
Plasma Amyloid-Beta Levels in a Pre-Symptomatic Dutch-Type Hereditary Cerebral Amyloid Angiopathy Pedigree: A Cross-Sectional and Longitudinal Investigation

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Plasma Aβ Measurements and APOE ε4 Genotype Status
2.3. Neuropsychological Assessments
2.4. Assessment of CSF Aβ Concentration and PET Aβ Load
2.5. Statistical Analysis
3. Results
3.1. Participant Characteristics
3.2. Cross-Sectional Differences in CSF Aβ and Brain Aβ between D-CAA Mutation Non-Carriers and Carriers
3.3. Cross-Sectional Differences in Plasma Aβ between D-CAA Mutation Non-Carriers and Carriers Employing the Ultrasensitive Simoa Technology
3.4. Cross-Sectional Comparison of Plasma Aβ between D-CAA Mutation Non-Carriers and Carriers Employing xMAP Technology
3.5. Longitudinal Changes in Plasma Aβ Concentrations Employing the Ultrasensitive Simoa Technology
3.6. Longitudinal Changes in Plasma Aβ Concentrations Employing the xMAP Technology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef] [Green Version]
- van Asch, C.J.; Luitse, M.J.; Rinkel, G.J.; van der Tweel, I.; Algra, A.; Klijn, C.J. Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: A systematic review and meta-analysis. Lancet Neurol. 2010, 9, 167–176. [Google Scholar] [CrossRef]
- Jellinger, K.A. Alzheimer disease and cerebrovascular pathology: An update. J. Neural Transm. 2002, 109, 813–836. [Google Scholar] [CrossRef]
- Kumar-Singh, S. Hereditary and sporadic forms of abeta-cerebrovascular amyloidosis and relevant transgenic mouse models. Int. J. Mol. Sci. 2009, 10, 1872–1895. [Google Scholar] [CrossRef] [Green Version]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef]
- Linn, J.; Halpin, A.; Demaerel, P.; Ruhland, J.; Giese, A.D.; Dichgans, M.; van Buchem, M.A.; Bruckmann, H.; Greenberg, S.M. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010, 74, 1346–1350. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, K.A.; Rosand, J.; Karluk, D.; Greenberg, S.M. Clinical diagnosis of cerebral amyloid angiopathy: Validation of the Boston Criteria. Neurology 2001, 56, 537–539. [Google Scholar] [CrossRef]
- Bakker, E.; van Broeckhoven, C.; Haan, J.; Voorhoeve, E.; van Hul, W.; Levy, E.; Lieberburg, I.; Carman, M.D.; Van Ommen, G.J.B.; Frangione, B.; et al. DNA diagnosis for hereditary cerebral hemorrhage with amyloidosis (Dutch type). Am. J. Hum. Genet. 1991, 49, 518–521. [Google Scholar]
