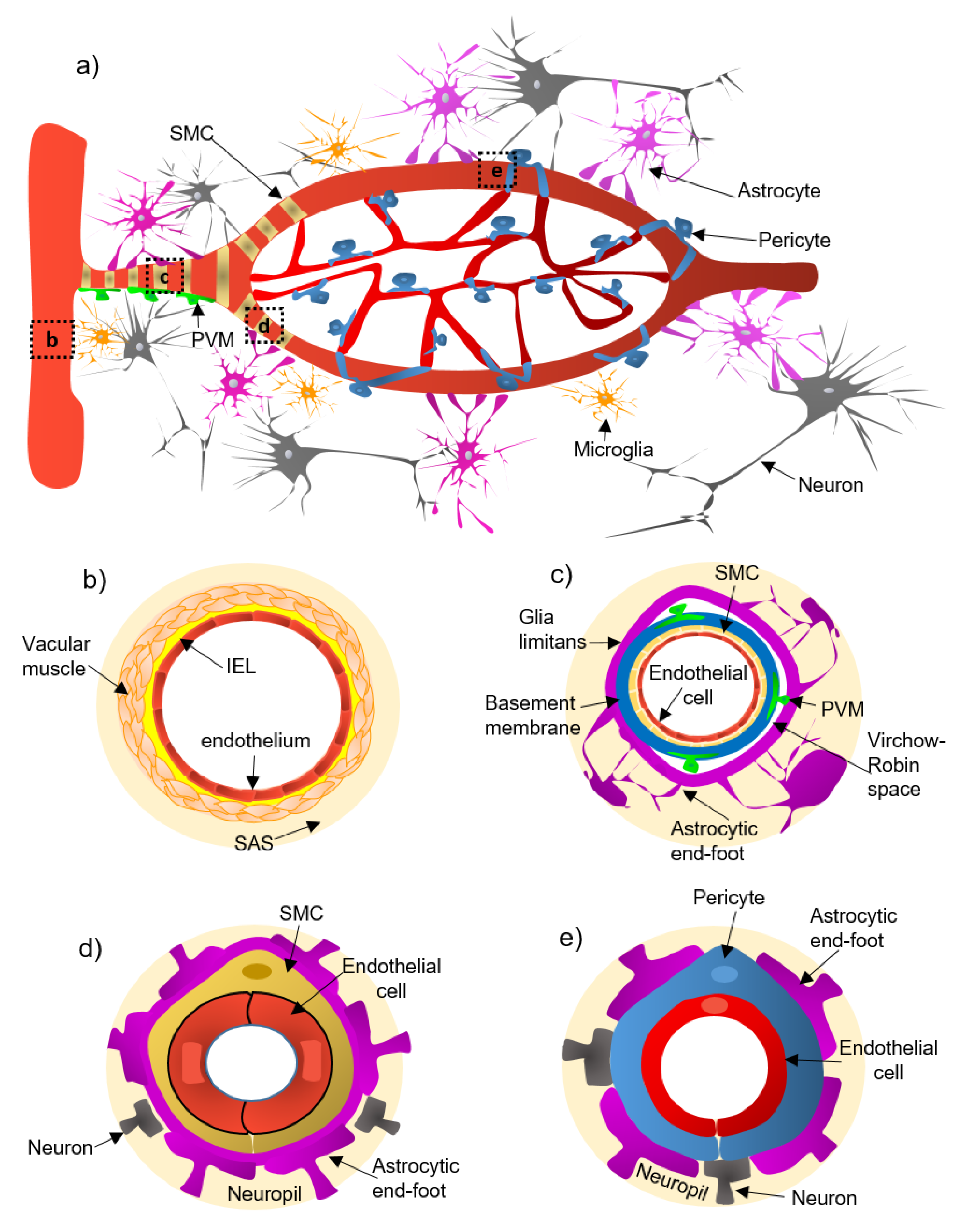
2. Cellular and Structural Components of the NVU Along the Cerebrovascular Tree
- (a)
- Pial arteries consist of multiple layers of SMCs, separated from the endothelium by a notable elastic lamina (Figure 1b), and innervated by nerve fibers formed from sensory and peripheral autonomic ganglia [18]. The pial arteries penetrating the brain are surrounded by the subarachnoid space (SAS; Figure 1b). Traditionally, the pial arteries have been associated with CBF and neuronal homeostasis [19].
- (b)
- Penetrating arterioles have several thin SMC layers that become a single layer (Figure 1c) [20]. The density of perivascular nerves is low at this level, and the elastic lamina becomes less prominent [20]. The perivascular space (also known as “Virchow-Robin space”) is delimited by the astrocytic end-foot (glia limitans) and the vascular basement membrane [21]. It comprises different cell types, including perivascular macrophages (PVMs), pial cells, Mato cells and mast cells, and collagen and nerve fibers [21] (Figure 1c). The perivascular space is an exchange pathway for the glymphatic system. The glymphatic system has been defined as a network of perivascular pathways that favors the exchange of both solutes and liquids between the cerebrospinal fluid (CSF) and the interstitial compartments, promoting clearance of metabolites, proteins, and debris from the brain interstitium [22,23,24]. This clearance depends mainly on the aquaporin-4 water channels (AQP4), found in astrocytic end-feet [25]. It has been demonstrated that the pial and penetrating arterioles can regulate arterial tone by the extrinsic and intrinsic innervations, respectively [26].
- (c)
- The intraparenchymal arterioles are formed when the arterioles invade deeper into the brain. Here, the glial limitans and the vascular basement membrane fuse, eliminating the Virchow-Robin space [21]. These arterioles have a single layer of SMCs, lack perivascular nerves, and are encapsulated by the astrocytic end-feet (Figure 1d) [20]. Endothelial cells extend their protrusions to SMCs and connect through gap junctions [27]. They are related to functional hyperemia, which ensures a rapid increase in the CBF rate to activated brain structures [28].
- (d)
- Capillaries are the smallest vessels in the brain and exchange molecules between blood and brain across the BBB [29]. Pericytes replace the SMCs, and mural cells are immersed in the endothelial basement membrane (Figure 1e) [30]. The border of the capillaries is enveloped by astrocytic end-feet, and the neural processes can be adjacent to the capillary basal lamina (Figure 1e) [31]. Pericytes and endothelial cells make direct interdigitated contacts where cytoplasmic protrusions (pegs) of one cell type insert into the opposing cell membrane (socket) of the other cell type [32].
3. The Physiological Characteristics of the Blood-Brain Barrier (BBB)
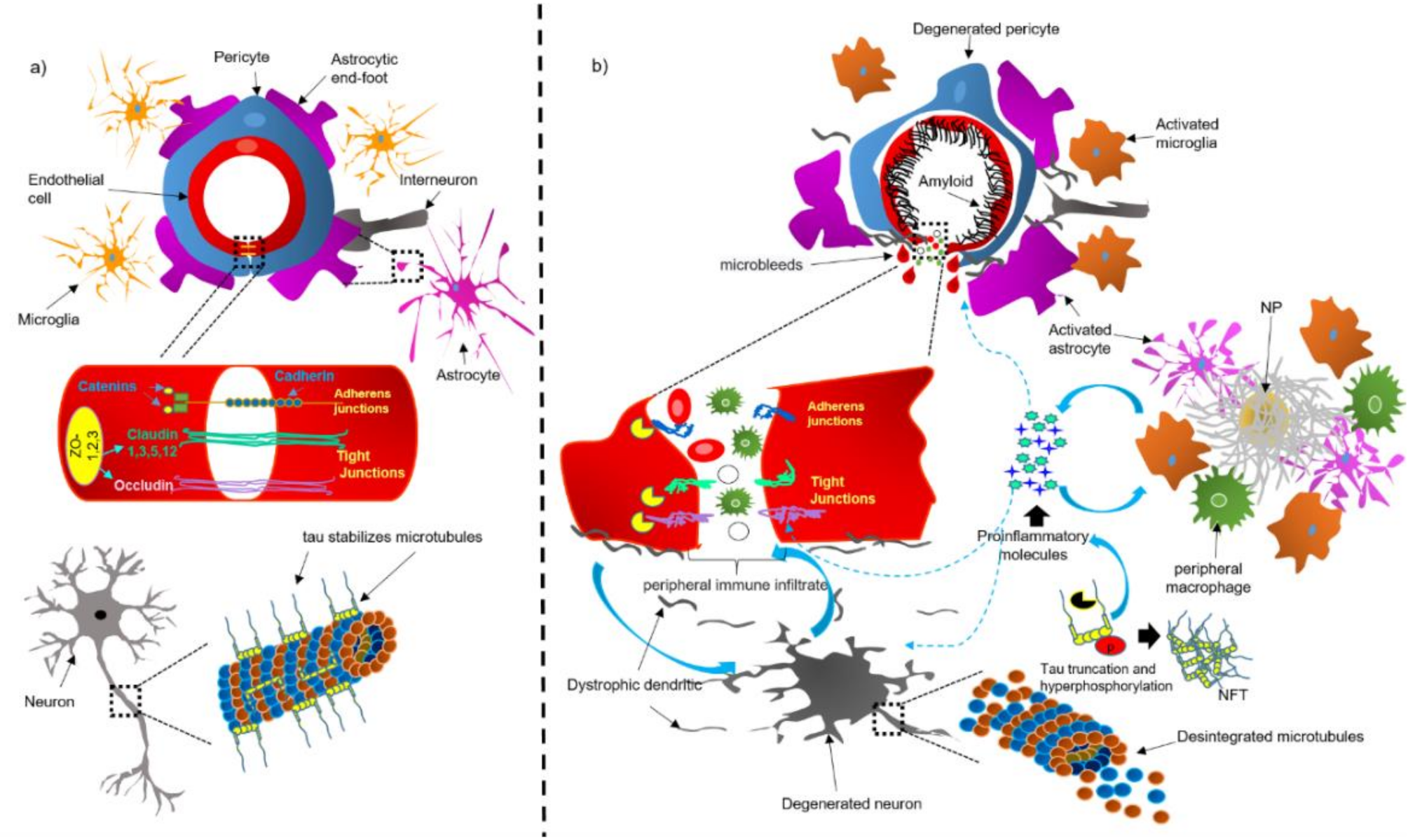
4. Dysfunction of the NVU and BBB in AD Brains
4.1. Bidirectional Pathological Association between Tau and the NVU and BBB
4.2. CAA Acts as a Trigger for Dysfunction of the NVU
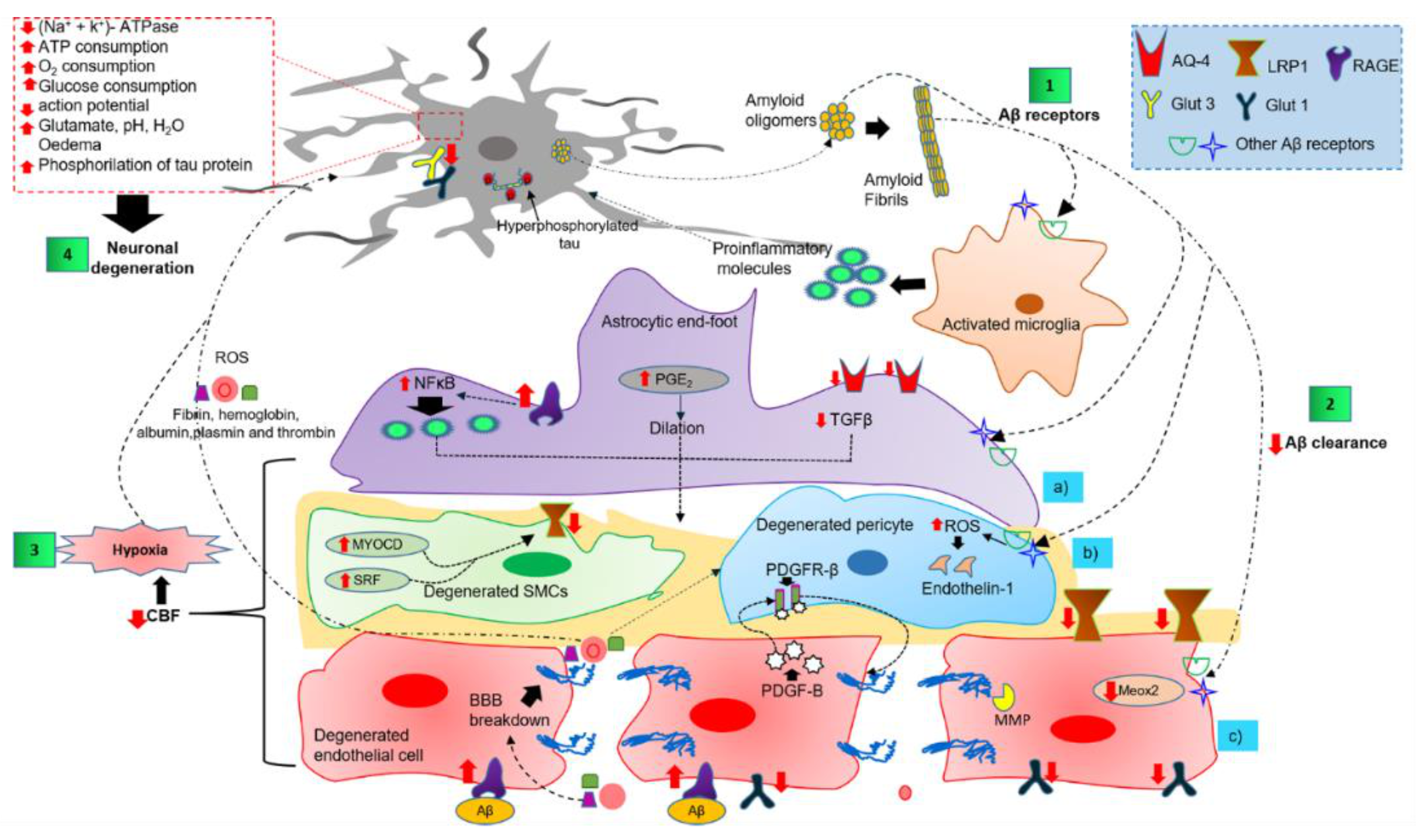
4.3. Dysfunction of Components of the NVU in AD
4.3.1. Perivascular Microglial Activation
4.3.2. Astrocytic End-Foot Dysfunction
4.3.3. Pericyte Degeneration
4.3.4. Endothelial Cell Degeneration and BBB Breakdown
4.3.5. Neuronal Cell Death Mechanisms
4.4. The Two-Hit Vascular Hypothesis and Dysfunction of the NVU
5. Current Status and Challenges for the Pharmacological Treatment of AD
5.1. General Limitations in AD Therapy
5.2. Therapeutics Associated with the Amyloidogenic Pathway
5.3. Drugs Targeting Tau Protein
5.4. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
6. Therapeutic Strategies Focused on the NVU
6.1. Vasculoprotective Effects of Anti-Diabetic, Lipid-Lowering, and Anti-Hypertensive Drugs
6.2. Drugs for Maintenance of BBB and NVU Integrity
6.3. Recent Approaches for Improving the NVU
7. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| a7nAChR | alpha-7 nicotinic acetylcholine receptor |
| ABCA1 | ATP binding cassette A1 |
| ABCB1 | ATP-binding cassette sub-family B member 1 |
| ACE | Angiotensin-converting enzyme |
| AD | Alzheimer’s disease |
| AJ | Adherent-junction |
| ApoE | Apolipoprotein E |
| APP | Amyloid precursor protein |
| AQP4 | Aquaporin-4 water channels |
| ARB | Angiotensin receptor blocker |
| AT1 | Angiotensin II type 1 receptor |
| Aβ | Amyloid β-peptide |
| BACE1 | Beta-site APP cleaving enzyme 1 |
| BBB | Blood-brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| BEC | Brain endothelial cell |
| b-FGF | Basic fibroblast growth factor |
| C.p. | Clinical phase |
| Cx | Complement factors |
| CAA | Cerebral amyloid angiopathy |
| CBF | Cerebral blood flow |
| CCB | Calcium channel blocker |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| CR | Complement receptor |
| CSF | Cerebrospinal fluid |
| DDS | Drug delivery systems |
| eNOS | Endothelial nitric oxide synthase |
| fAβ | Fibrillar Aβ |
| Fc γ R | Fc γ receptor |
| Fyn | Proto-oncogene tyrosine-protein kinase Fyn |
| GDNF | Glial cell-derived neurotrophic factor |
| GLP-1 | Glucagon-like peptide 1 |
| GLUT | Glucose transporter |
| GSK3β | Glycogen synthase kinase 3β |
| HCHWA-D | Hereditary cerebral hemorrhage with amyloidosis-Dutch type |
| HImAb | Human insulin mAb |
| HMG-CoA | 3-Hydroxy-3-methylglutaryl-CoA |
| HSA | Human serum albumin |
| IEL | Internal elastic lamina |
| IGF-1 | Insulin-like growth factor 1 |
| IgG | Immunoglobulin G |
| IL-1α | Interleukin-1 alpha |
| IL-1 β | Interleukin-1 beta |
| IL-1R1 | Interleukin 1 receptor type 1 |
| iNos | inducible nitric oxide synthase |
| IR | Insulin receptor |
| ISF | Interstitial fluid |
| LAMs | Leukocyte adhesion molecules |
| LRP1 | Low-density lipoprotein receptor-related protein 1 |
| mAb | Monoclonal antibody |
| mAβ | Monomeric Aβ |
| MEOX2 | Mesenchyme homeobox gene 2 |
| MMP | Matrix metalloproteinase |
| MTJ | Molecular Trojan horse |
| NF-kB | Nuclear factor-κB |
| NFT | Neurofibrillary tangle |
| NGF | Nerve growth factor |
| NMDAR | N-Methyl-d-aspartate receptors |
| NP | Neuritic plaque |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| NVU | Neurovascular unit |
| oAβ | Oligomeric Aβ |
| p47PHOX | Neutrophil cytosol factor 1 |
| Pcp | Preclinical phase |
| PDGF-B | Platelet-derived growth factor subunit B |
| PDGFRβ | Platelet-derived growth factor receptor-β |
| PGE2 | Prostaglandin-E2 |
| P-gp | P-glycoprotein |
| PHF | Paired helical filament |
| PI3K | Phosphatidylinositol 3-kinase |
| PVMs | Perivascular macrophages |
| RAGE | Receptor for advanced glycation endproducts |
| RMT | Receptor-mediated transcytosis |
| ROS | Reactive oxygen species |
| sAPPα | Secreted ectodomain APP alpha |
| SAS | Subarachnoid space |
| SEC-R | Serpin-enzyme complex receptor |
| sLRP | Soluble low-density lipoprotein receptor-related protein |
| SMC | Smooth muscle cell |
| SRA | Class A scavenger receptor |
| SRB2 | Scavenger receptor class B member 2 |
| SREBP2 | Sterol response element-binding protein 2 |
| SRF | Serum response factor |
| TfR | Human transferrin receptor |
| TGF-β | Transforming growth factor-beta |
| TJ | Tight-junction |
| TLR | Toll-like receptor |
| TNF-α | Tumoral necrosis factor-α |
| TNFR | Tumoral necrosis factor receptor |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| VEGF | Vascular endothelial growth factor |
| ZO-1 | Zonula occludens-1 |
| γ-PPAR | Peroxisome proliferator-activated receptor-γ |
References
- Harder, D.R.; Zhang, C.; Gebremedhin, D. Astrocytes function in matching blood flow to metabolic activity. News Physiol. Sci. 2002, 17, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood-brain barrier in health and disease. Epilepsia 2012, 53, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit—Concept review. Acta Physiol. (Oxford) 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Filosa, J.A. Vascular tone and neurovascular coupling: Considerations toward an improved in vitro model. Front. Neuroenerg. 2010, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Fields, R.D.; Stevens-Graham, B. New insights into neuron-glia communication. Science 2002, 298, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Ji, C.; Shao, A. Neurovascular unit dysfunction and neurodegenerative disorders. Front. Neurosci. 2020, 14, 334. [Google Scholar] [CrossRef]
- Mucke, L. Neuroscience: Alzheimer’s disease. Nature 2009, 461, 895–897. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular defects and faulty amyloid-beta vascular clearance in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, S87–S100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, Y.; Kanekiyo, T. Blood-brain barrier dysfunction and the pathogenesis of Alzheimer’s disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Lin, Y.Y.; Biemann, K.; Iqbal, K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch. Biochem. Biophys. 1993, 301, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Zhou, M.L.; Abousaleh, F.; Brendza, R.P.; Dietrich, H.H.; Koenigsknecht-Talboo, J.; Cirrito, J.R.; Milner, E.; Holtzman, D.M.; Zipfel, G.J. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: Contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J. Neurosci. 2008, 28, 13542–13550. [Google Scholar] [CrossRef]
- Chen, S.J.; Tsai, H.H.; Tsai, L.K.; Tang, S.C.; Lee, B.C.; Liu, H.M.; Yen, R.F.; Jeng, J.S. Advances in cerebral amyloid angiopathy imaging. Ther. Adv. Neurol. Disord. 2019, 12, 1–11. [Google Scholar] [CrossRef]
- Xu, F.; Fu, Z.; Dass, S.; Kotarba, A.E.; Davis, J.; Smith, S.O.; van Nostrand, W.E. Cerebral vascular amyloid seeds drive amyloid beta-protein fibril assembly with a distinct anti-parallel structure. Nat. Commun. 2016, 7, 13527. [Google Scholar] [CrossRef] [Green Version]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Hamel, E. Perivascular nerves and the regulation of cerebrovascular tone. J. Appl. Physiol. 2006, 100, 1059–1064. [Google Scholar] [CrossRef] [Green Version]
- Sekiguchi, Y.; Takuwa, H.; Kawaguchi, H.; Kikuchi, T.; Okada, E.; Kanno, I.; Ito, H.; Tomita, Y.; Itoh, Y.; Suzuki, N.; et al. Pial arteries respond earlier than penetrating arterioles to neural activation in the somatosensory cortex in awake mice exposed to chronic hypoxia: An additional mechanism to proximal integration signaling? J. Cereb. Blood Flow Metab. 2014, 34, 1761–1770. [Google Scholar] [CrossRef]
- Roggendorf, W.; Cervos-Navarro, J. Ultrastructure of arterioles in the cat brain. Cell Tissue Res. 1977, 178, 495–515. [Google Scholar] [CrossRef]
- Zhang, E.T.; Inman, C.B.; Weller, R.O. Interrelationships of the pia mater and the perivascular (Virchow-Robin) spaces in the human cerebrum. J. Anat. 1990, 170, 111–123. [Google Scholar]
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The glymphatic system: A beginner’s guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangroo Thrane, V.; Thrane, A.S.; Plog, B.A.; Thiyagarajan, M.; Iliff, J.J.; Deane, R.; Nagelhus, E.A.; Nedergaard, M. Paravascular microcirculation facilitates rapid lipid transport and astrocyte signaling in the brain. Sci. Rep. 2013, 3, 2582. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Goldman, S.A. Glymphatic failure as a final common pathway to dementia. Science 2020, 370, 50–56. [Google Scholar] [CrossRef]
- Braun, M.; Iliff, J.J. The impact of neurovascular, blood-brain barrier, and glymphatic dysfunction in neurodegenerative and metabolic diseases. Int. Rev. Neurobiol. 2020, 154, 413–436. [Google Scholar] [PubMed]
- Hotta, H.; Masamoto, K.; Uchida, S.; Sekiguchi, Y.; Takuwa, H.; Kawaguchi, H.; Shigemoto, K.; Sudo, R.; Tanishita, K.; Ito, H.; et al. Layer-specific dilation of penetrating arteries induced by stimulation of the nucleus basalis of Meynert in the mouse frontal cortex. J. Cereb. Blood Flow Metab. 2013, 33, 1440–1447. [Google Scholar] [CrossRef] [Green Version]
- Longden, T.A.; Hill-Eubanks, D.C.; Nelson, M.T. Ion channel networks in the control of cerebral blood flow. J. Cereb. Blood Flow Metab. 2016, 36, 492–512. [Google Scholar] [CrossRef] [Green Version]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Damisah, E.C.; Hill, R.A.; Tong, L.; Murray, K.N.; Grutzendler, J. A fluoro-Nissl dye identifies pericytes as distinct vascular mural cells during in vivo brain imaging. Nat. Neurosci. 2017, 20, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuevas, P.; Gutierrez-Diaz, J.A.; Reimers, D.; Dujovny, M.; Diaz, F.G.; Ausman, J.I. Pericyte endothelial gap junctions in human cerebral capillaries. Anat. Embryol. (Berlin) 1984, 170, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and dysfunction of the blood-brain barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Smith, Q.R. A review of blood-brain barrier transport techniques. Methods Mol. Med. 2003, 89, 193–208. [Google Scholar] [PubMed]
- Ohtsuki, S.; Terasaki, T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm. Res. 2007, 24, 1745–1758. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Deeken, J.F.; Loscher, W. The blood-brain barrier and cancer: Transporters, treatment, and Trojan horses. Clin. Cancer Res. 2007, 13, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [Green Version]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Michalicova, A.; Banks, W.A.; Legath, J.; Kovac, A. Tauopathies—focus on changes at the neurovascular unit. Curr. Alzheimer Res. 2017, 14, 790–801. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Novak, M.; Kabat, J.; Wischik, C.M. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993, 12, 365–370. [Google Scholar] [CrossRef]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1996, 777, 121–131. [Google Scholar] [CrossRef]
- Alonso, A.D.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Luna-Munoz, J.; Garcia-Sierra, F.; Falcon, V.; Menendez, I.; Chavez-Macias, L.; Mena, R. Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz-50 antibody in Alzheimer’s disease. J. Alzheimers Dis. 2005, 8, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Frauen, H.D.; Zhang, B.; Nordhues, B.A.; Bijan, S.; Lin, Y.C.; Zamudio, F.; Hernandez, L.D.; Sabbagh, J.J.; Selenica, M.L.; et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic brain injury and Alzheimer’s disease: The cerebrovascular link. EbioMed. 2018, 28, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Qiao, P.F.; Wan, C.Q.; Cai, M.; Zhou, N.K.; Li, Q. Role of blood-brain barrier in Alzheimer’s disease. J. Alzheimers Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef]
- Shabir, O.; Berwick, J.; Francis, S.E. Neurovascular dysfunction in vascular dementia, Alzheimer’s and atherosclerosis. BMC Neurosci. 2018, 19, 62. [Google Scholar] [CrossRef] [Green Version]
- Gatti, L.; Tinelli, F.; Scelzo, E.; Arioli, F.; Di Fede, G.; Obici, L.; Pantoni, L.; Giaccone, G.; Caroppo, P.; Parati, E.A.; et al. Understanding the Pathophysiology of Cerebral Amyloid Angiopathy. Int. J. Mol. Sci. 2020, 21, 3435. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.; Thal, D.R.; van Nostrand, W. Review: Sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 2011, 37, 75–93. [Google Scholar] [CrossRef]