- Maat-Schieman, M.L.; Duinen, S.G.; Bornebroek, M.; Haan, J.; Roos, R.A. Hereditary Cerebral Hemorrhage with Amyloidosis-Dutch type (HCHWA-D): II-A Review of Histopathological Aspects. Brain Pathol. 1996, 6, 115–120. [Google Scholar] [CrossRef]
- Bornebroek, M.; Haan, J.; Maat-Schieman, M.L.; van Duinen, S.G.; Roos, R.A. Hereditary Cerebral Hemorrhage with Amyloidosis-Dutch Type (HCHWA-D): I-A Review of Clinical, Radiologic and Genetic Aspects. Brain Pathol. 1996, 6, 111–114. [Google Scholar] [CrossRef]
- Verbeek, M.M.; Kremer, B.P.H.; Rikkert, M.O.; van Domburg, P.H.M.F.; Skehan, M.E.; Greenberg, S.M. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann. Neurol. 2009, 66, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Etten, E.S.; Verbeek, M.M.; van Der Grond, J.; Zielman, R.; van Rooden, S.; van Zwet, E.W.; van Opstal, A.M.; Haan, J.; Greenberg, S.M.; van Buchem, M.A.; et al. β-Amyloid in CSF. Neurology 2017, 88, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, A.P.; Kloet, R.W.; Sohrabi, H.R.; van der Weerd, L.; van Rooden, S.; Wermer, M.J.H.; Moursel, L.G.; Yaqub, M.; Van Berckel, B.N.M.; Chatterjee, P.; et al. Amyloid imaging of dutch-type hereditary cerebral amyloid angiopathy carriers. Ann. Neurol. 2019, 86, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Fagan, A.M.; Xiong, C.; McKay, M.; Bhatnagar, A.; Wu, Y.; Singh, A.K.; Taddei, K.; Martins, I.; Gardener, S.L.; et al. Presymptomatic Dutch-Type Hereditary Cerebral Amyloid Angiopathy-Related Blood Metabolite Alterations. J. Alzheimer’s Dis. 2021, 79, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Cho, H.-S.; O’Donnell, H.C.; Rosand, J.; Segal, A.Z.; Younkin, L.H.; Younkin, S.G.; Rebeck, G.W. Plasma beta-Amyloid Peptide, Transforming Growth Factor-beta1, and Risk for Cerebral Amyloid Angiopathy. Ann. N. Y. Acad. Sci. 2000, 903, 144–149. [Google Scholar] [CrossRef]
- Bornebroek, M.; De Jonghe, C.; Haan, J.; Kumar-Singh, S.; Younkin, S.; Roos, R.; Van Broeckhoven, C. Hereditary cerebral hemorrhage with amyloidosis dutch type (AβPP 693): Decreased plasma amyloid-β 42 concentration. Neurobiol. Dis. 2003, 14, 619–623. [Google Scholar] [CrossRef]
- Hernández-Guillamon, M.; Delgado, P.; Penalba, A.; Rodriguez-Luna, D.; Molina, C.A.; Rovira, Á.; Álvarez-Sabín, J.; Boada, M.; Montaner, J. Plasma ß-Amyloid Levels in Cerebral Amyloid Angiopathy-Associated Hemorrhagic Stroke. Neurodegener. Dis. 2012, 10, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Pedrini, S.; Stoops, E.; Goozee, K.; Villemagne, V.L.; Asih, P.R.; Verberk, I.M.W.; Dave, P.; Taddei, K.; Sohrabi, H.R.; et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Transl. Psychiatry 2021, 11, 27. [Google Scholar] [CrossRef]