- Bugiani, O.; Giaccone, G.; Rossi, G.; Mangieri, M.; Capobianco, R.; Morbin, M.; Mazzoleni, G.; Cupidi, C.; Marcon, G.; Giovagnoli, A.; et al. Hereditary cerebral hemorrhage with amyloidosis associated with the E693K mutation of APP. Arch. Neurol. 2010, 67, 987–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maat-Schieman, M.; Roos, R.; van Duinen, S. Hereditary cerebral hemorrhage with amyloidosis-Dutch type. Neuropathology 2005, 25, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Maat-Schieman, M.L.; Yamaguchi, H.; Hegeman-Kleinn, I.M.; Welling-Graafland, C.; Natte, R.; Roos, R.A.; van Duinen, S.G. Glial reactions and the clearance of amyloid beta protein in the brains of patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type. Acta Neuropathol. 2004, 107, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Maat-Schieman, M.L.; Yamaguchi, H.; van Duinen, S.G.; Natte, R.; Roos, R.A. Age-related plaque morphology and C-terminal heterogeneity of amyloid beta in Dutch-type hereditary cerebral hemorrhage with amyloidosis. Acta Neuropathol. 2000, 99, 409–419. [Google Scholar] [CrossRef]
- Maat-Schieman, M.L.; van Duinen, S.G.; Rozemuller, A.J.; Haan, J.; Roos, R.A. Association of vascular amyloid beta and cells of the mononuclear phagocyte system in hereditary cerebral hemorrhage with amyloidosis (Dutch) and Alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Tagliavini, F.; Giaccone, G.; Bugiani, O.; Frangione, B. Ubiquitinated neurites are associated with preamyloid and cerebral amyloid beta deposits in patients with hereditary cerebral hemorrhage with amyloidosis Dutch type. Acta Neuropathol. 1993, 85, 267–271. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; El Khoury, J.; Medeiros, L.A.; Terada, K.; Geula, C.; Luster, A.D.; Freeman, M.W. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J. Biol. Chem. 2002, 277, 47373–47379. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, M.I.; Chu, S.; Pierce, A.L.; Brubaker, W.D.; Hauhart, R.E.; Mastroeni, D.; Clarke, E.V.; Rogers, J.; Atkinson, J.P.; Tenner, A.J. Analysis of the putative role of CR1 in Alzheimer’s disease: Genetic association, expression and function. PLoS ONE 2016, 11, e0149792. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.C.; Kretzschmar, G.C.; Dos Santos, A.C.M.; Camargo, C.M.; Nisihara, R.M.; Farias, T.D.J.; Franke, A.; Wittig, M.; Schmidt, E.; Busch, H.; et al. Complement Receptor 1 (CR1, CD35) Polymorphisms and soluble CR1: A proposed anti-inflammatory role to quench the fire of “Fogo Selvagem” Pemphigus Foliaceus. Front. Immunol. 2019, 10, 2585. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, B.; Frost, J.L.; Hong, S.; Jin, M.; Ostaszewski, B.; Shankar, G.M.; Costantino, I.M.; Carroll, M.C.; Mayadas, T.N.; et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia 2012, 60, 993–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Hu, X.; Qian, L.; Chen, S.H.; Zhou, H.; Wilson, B.; Miller, D.S.; Hong, J.S. Microglial MAC1 receptor and PI3K are essential in mediating beta-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J. Neuroinflamm. 2011, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef] [PubMed]
- An, X.Q.; Xi, W.; Gu, C.Y.; Huang, X. Complement protein C5a enhances the beta-amyloid-induced neuro-inflammatory response in microglia in Alzheimer’s disease. Med. Sci. (Paris) 2018, 34, 116–120. [Google Scholar] [CrossRef]
- Ager, R.R.; Fonseca, M.I.; Chu, S.H.; Sanderson, S.D.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer’s disease. J. Neurochem. 2010, 113, 389–401. [Google Scholar] [CrossRef] [Green Version]
- El Khoury, J.; Hickman, S.E.; Thomas, C.A.; Cao, L.; Silverstein, S.C.; Loike, J.D. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 1996, 382, 716–719. [Google Scholar] [CrossRef]
- Yang, C.N.; Shiao, Y.J.; Shie, F.S.; Guo, B.S.; Chen, P.H.; Cho, C.Y.; Chen, Y.J.; Huang, F.L.; Tsay, H.J. Mechanism mediating oligomeric Abeta clearance by naive primary microglia. Neurobiol. Dis. 2011, 42, 221–230. [Google Scholar] [CrossRef]
- Ganote, C.E.; Sims, M.A. Parallel temperature dependence of contracture-associated enzyme release due to anoxia, 2,4-dinitrophenol (DNP), or caffeine and the calcium paradox. Am. J. Pathol. 1984, 116, 94–106. [Google Scholar]
- Marshall, R.J. Bayesian analysis of case-control studies. Stat. Med. 1988, 7, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Marutle, A.; Nordberg, A. Modulation of alpha7 nicotinic acetylcholine receptor and fibrillar amyloid-beta interactions in Alzheimer’s disease brain. J. Alzheimers Dis. 2013, 33, 841–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oz, M.; Lorke, D.E.; Yang, K.H.; Petroianu, G. On the interaction of beta-amyloid peptides and alpha7-nicotinic acetylcholine receptors in Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; Van Bronswijk, W.; Martins, R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J. Neurosci. 2002, 22, RC221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, K.; Behrens, M.; Choi, D.; Manias, K.; Perlmutter, D.H. The serpin-enzyme complex receptor recognizes soluble, nontoxic amyloid-beta peptide but not aggregated, cytotoxic amyloid-beta peptide. J. Biol. Chem. 1996, 271, 18032–18044. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.Y.; McLaurin, J. Mechanisms of amyloid-beta peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int. J. Alzheimers Dis. 2010, 2011, 548380. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.L.; Koike, M.; Spusta, S.C.; Niemi, E.C.; Yenari, M.; Nakamura, M.C.; Seaman, W.E. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 2009, 109, 1144–1156. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Zhao, Z.; Sagare, A.P.; Wu, Y.; Wang, M.; Owens, N.C.; Verghese, P.B.; Herz, J.; Holtzman, D.M.; Zlokovic, B.V. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 2018, 13, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelmus, M.M.; Otte-Holler, I.; van Triel, J.J.; Veerhuis, R.; Maat-Schieman, M.L.; Bu, G.; de Waal, R.M.; Verbeek, M.M. Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am. J. Pathol. 2007, 171, 1989–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Pol, J.; Vandenhaute, E.; Boucau, M.C.; Candela, P.; Dehouck, L.; Cecchelli, R.; Dehouck, M.P.; Fenart, L.; Gosselet, F. Brain pericytes ABCA1 expression mediates cholesterol efflux but not cellular amyloid-beta peptide accumulation. J. Alzheimers Dis. 2012, 30, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhnke, D.; Jedlitschky, G.; Grube, M.; Krohn, M.; Jucker, M.; Mosyagin, I.; Cascorbi, I.; Walker, L.C.; Kroemer, H.K.; Warzok, R.W.; et al. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s amyloid-beta peptides-implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 2007, 17, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Rosenegger, D.G.; Tran, C.H.; Wamsteeker Cusulin, J.I.; Gordon, G.R. Tonic local brain blood flow control by astrocytes independent of phasic neurovascular coupling. J. Neurosci. 2015, 35, 13463–13474. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef]
- Bakker, E.N.; Bacskai, B.J.; Arbel-Ornath, M.; Aldea, R.; Bedussi, B.; Morris, A.W.; Weller, R.O.; Carare, R.O. Lymphatic clearance of the brain: Perivascular, paravascular and significance for neurodegenerative diseases. Cell Mol. Neurobiol. 2016, 36, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Ulv Larsen, S.M.; Landolt, H.P.; Berger, W.; Nedergaard, M.; Knudsen, G.M.; Holst, S.C. Haplotype of the astrocytic water channel AQP4 is associated with slow wave energy regulation in human NREM sleep. PLoS Biol. 2020, 18, e3000623. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Murchison, C.F.; Westaway, S.K.; Simon, M.J.; Erten-Lyons, D.; Kaye, J.A.; Quinn, J.F.; Iliff, J.J. The effects of noncoding aquaporin-4 single-nucleotide polymorphisms on cognition and functional progression of Alzheimer’s disease. Alzheimers Dement. (NY) 2017, 3, 348–359. [Google Scholar] [CrossRef]
- Zeppenfeld, D.M.; Simon, M.; Haswell, J.D.; D’Abreo, D.; Murchison, C.; Quinn, J.F.; Grafe, M.R.; Woltjer, R.L.; Kaye, J.; Iliff, J.J. Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol. 2017, 74, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcendor, D.J. Interactions between smyloid-beta proteins and human brain pericytes: Implications for the pathobiology of Alzheimer’s disease. J. Clin. Med. 2020, 9, 1490. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009, 8, 205–216. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, H.; Chow, N.; Sallstrom, J.; Bell, R.D.; Deane, R.; Brooks, A.I.; Kanagala, S.; Rubio, A.; Sagare, A.; et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 2005, 11, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Ogunshola, O.O.; Antoniou, X. Contribution of hypoxia to Alzheimer’s disease: Is HIF-1alpha a mediator of neurodegeneration? Cell Mol. Life Sci. 2009, 66, 3555–3563. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [Green Version]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A., 3rd; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Deane, R.; Chow, N.; Long, X.; Sagare, A.; Singh, I.; Streb, J.W.; Guo, H.; Rubio, A.; Van Nostrand, W.; et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat. Cell Biol. 2009, 11, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular dysfunction and faulty amyloid beta-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.M.; Kwan, J.; Malek-Ahmadi, M.; Maarouf, C.L.; Kokjohn, T.A.; Belden, C.; Sabbagh, M.N.; Beach, T.G.; Roher, A.E. Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer’s disease. PLoS ONE 2012, 7, e36893. [Google Scholar] [CrossRef]
- Li, J.M.; Huang, L.L.; Liu, F.; Tang, B.S.; Yan, X.X. Can brain impermeable BACE1 inhibitors serve as anti-CAA medicine? BMC Neurol. 2017, 17, 163. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers. Res. Ther. 2014, 6, 89. [Google Scholar] [CrossRef] [Green Version]
- Chris Min, K.; Dockendorf, M.F.; Palcza, J.; Tseng, J.; Ma, L.; Stone, J.A.; Kleijn, H.J.; Hodsman, P.; Masuo, K.; Tanen, M.; et al. Pharmacokinetics and Pharmacodynamics of the BACE1 Inhibitor verubecestat (MK-8931) in healthy Japanese adults: A randomized, placebo-controlled study. Clin. Pharmacol. Ther. 2019, 105, 1234–1243. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Riviere, M.E.; Caputo, A.; Sovago, J.; Maguire, R.P.; Farlow, M.; Marotta, G.; Sanchez-Valle, R.; Scheltens, P.; Ryan, J.M.; et al. Active Abeta immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimers Dement. (NY) 2017, 3, 10–22. [Google Scholar] [CrossRef]
- Folch, J.; Ettcheto, M.; Petrov, D.; Abad, S.; Pedros, I.; Marin, M.; Olloquequi, J.; Camins, A. Review of the advances in treatment for Alzheimer disease: Strategies for combating beta-amyloid protein. Neurologia 2018, 33, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavalu, K.; Zhang, C.; Zhang, X.; Tanzi, R.E.; Sisodia, S.S. Age-dependent, non-cell-autonomous deposition of amyloid from synthesis of beta-amyloid by cells other than excitatory neurons. J. Neurosci. 2014, 34, 3668–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henley, D.B.; May, P.C.; Dean, R.A.; Siemers, E.R. Development of semagacestat (LY450139), a functional gamma-secretase inhibitor, for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2009, 10, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; Salloway, S.; van Dyck, C.H.; Dubois, B.; Andreasen, N.; Brody, M.; Curtis, C.; Soininen, H.; Thein, S.; Shiovitz, T.; et al. Targeting prodromal Alzheimer disease with avagacestat: A randomized clinical trial. JAMA Neurol. 2015, 72, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cui, J. (-)-Epigallocatechin-3-gallate provides neuroprotection via AMPK activation against traumatic brain injury in a mouse model. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Koyama, N.; Tan, J.; Segawa, T.; Maeda, M.; Town, T. Combined treatment with the phenolics (-)-epigallocatechin-3-gallate and ferulic acid improves cognition and reduces Alzheimer-like pathology in mice. J. Biol. Chem. 2019, 294, 2714–2731. [Google Scholar] [CrossRef] [Green Version]
- Raghuvanshi, R.; Bharate, S.B. Preclinical and clinical studies on bryostatins, a class of marine-derived protein kinase C modulators: A mini-review. Curr. Top Med. Chem. 2020, 20, 1124–1135. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in Alzheimer disease: An update. J. Cent. Nerv. Syst. Dis. 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Talaga, P. Beta-amyloid aggregation inhibitors for the treatment of Alzheimer’s disease: Dream or reality? Mini Rev. Med. Chem. 2001, 1, 175–186. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Hernandez, F.; Del Rio, J.; Moreno, F.J.; Avila, J. Tramiprosate, a drug of potential interest for the treatment of Alzheimer’s disease, promotes an abnormal aggregation of tau. Mol. Neurodegener. 2007, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, M.N.; Agro, A.; Bell, J.; Aisen, P.S.; Schweizer, E.; Galasko, D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2011, 25, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; Sanchez-Lopez, E.; Garcia, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current research therapeutic strategies for Alzheimer’s disease treatment. Neural. Plast. 2016, 2016, 8501693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, M. An overview on the clinical development of tau-based therapeutics. Int. J. Mol. Sci. 2018, 19, 1160. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.S.; Hebron, M.L.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, J.N.; Anjum, M.; Pagan, F.; Torres-Yaghi, Y.; Shi, W.; et al. Nilotinib effects on safety, tolerability, and biomarkers in Alzheimer’s disease. Ann. Neurol. 2020, 88, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, F.; Sadowsky, C.; Holstein, A.; Leterme Gle, P.; Peng, Y.; Jackson, N.; Fox, N.C.; Ketter, N.; Liu, E.; Ryan, J.M.; et al. Two phase 2 multiple ascending-dose studies of vanutide cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer’s disease. J. Alzheimers Dis. 2016, 51, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Harigaya, Y.; Saido, T.C.; Eckman, C.B.; Prada, C.M.; Shoji, M.; Younkin, S.G. Amyloid beta protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer’s disease brain. Biochem. Biophys. Res. Commun. 2000, 276, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Overman, M.J.; Cotman, C.W. Amino-terminal deletions enhance aggregation of beta-amyloid peptides in vitro. J. Biol. Chem. 1995, 270, 23895–23898. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, D.M.; Colton, C.A. Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS Neurol. Disord. Drug Targets 2009, 8, 50–64. [Google Scholar] [CrossRef] [Green Version]
- Budd Haeberlein, S.; O’Gorman, J.; Chiao, P.; Bussiere, T.; von Rosenstiel, P.; Tian, Y.; Zhu, Y.; von Hehn, C.; Gheuens, S.; Skordos, L.; et al. Clinical development of aducanumab, an anti-Abeta human monoclonal antibody being investigated for the treatment of early Alzheimer’s disease. J. Prev. Alzheimers Dis. 2017, 4, 255–263. [Google Scholar]
- Crehan, H.; Liu, B.; Kleinschmidt, M.; Rahfeld, J.U.; Le, K.X.; Caldarone, B.J.; Frost, J.L.; Hettmann, T.; Hutter-Paier, B.; O’Nuallain, B.; et al. Effector function of anti-pyroglutamate-3 Abeta antibodies affects cognitive benefit, glial activation and amyloid clearance in Alzheimer’s-like mice. Alzheimers. Res. Ther. 2020, 12, 12. [Google Scholar] [CrossRef]