- Verberk, I.M.W.; Thijssen, E.; Koelewijn, J.; Mauroo, K.; Vanbrabant, J.; De Wilde, A.; Zwan, M.D.; Verfaillie, S.C.J.; Ossenkoppele, R.; Barkhof, F.; et al. Combination of plasma amyloid beta(1-42/1-40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimer’s Res. Ther. 2020, 12, 118. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Morris, J.C.; Ernesto, C.; Schafer, K.; Coats, M.; Leon, S.; Sano, M.; Thal, L.J.; Woodbury, P. Clinical Dementia Rating training and reliability in multicenter studies: The Alzheimer’s Disease Cooperative Study experience. Neurology 1997, 48, 1508–1510. [Google Scholar] [CrossRef]
- Su, Y.; Blazey, T.M.; Snyder, A.Z.; Raichle, M.E.; Marcus, D.S.; Ances, B.M.; Bateman, R.J.; Cairns, N.J.; Aldea, P.; Cash, L.; et al. Partial volume correction in quantitative amyloid imaging. NeuroImage 2015, 107, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, N.-F.; Chao, S.-P.; Huang, L.-K.; Chan, L.; Chen, Y.-R.; Chiou, H.-Y.; Hu, C.-J. Plasma Amyloid Beta and Tau Levels Are Predictors of Post-stroke Cognitive Impairment: A Longitudinal Study. Front. Neurol. 2019, 10, 715. [Google Scholar] [CrossRef] [PubMed]
- Monro, O.R. Substitution at codon 22 reduces clearance of Alzheimer’s amyloid-β peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol. Aging 2002, 23, 405–412. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. LRP1 and cerebral amyloid angiopathy. Futur. Neurol. 2009, 4, 55–65. [Google Scholar] [CrossRef]
- Yan, S.S.; Chen, D.; Yan, S.; Guo, L.; Du, H.; Chen, J.X. RAGE is a key cellular target for Abeta-induced perturbation in Alzheimer’s disease. Front. Biosci. 2012, 4, 240–250. [Google Scholar] [CrossRef]
- Maat-Schieman, M.L.C.; Van Duinen, S.G.; Haan, J.; Roos, R.A.C. Morphology of cerebral plaque-like lesions in hereditary cerebral hemorrhage with amyloidosis (Dutch). Acta Neuropathol. 1992, 84, 674–679. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| D-CAA NC | D-CAA MC | p | pa | |
|---|---|---|---|---|
| T1 | ||||
| N | 8 | 9 | - | - |
| Sex (M/F) | 3/5 | 3/6 | 1.00 | - |
| Age (Mean ± SD) | 43.5 ± 6.57 | 44.11 ± 4.31 | 0.822 | - |
| EYO (years; Mean ± SD) | −6.09 ± 6.43 | −5.27 ± 4.30 | 0.759 | - |
| APOE ε4 (%) | 37.5 | 22.2 | 0.620 | - |
| MMSE (Mean ± SD) | 28.63 ± 1.60 | 28.67 ± 1.41 | 0.955 | - |
| CDR global = 0 (%) | 100 | 100 | - | - |