- Samadi, H.; Sultzer, D. Solanezumab for Alzheimer’s disease. Expert Opin. Biol. Ther. 2011, 11, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Dang, Y.; Ostaszewski, B.; Mengel, D.; Steffen, V.; Rabe, C.; Bittner, T.; Walsh, D.M.; Selkoe, D.J. Target engagement in an alzheimer trial: Crenezumab lowers amyloid beta oligomers in cerebrospinal fluid. Ann. Neurol. 2019, 86, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Pradier, L.; Blanchard-Bregeon, V.; Bohme, A.; Debeir, T.; Menager, J.; Benoit, P.; Barneoud, P.; Taupin, V.; Bertrand, P.; Dugay, P.; et al. SAR228810: An antibody for protofibrillar amyloid beta peptide designed to reduce the risk of amyloid-related imaging abnormalities (ARIA). Alzheimers. Res. Ther. 2018, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, N.; Simeoni, M.; Ostlund, H.; Lisjo, P.I.; Fladby, T.; Loercher, A.E.; Byrne, G.J.; Murray, F.; Scott-Stevens, P.T.; Wallin, A.; et al. First administration of the Fc-attenuated anti-beta amyloid antibody GSK933776 to patients with mild Alzheimer’s disease: A randomized, placebo-controlled study. PLoS ONE 2015, 10, e0098153. [Google Scholar] [CrossRef] [PubMed]
- Landen, J.W.; Andreasen, N.; Cronenberger, C.L.; Schwartz, P.F.; Borjesson-Hanson, A.; Ostlund, H.; Sattler, C.A.; Binneman, B.; Bednar, M.M. Ponezumab in mild-to-moderate Alzheimer’s disease: Randomized phase II PET-PIB study. Alzheimers Dement. (NY) 2017, 3, 393–401. [Google Scholar] [CrossRef]
- Sollvander, S.; Nikitidou, E.; Gallasch, L.; Zysk, M.; Soderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. The Abeta protofibril selective antibody mAb158 prevents accumulation of Abeta in astrocytes and rescues neurons from Abeta-induced cell death. J. Neuroinflamm. 2018, 15, 98. [Google Scholar] [CrossRef]
- Loeffler, D.A. Intravenous immunoglobulin and Alzheimer’s disease: What now? J. Neuroinflamm. 2013, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Sano, M. Tarenflurbil: Mechanisms and myths. Arch. Neurol. 2010, 67, 750–752. [Google Scholar] [CrossRef]
- Sivilia, S.; Lorenzini, L.; Giuliani, A.; Gusciglio, M.; Fernandez, M.; Baldassarro, V.A.; Mangano, C.; Ferraro, L.; Pietrini, V.; Baroc, M.F.; et al. Multi-target action of the novel anti-Alzheimer compound CHF5074: In vivo study of long term treatment in Tg2576 mice. BMC Neurosci. 2013, 14, 44. [Google Scholar] [CrossRef] [Green Version]
- Wijesuriya, H.C.; Bullock, J.Y.; Faull, R.L.; Hladky, S.B.; Barrand, M.A. ABC efflux transporters in brain vasculature of Alzheimer’s subjects. Brain Res. 2010, 1358, 228–238. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Molecular Trojan horses for blood-brain barrier drug delivery. Discov. Med. 2006, 6, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Park, K. Drug delivery of the future: Chasing the invisible gorilla. J. Control. Release 2016, 240, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, C.; Praca, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, M.J.; Andrade, S.; Loureiro, J.A.; do Carmo Pereira, M. Nanotechnology to improve the Alzheimer’s disease therapy with natural compounds. Drug Deliv. Transl. Res. 2020, 10, 380–402. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Maeng, H.J.; Yu, K.H.; Lee, K.R.; Tsuruo, T.; Kim, D.D.; Shim, C.K.; Chung, S.J. Evidence of carrier-mediated transport in the penetration of donepezil into the rat brain. J. Pharm. Sci. 2010, 99, 1548–1566. [Google Scholar] [CrossRef]
- Brousseau, G.; Rourke, B.P.; Burke, B. Acetylcholinesterase inhibitors, neuropsychiatric symptoms, and Alzheimer’s disease subtypes: An alternate hypothesis to global cognitive enhancement. Exp. Clin. Psychopharmacol. 2007, 15, 546–554. [Google Scholar] [CrossRef]
- Ishiwata, K.; Kawamura, K.; Yanai, K.; Hendrikse, N.H. In vivo evaluation of P-glycoprotein modulation of 8 PET radioligands used clinically. J. Nucl. Med. 2007, 48, 81–87. [Google Scholar]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Cheng, J.; Hou, L. Drug development for Alzheimer’s disease: Microglia induced neuroinflammation as a target? Int. J. Mol. Sci. 2019, 20, 558. [Google Scholar] [CrossRef] [Green Version]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. gamma-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Lemere, C.A. Immunotherapy for Alzheimer’s disease: Hoops and hurdles. Mol. Neurodegener. 2013, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Pagliari, P.; Nakaoke, R.; Morley, J.E. Effects of a behaviorally active antibody on the brain uptake and clearance of amyloid beta proteins. Peptides 2005, 26, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Terrell, B.; Farr, S.A.; Robinson, S.M.; Nonaka, N.; Morley, J.E. Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides 2002, 23, 2223–2226. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Curran, G.L.; Berg, C.T. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc. Natl. Acad. Sci. USA 1994, 91, 5705–5709. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, G.P.; Sterzl, I.; Nilaver, G. Localization patterns for immunoglobulins and albumins in the brain suggest diverse mechanisms for their transport across the blood-brain barrier (BBB). Prog. Brain Res. 1992, 91, 149–154. [Google Scholar]
- Banks, W.A. Are the extracellular pathways a conduit for the delivery of therapeutics to the brain? Curr. Pharm. Des. 2004, 10, 1365–1370. [Google Scholar] [CrossRef]
- Schlachetzki, F.; Zhu, C.; Pardridge, W.M. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J. Neurochem. 2002, 81, 203–206. [Google Scholar] [CrossRef]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melis, V.; Magbagbeolu, M.; Rickard, J.E.; Horsley, D.; Davidson, K.; Harrington, K.A.; Goatman, K.; Goatman, E.A.; Deiana, S.; Close, S.P.; et al. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav. Pharmacol. 2015, 26, 353–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Asuni, A.A.; Boutajangout, A.; Quartermain, D.; Sigurdsson, E.M. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 2007, 27, 9115–9129. [Google Scholar] [CrossRef] [PubMed]
- Boutajangout, A.; Ingadottir, J.; Davies, P.; Sigurdsson, E.M. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J. Neurochem. 2011, 118, 658–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kfoury, N.; Holmes, B.B.; Jiang, H.; Holtzman, D.M.; Diamond, M.I. Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 2012, 287, 19440–19451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontsekova, E.; Zilka, N.; Kovacech, B.; Novak, P.; Novak, M. First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers. Res. Ther. 2014, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitner, J.C.; Haneuse, S.J.; Walker, R.; Dublin, S.; Crane, P.K.; Gray, S.L.; Larson, E.B. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 2009, 72, 1899–1905. [Google Scholar] [CrossRef] [Green Version]
- Daniels, M.J.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tan, L.; Wang, H.F.; Tan, C.C.; Meng, X.F.; Wang, C.; Tang, S.W.; Yu, J.T. Anti-inflammatory drugs and risk of Alzheimer’s disease: An updated systematic review and meta-analysis. J. Alzheimers Dis. 2015, 44, 385–396. [Google Scholar] [CrossRef]
- Akinyemi, R.O.; Mukaetova-Ladinska, E.B.; Attems, J.; Ihara, M.; Kalaria, R.N. Vascular risk factors and neurodegeneration in ageing related dementias: Alzheimer’s disease and vascular dementia. Curr. Alzheimer Res. 2013, 10, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Sebastiao, I.; Candeias, E.; Santos, M.S.; de Oliveira, C.R.; Moreira, P.I.; Duarte, A.I. Insulin as a bridge between yype 2 diabetes and Alzheimer disease—how anti-diabetics could be a solution for dementia. Front. Endocrinol. (Lausanne) 2014, 5, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.M.; Torres-Aleman, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Hughes, T.M.; Craft, S. The role of insulin in the vascular contributions to age-related dementia. Biochim. Biophys. Acta 2016, 1862, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Barazzoni, R.; Zanetti, M.; Gortan Cappellari, G.; Semolic, A.; Boschelle, M.; Codarin, E.; Pirulli, A.; Cattin, L.; Guarnieri, G. Fatty acids acutely enhance insulin-induced oxidative stress and cause insulin resistance by increasing mitochondrial reactive oxygen species (ROS) generation and nuclear factor-kappaB inhibitor (IkappaB)-nuclear factor-kappaB (NFkappaB) activation in rat muscle, in the absence of mitochondrial dysfunction. Diabetologia 2012, 55, 773–782. [Google Scholar]
- Cersosimo, E.; DeFronzo, R.A. Insulin resistance and endothelial dysfunction: The road map to cardiovascular diseases. Diabetes Metab. Res. Rev. 2006, 22, 423–436. [Google Scholar] [CrossRef]
- Bhat, N.R. Vasculoprotection as a convergent, multi-targeted mechanism of anti-AD therapeutics and interventions. J. Alzheimers Dis. 2015, 46, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Asraf, K.; Torika, N.; Apte, R.N.; Fleisher-Berkovich, S. Microglial activation is modulated by captopril: In vitro and in vivo studies. Front. Cell Neurosci. 2018, 12, 116. [Google Scholar] [CrossRef]
- Ongali, B.; Nicolakakis, N.; Tong, X.K.; Aboulkassim, T.; Papadopoulos, P.; Rosa-Neto, P.; Lecrux, C.; Imboden, H.; Hamel, E. Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol. Dis. 2014, 68, 126–136. [Google Scholar] [CrossRef]
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta 2011, 1812, 1584–1590. [Google Scholar] [CrossRef] [Green Version]
- Yildirim Simsir, I.; Soyaltin, U.E.; Cetinkalp, S. Glucagon like peptide-1 (GLP-1) likes Alzheimer’s disease. Diabetes Metab. Syndr. 2018, 12, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.K.; Chiang, C.; Welsh-Bohmer, K.A.; Brannan, S.K.; Culp, M.; O’Neil, J.; Runyan, G.; Harrigan, P.; Plassman, B.L.; Lutz, M.; et al. The TOMMORROW study: Design of an Alzheimer’s disease delay-of-onset clinical trial. Alzheimers Dement. (NY) 2019, 5, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.W.; Willett, K.C.; Desilets, A.R. Rosiglitazone and pioglitazone for the treatment of Alzheimer’s disease. Ann. Pharmacother. 2011, 45, 1416–1424. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geifman, N.; Brinton, R.D.; Kennedy, R.E.; Schneider, L.S.; Butte, A.J. Evidence for benefit of statins to modify cognitive decline and risk in Alzheimer’s disease. Alzheimers. Res. Ther. 2017, 9, 10. [Google Scholar] [CrossRef]
- Simons, M.; Schwarzler, F.; Lutjohann, D.; von Bergmann, K.; Beyreuther, K.; Dichgans, J.; Wormstall, H.; Hartmann, T.; Schulz, J.B. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann. Neurol. 2002, 52, 346–350. [Google Scholar] [CrossRef]
- Manocha, G.; Ghatak, A.; Puig, K.; Combs, C. Anti-alpha4beta1 integrin antibodies attenuated brain inflammatory changes in a mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2018, 15, 1123–1135. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Corrigendum: Anakinra therapy for non-cancer inflammatory diseases. Front. Pharmacol. 2019, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Garcez, M.L.; Zugno, A.I. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 2015, 6, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Revilla, S.; Ursulet, S.; Alvarez-Lopez, M.J.; Castro-Freire, M.; Perpina, U.; Garcia-Mesa, Y.; Bortolozzi, A.; Gimenez-Llort, L.; Kaliman, P.; Cristofol, R.; et al. Lenti-GDNF gene therapy protects against Alzheimer’s disease-like neuropathology in 3xTg-AD mice and MC65 cells. CNS Neurosci. Ther. 2014, 20, 961–972. [Google Scholar] [CrossRef]
- Harris, R.; Miners, J.S.; Allen, S.; Love, S. VEGFR1 and VEGFR2 in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 741–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Religa, P.; Cao, R.; Religa, D.; Xue, Y.; Bogdanovic, N.; Westaway, D.; Marti, H.H.; Winblad, B.; Cao, Y. VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci. Rep. 2013, 3, 2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, S.; Behbahani, H.; Eriksdotter, M. Innovative therapy for Alzheimer’s disease-with focus on biodelivery of NGF. Front. Neurosci. 2019, 13, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolakakis, N.; Aboulkassim, T.; Ongali, B.; Lecrux, C.; Fernandes, P.; Rosa-Neto, P.; Tong, X.K.; Hamel, E. Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J. Neurosci. 2008, 28, 9287–9296. [Google Scholar] [CrossRef] [PubMed]
- Zolezzi, J.M.; Bastias-Candia, S.; Santos, M.J.; Inestrosa, N.C. Alzheimer’s disease: Relevant molecular and physiopathological events affecting amyloid-beta brain balance and the putative role of PPARs. Front. Aging Neurosci. 2014, 6, 176. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, P.; Rosa-Neto, P.; Rochford, J.; Hamel, E. Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of Alzheimer’s disease with combined amyloid-beta and cerebrovascular pathology. PLoS ONE 2013, 8, e68612. [Google Scholar] [CrossRef]
- Tong, X.K.; Lecrux, C.; Rosa-Neto, P.; Hamel, E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. J. Neurosci. 2012, 32, 4705–4715. [Google Scholar] [CrossRef] [Green Version]
- Silva, I.V.G.; de Figueiredo, R.C.; Rios, D.R.A. Effect of different classes of antihypertensive drugs on endothelial function and inflammation. Int. J. Mol. Sci. 2019, 20, 3458. [Google Scholar] [CrossRef] [Green Version]
- Nimmrich, V.; Eckert, A. Calcium channel blockers and dementia. Br. J. Pharmacol. 2013, 169, 1203–1210. [Google Scholar] [CrossRef] [Green Version]
- Saavedra, J.M. Angiotensin II AT(1) receptor blockers as treatments for inflammatory brain disorders. Clin. Sci. (Lond.) 2012, 123, 567–590. [Google Scholar] [CrossRef] [Green Version]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sanchez-Lopez, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesus, G.; Beas-Zarate, C.; et al. Memantine for the treatment of dementia: A review on its current and future Applications. J. Alzheimers Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.Z.; Yang, D.D.; Zhao, Z.; Yan, H.; Ji, J.; Sun, X.L. Memantine mediates neuroprotection via regulating neurovascular unit in a mouse model of focal cerebral ischemia. Life Sci. 2016, 150, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Mathiisen, T.M.; Ottersen, O.P. Aquaporin-4 in the central nervous system: Cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 2004, 129, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rosell, A.; Lo, E.H. Targeting extracellular matrix proteolysis for hemorrhagic complications of tPA stroke therapy. CNS Neurol. Disord. Drug Targets 2008, 7, 235–242. [Google Scholar] [CrossRef]
- Blamire, A.M.; Anthony, D.C.; Rajagopalan, B.; Sibson, N.R.; Perry, V.H.; Styles, P. Interleukin-1beta -induced changes in blood-brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: A magnetic resonance study. J. Neurosci. 2000, 20, 8153–8159. [Google Scholar] [CrossRef] [Green Version]
- Kary, S.; Burmester, G.R. Anakinra: The first interleukin-1 inhibitor in the treatment of rheumatoid arthritis. Int. J. Clin. Pract. 2003, 57, 231–234. [Google Scholar]
- Hue, C.D.; Cho, F.S.; Cao, S.; Dale Bass, C.R.; Meaney, D.F.; Morrison, B., 3rd. Dexamethasone potentiates in vitro blood-brain barrier recovery after primary blast injury by glucocorticoid receptor-mediated upregulation of ZO-1 tight junction protein. J. Cereb. Blood Flow Metab. 2015, 35, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B.; Kappos, L. Natalizumab: Targeting alpha4-integrins in multiple sclerosis. Neurodegener. Dis. 2008, 5, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [Green Version]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Hui, E.K.; Lu, J.Z.; Zhou, Q.H.; Pardridge, W.M. Selective targeting of a TNFR decoy receptor pharmaceutical to the primate brain as a receptor-specific IgG fusion protein. J. Biotechnol. 2010, 146, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Koo, Y.E.; Reddy, G.R.; Bhojani, M.; Schneider, R.; Philbert, M.A.; Rehemtulla, A.; Ross, B.D.; Kopelman, R. Brain cancer diagnosis and therapy with nanoplatforms. Adv. Drug Deliv. Rev. 2006, 58, 1556–1577. [Google Scholar] [CrossRef]
- Dadparvar, M.; Wagner, S.; Wien, S.; Kufleitner, J.; Worek, F.; von Briesen, H.; Kreuter, J. HI 6 human serum albumin nanoparticles--development and transport over an in vitro blood-brain barrier model. Toxicol. Lett. 2011, 206, 60–66. [Google Scholar] [CrossRef]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Pereira, M.C. Interaction of natural compounds with biomembrane models: A biophysical approach for the Alzheimer’s disease therapy. Coll. Surf. B Biointerfaces 2019, 180, 83–92. [Google Scholar] [CrossRef]
- Surnar, B.; Basu, U.; Banik, B.; Ahmad, A.; Marples, B.; Kolishetti, N.; Dhar, S. Nanotechnology-mediated crossing of two impermeable membranes to modulate the stars of the neurovascular unit for neuroprotection. Proc. Natl. Acad. Sci. USA 2018, 115, E12333–E12342. [Google Scholar] [CrossRef]
- Liu, X.; Ye, M.; An, C.; Pan, L.; Ji, L. The effect of cationic albumin-conjugated PEGylated tanshinone IIA nanoparticles on neuronal signal pathways and neuroprotection in cerebral ischemia. Biomaterials 2013, 34, 6893–6905. [Google Scholar] [CrossRef]
- Liu, X.; An, C.; Jin, P.; Liu, X.; Wang, L. Protective effects of cationic bovine serum albumin-conjugated PEGylated tanshinone IIA nanoparticles on cerebral ischemia. Biomaterials 2013, 34, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Aktas, Y.; Gursoy-Ozdemir, Y.; Bodur, E.; Yemisci, M.; Caban, S.; Vural, A.; Pinarbasli, O.; Capan, Y.; Fernandez-Megia, E.; et al. A nanomedicine transports a peptide caspase-3 inhibitor across the blood-brain barrier and provides neuroprotection. J. Neurosci. 2009, 29, 13761–13769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, C.; Taylor, M.; Fullwood, N.; Allsop, D. Liposome delivery systems for the treatment of Alzheimer’s disease. Int. J. Nanomed. 2018, 13, 8507–8522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangilder, R.L.; Rosen, C.L.; Barr, T.L.; Huber, J.D. Targeting the neurovascular unit for treatment of neurological disorders. Pharmacol. Ther. 2011, 130, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Singh-Moon, R.P.; Ellis, J.A.; Chaudhuri, D.B.; Wang, M.; Reif, R.; Bruce, J.N.; Bigio, I.J.; Straubinger, R.M. Cerebral hypoperfusion-assisted intra-arterial deposition of liposomes in normal and glioma-bearing rats. Neurosurgery 2015, 76, 92–100. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Aβ Receptor and Ligand | NVU Cells | Pathological Effects | Ref. |
|---|---|---|---|
| CR1 (CD35) fAβ | Microglia, astrocytes, neurons | fAβ/CR1 interaction results in C3b/C4b activation. Aβ clearance from the brain via blood cell expresses CR1 in its surface and later metabolism in the liver and/or spleen. | [70,71] |
| CR3 (Mac-1) fAβ | Microglia | Interaction leads to an increased PI3K/p47PHOX activity (neurotoxicity by superoxide) or NF-kB (inflammatory factors production). | [72,73] |
| TLR4/6 fAβ | Microglia, astrocytes | CD36/TLR4/6 complex mediates Aβ internalization, followed by ROS and proinflammatory production and phagocytosis. | [74,75] |
| C5aR (CD88) fAβ, oAβ | C5a/C5aR binding in response to fAβ/oAβ induces TNFα production. | [76,77] | |
| SRA 1/2 fAβ, oAβ | Aβ/SRA interaction results in NF-kB activation and consequently, the secretion of ROS, TNF-a, complement components, among other pro-inflammatory substances. | [78,79] | |
| SRB2 (CD36) fAβ, oAβ | Microglia, BECs, neurons | CD36/a3b1-integrin/CD47 complex regulates fAβ interaction in microglia cells and triggers ROS production, pro-inflammatory cytokines release, and phagocytosis. | [79,80] |
| RAGE mAβ, fAβ | Aβ/RAGE/p38 and ERK1/2 signaling pathways trigger oxidative stress, NF-kB activation, proinflammatory molecules production, triggering NVU damage. | [81,82] | |
| a7nAChR mAβ, fAβ | Neurons, SMCs, astrocytes | a7nAChR may mediate Aβ internalization. Aβ could activate the JNK/ERK2/MAPK pathway, which results in cell death by apoptosis. | [83,84] |
| IR mAβ, oAβ | neurons | Aβ/IR binding triggers impaired insulin signaling, which could cause neuronal dysfunction and memory deficits. | [85,86] |
| SEC-R mAβ | Neurons, glia | Interaction mediates endocytosis and degradation of Aβ by recognizing its 25–35 region. | [87,88] |
| TREM2 oAβ | Microglia, neurons | Decreased TREM2 leads to Aβ accumulation. TREM2/Aβ linking could trigger neuronal phagocytosis or apoptosis. | [89,90] |
| LRP1 mAβ | Pericytes, astrocyte, microglia, neurons | LRP1 is widely expressed in NVU cells and mediates the Aβ transport across the BBB. LRP1 controls the Aβ uptake and its subsequent trafficking to the lysosome for degradation. | [91,92] |
| ABCA1 * Aβ | BECs, pericytes | ABCA1/ApoE/LRP1 complex contributes to brain Aβ transport/clearance. Abca1 gene deficiency promotes Aβ accumulation in an AD mice model. | [93] |
| ABCB1 * Aβ | ABCB1/LRP1 transports the Aβ peptides across the BBB. ABCB1 is considered a marker for BBB maturity and functionality. | [94] |
| Drug/CT Identifier | Clinical Phase | Effects on the NVU | |
|---|---|---|---|
| (a) BACE 1 inhibitors | |||
| E2609/NCT01294540 ¥ | I | Effect on the neuron and BEC. They reduce extracellular Aβ accumulation. Inhibition of the endothelial BACE1 activity may reduce Aβ release into the SMCs and protect vascular wall integrity [117]. | [118] |
| Verubecestat/NCT01739348 ¥ | III | [119] | |
| Umibecestat/NCT01097096 ¥ | III | [120] | |
| LY2886721/NCT01561430 ¥ | III | [121] | |
| (b) Gamma-secretase inhibitors and modulators | |||
| Semagacestat/NCT00594568 ¥ | III | Effect on NVU cells. They block Aβ-peptide secretion, precursor agent of NPs, and neuronal dysfunction. According to an animal AD model, some of these drugs can interact with astrocytes and microglia, preventing the Aβ secretion and decreasing the total brain amyloid load [122]. | [123] |
| Avagacestat/NCT00810147 ¥ | II | [124] | |
| Pintol/NCT00470418 * | II | [121] | |
| (c) Alpha-secretase enhancers | |||
| Epigallocatechin gallate/NCT00951834, NCT03978052 Φ | II/III | Effect on NVU cells. Some of these drugs have been shown to improve NVU in animal models, reducing vascular Aβ deposits, neuroinflammation, oxidative stress, synaptotoxicity, and favoring angiogenesis and antioxidant and anti-inflammatory effects [125,126]. | [121] |
| Bryostatin 1/NCT04538066 Φ | II | [127] | |
| Etazolate/NCT00880412 * | II | [128] | |
| (d) β-Amyloid aggregation inhibitors | |||
| Scyllo-inositol (ELND005) ¥ | II | Effect on NVU cells. The Aβ polymerization process is a key event involved in AD. Some of these drugs could inhibit Aβ self-association [129]. Therefore, they could inhibit vascular insoluble amyloid deposits and improve the functionality of the NVU in AD. | [130] |
| Clioquinol (PBT1) & | III | [121] | |
| Colostrinin * | III | [121] | |
| (e) Modulators of beta-amyloid peptide transport-RAGE inhibitors | |||
| Azeliragon (PF-04494700)/NCT03980730 Φ | II | Effect on microglia, BEC, and neurons. They regulate the Aβ clearance and improve the BBB functionality, neuroinflammatory environment, and CBF. | [131] |
| TTP4000/NCT01548430 * | I | [132] | |
| (f) Tau aggregation inhibitors | |||
| Rember/NCT00515333 * | II | Effect on the neuron. Effects on other NVU cells unknown. They prevent oligomeric tau aggregation and NFT formation and disrupt aggregated tau. | [133] |
| LMTM/NCT01689246, NCT03539380, NCT03446001 Φ | III | [133] | |
| (g) Kinases inhibitors | |||
| Tideglusib (GSK3β inhibitor)/NCT00948259, NCT01350362 * | III | Effect on the neuron. No effect on other NVU cells. Inhibition of kinases leads to tau phosphorylation, loss of affinity for microtubules, NFT formation, and neuronal death. | [133] |
| Saracatinib (Fyn inhibitor)/NCT01864655, NCT02167256 * | II | [133] | |
| Nilotinib (tyrosine kinase inhibitor)/NCT02947893Φ | II | [134] | |
| (h) Microtubule Stabilizers | |||
| Epothilone D/NCT01492374 ¥ | I | Effect on the neuron. No impact on other NVU cells. They stabilize microtubules and reduce tau pathology and hippocampal neuronal loss. | [133] |
| Abetotexate/NCT01966666 * | I | [133] | |
| (i) Active immunotherapy anti-tau protein | |||
| AADvac-1/NCT01850238 * | I | Effect on the neuron. No effect on other NVU cells. It activates T cells and triggers an immune response to eliminate abnormally phosphorylated tau. | [133] |
| (j) Active immunity (anti-Aβ peptide polyclonal antibody) | |||
| Vanutide cridificar/NCT00479557, NCT01227564 * | IIa | Effect on the neuron. It induces antibodies against Aβ, preventing Aβ deposition, promoting plaque clearance, and improving cognitive functions. | [135] |
| (k) Passive immunotherapy (anti-tau antibody) | |||
| R07105705 (RG6100)/NCT02820896, NCT0328914 Φ | II | Effect on the neuron. No effect on other NVU cells. It recognizes abnormal tau and blocks its transmission from one neuron to another. | [133] |
| (l) Passive immunity (anti-Aβ monoclonal antibodies) | |||
| Aducanumab (IgG1)/NCT04241068 Φ | III | Effect on the neuron and possibly on other cells of the NVU. Most of these drugs are directed against epitopes of aggregated forms of Aβ, including soluble monomers, oligomers, and insoluble fibrils. Besides, they favor the central Aβ clearance (except Ponezumab that promotes peripheral release), having a favorable effect on neurons and the other NVU cells. Donanemab, unlike the others, has an affinity for the Aβp3-42 conformation, which is more toxic than Aβ1-40 or 1–42 [136,137]. Furthermore, IgG1 or IgG2 carries a high risk of Fc γ receptor (FcγRs)-mediated overactivation of microglial cells, due to the binding with C1q that can contribute to an inappropriate pro-inflammatory response leading to vasogenic edema and cerebral microbleeds, an effect that does not occur with IgG4. In general, both active and passive immunotherapy tend to have harmful effects on the NVU, increasing the CAA and causing microhemorrhages [138]. | [139] |
| Gantenerumab (IgG1)/NCT01760005 Φ | III | [121] | |
| Donanemab (IgG1)/NCT03367403Φ | II | [140] | |
| Solanezumab (IgG)/NCT00905372, NCT01760005 Φ | III | [141] | |
| Crenezumab (IgG4)/NCT03977584, NCT01998841 Φ | II | [142] | |
| Sar228810 (IgG4)/NCT01485302 * | I | [143] | |
| GSK933776A (IgG1)/NCT00459550 * | I | [144] | |
| Ponezumab (IgG2)/ NCT00722046, NCT00945672 * | II | [145] | |
| BAN-2401 (IgG1)/NLT01767311 NCT03887455, NCT04468659 Φ | IIb | [146] | |
| (m) Passive immunity (Intravenous immunoglobulin G) | |||
| Octagam IVIgG/NCT01300728 Φ | III | Effects on the NVU cells. They promote central Aβ clearance, block the RAGE receptor, increases sLRP levels, anti-inflammatory effects, and selectively target aggregated Aβ forms (monomers and oligomers). | [147] |
| Gammagard IVIgG/NCT00818662 * | III | [147] | |
| (n) Nonsteroidal anti-inflammatory drugs (NSAIDs) | |||
| r-Flurbiprofen ζ | III | Effects on the NVU cells. γ-secretase inhibitor acts restoring neurogenesis, reorganizing the astrocytic cytoskeleton, reducing pathological tau, rescuing synaptic plasticity, acting on microglia to counteract neuroinflammation | [148] |
| Itanapraced/NCT01303744 ζ | II | [121,149] | |
| Drug/CT Identifier | Clinical Phase | Effects on the NVU | Ref |
|---|---|---|---|
| (a) Antihypertensive drugs | |||
| Captopril (ACE inhibitor) | Pcp | Effects on the NVU cells. It inhibits Aβ production caused by ACE, regulates pro-inflammatory molecules, and inhibits ROS. | [188] |
| Losartán (ARBs)/NCT02913664 Φ | II | Effects on the NVU cells. It inhibits ROS production and reduces cerebrovascular/neuropathological changes. | [189] |
| Amlodipine/NCT02913664 Φ Nilvadipine/NCT02017340 * (Calcium-channel blockers). | II III | Effects on the NVU cells. They attenuate the neuronal deterioration induced by Aβ, decrease cerebral hypoperfusion due to vasodilator effects. | [190] |
| (b) Antidiabetic drugs | |||
| Exenatide/NCT01255163 ζ Liraglutide/NCT01843075 Φ (GLP-1 agonists) | II | Neuron and microglia effects. They promote neuronal survival, synaptogenesis, neurogenesis, anti-inflammation, and protecting against oxidative injury. In AD mouse models reduce Aβ oligomers and plaque load, and microglial activation, improving memory. Also, elicit vasculoprotective effects (protect against hypoxia injury). | [191] |
| Pioglitazone/NCT01456117 † NCT00982202 (γ-PPAR) † Rosiglitazone/NCT00550420 † NCT00265148 (γ-PPAR) * | III | Effects on the NVU cells. They reduce Aβ levels (enhanced phagocytosis of Aβ deposits), oxidative stress, mitochondrial dysfunction, and neuroinflammation induced by glial cells. They improve cerebral blood flow, and lead to cognitive improvement. | [192] |
| [193] | |||
| Intranasal insulin/NCT00438568 NCT01547169, NCT01436045 * | II | Neuron effects. They modulate the Aβ levels, protect against synapses damage by Aβ oligomers, and modulate memory consolidation. | [194] |
| Simvastatine (HMG-CoA)/NCT00053599, NCT00939822, NCT00486044, NCT00303277 * | II | Effects on the NVU cells. They have neuroprotective and pleiotropic impact, improve the vascular system, eNOS activation, and antioxidant effects. | [195,196] |
| (c) Novel drugs | |||
| Natalizumab (antibodies against the α4β1 integrin receptor) | Pcp | Effects on the NVU cells. It acts modulating the peripheral immune system infiltrate into the brain and decrease the proinflammatory environment. | [197] |
| Anakinra (IL-1 receptor antagonist) | Pcp | Probable effects on microglia cells. It blocks the interaction between IL-1β with its receptor (IL-1R1), then could decrease neuroinflammation in AD patients. | [198] |
| GDNF/lentiviral vector | Pcp | It has neuroprotector effects against Aβ, protecting neurons and astrocytes. | [199,200] |
| VEGF | Pcp | Effects on the NVU cells. It modulates angiogenesis, vascular permeability, vascular remodeling, vascular survival, neurotrophic activity, and anti-inflammation. | [201,202] |
| NGF and NGF mimetics/NCT03069014 *(Intracerebral infusion, nasal/intraocular administration) | II | Effects on the neuron and microglia. They regulate differentiation, growth, survival, and plasticity of cholinergic neurons. They have a direct role in modulating microglial cells toward a non-inflammatory phenotype. | [203] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soto-Rojas, L.O.; Pacheco-Herrero, M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Villegas-Rojas, M.M.; Harrington, C.R.; de la Cruz, F.; Garcés-Ramírez, L.; Luna-Muñoz, J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2022. https://doi.org/10.3390/ijms22042022
Soto-Rojas LO, Pacheco-Herrero M, Martínez-Gómez PA, Campa-Córdoba BB, Apátiga-Pérez R, Villegas-Rojas MM, Harrington CR, de la Cruz F, Garcés-Ramírez L, Luna-Muñoz J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(4):2022. https://doi.org/10.3390/ijms22042022
Chicago/Turabian StyleSoto-Rojas, Luis O., Mar Pacheco-Herrero, Paola A. Martínez-Gómez, B. Berenice Campa-Córdoba, Ricardo Apátiga-Pérez, Marcos M. Villegas-Rojas, Charles R. Harrington, Fidel de la Cruz, Linda Garcés-Ramírez, and José Luna-Muñoz. 2021. "The Neurovascular Unit Dysfunction in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 4: 2022. https://doi.org/10.3390/ijms22042022
APA StyleSoto-Rojas, L. O., Pacheco-Herrero, M., Martínez-Gómez, P. A., Campa-Córdoba, B. B., Apátiga-Pérez, R., Villegas-Rojas, M. M., Harrington, C. R., de la Cruz, F., Garcés-Ramírez, L., & Luna-Muñoz, J. (2021). The Neurovascular Unit Dysfunction in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(4), 2022. https://doi.org/10.3390/ijms22042022