| Stroke (%) | 0 | 0 | - | - |
| CSF Aβ1-40 (pg/mL; Mean ± SD) # | 9882.92 ± 3076.97 | 6971.35 ± 1088.65 | 0.051 | 0.039 |
| CSF Aβ1-42 (pg/mL; Mean ± SD) # | 618.63 ± 206.81 | 311.67 ± 154.85 | 0.012 | 0.007 |
| Brain Aβ load (SUVR, Mean ± SD) | 0.994 ± 0.064 | 1.268 ± 0.248 | 0.009 | 0.011 |
| T2 | ||||
| N | 7 | 8 | - | - |
| Sex (M/F) | 2/5 | 2/6 | 1.00 | - |
| Age (Mean ± SD) | 46.57 ± 7.21 | 46.25 ± 4.65 | 0.830 | - |
| EYO (years; Mean ± SD) | −2.97 ± 7.02 | −2.12 ± 4.75 | 0.785 | - |
| APOE ε4 (%) | 42.86 | 25 | 0.608 | - |
| MMSE (Mean ± SD) | 28.71 ± 1.38 | 28.63 ± 0.92 | 0.883 | - |
| CDR global = 0 (%) | 100 | 100 | - | - |
| Stroke (%) | 0 | 0 | - | - |
| CSF Aβ1-40 (pg/mL; Mean ± SD) # | 7483.03 ± 1454.50 | 3933.53 ± 1993.14 | - | - |
| CSF Aβ1-42 (pg/mL; Mean ± SD) # | 1078.44 ± 162.86 | 346.61 ± 147.11 | - | - |
| Brain Aβ load (SUVR, Mean ± SD) | 1.06 ± 0.034 | 1.35 ± 0.25 | 0.009 | 0.001 |
| D-CAA NC | D-CAA MC | p | pa | |
|---|---|---|---|---|
| T1 | n= 8 | n= 9 | ||
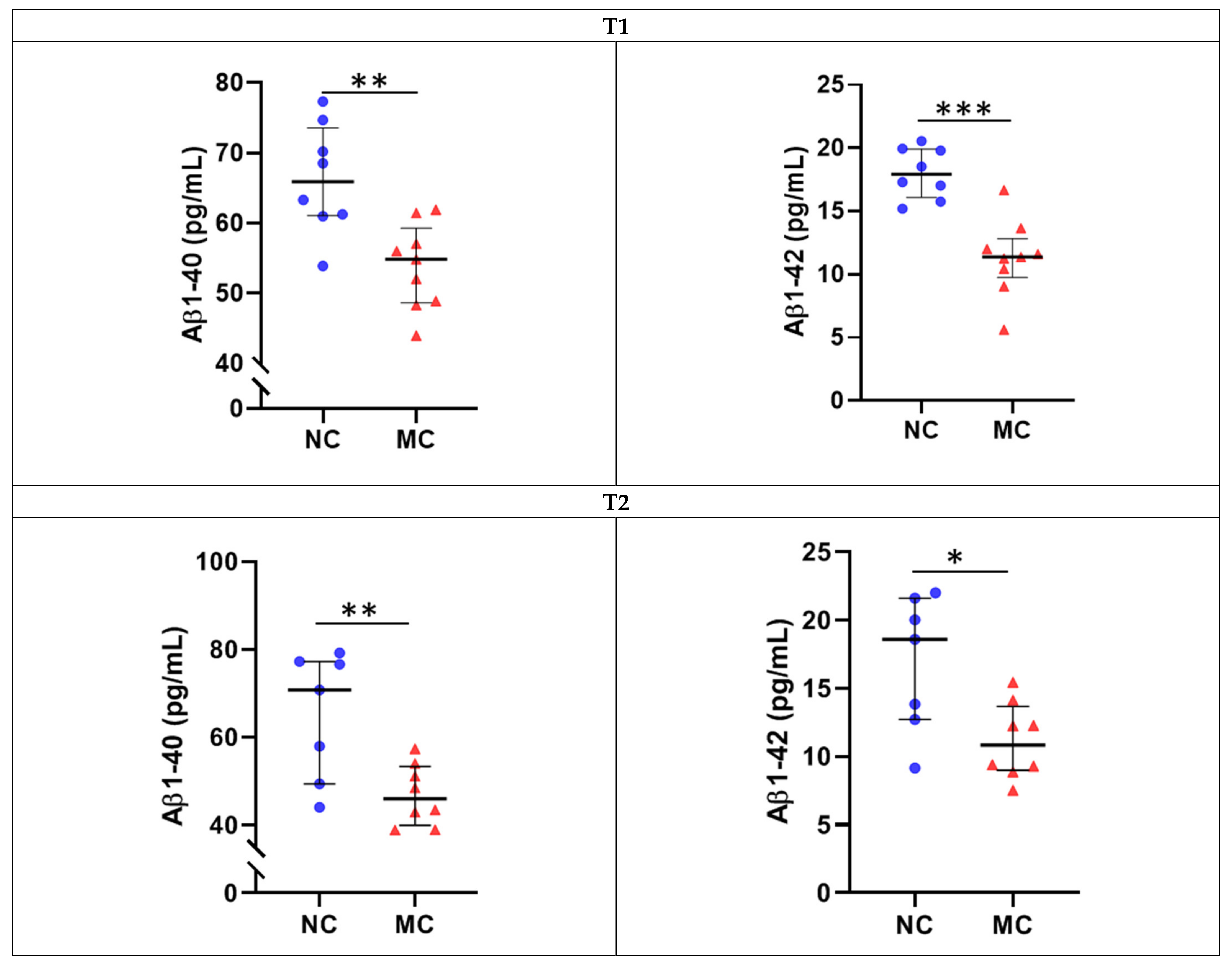
| Aβ1-40 | 66.30 ± 7.83 | 53.87 ± 6.07 | 0.002 | 0.001 |
| Aβ1-42 | 18.02 ± 2.01 | 11.31 ± 3.02 | 0.00009 | 0.0004 |
| T2 | n= 7 | n= 8 | ||
| Aβ1-40 | 65.04 ± 14.45 | 46.92 ± 6.93 | 0.008 ⱡ | 0.001 ⱡ |
| Aβ1-42 | 16.85 ± 4.96 | 11.16 ± 2.79 | 0.020 ⱡ | 0.016 |
| D-CAA NC (n = 7) | D-CAA MC (n = 8) | Time | Time (Adjusted for Covariates) | Time * Mutation | Time * Mutation (Adjusted for Covariates) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T1 | T2 | T1 | T2 | p | Pairwise | p | Pairwise | p | p | |||
| pNC | pMC | pNC | pMC | |||||||||
| Aβ1-40 | 67.01 ± 8.17 | 65.04 ± 14.45 | 52.86 ± 5.62 | 46.92 ± 6.93 | 0.057 ⱡ | 0.449 ⱡ | 0.045ⱡ | 0.028ⱡ | 0.227 ⱡ | 0.041ⱡ | 0.362 ⱡ | 0.526 ⱡ |
| Aβ1-42 | 18.12 ± 2.14 | 16.85 ± 4.96 | 10.63 ± 2.41 | 11.16 ± 2.79 | 0.665 ⱡ | 0.290 ⱡ | 0.605 ⱡ | 0.584 | 0.159 | 0.436 | 0.264 ⱡ | 0.130 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, P.; Tegg, M.; Pedrini, S.; Fagan, A.M.; Xiong, C.; Singh, A.K.; Taddei, K.; Gardener, S.; Masters, C.L.; Schofield, P.R.; et al. Plasma Amyloid-Beta Levels in a Pre-Symptomatic Dutch-Type Hereditary Cerebral Amyloid Angiopathy Pedigree: A Cross-Sectional and Longitudinal Investigation. Int. J. Mol. Sci. 2021, 22, 2931. https://doi.org/10.3390/ijms22062931
Chatterjee P, Tegg M, Pedrini S, Fagan AM, Xiong C, Singh AK, Taddei K, Gardener S, Masters CL, Schofield PR, et al. Plasma Amyloid-Beta Levels in a Pre-Symptomatic Dutch-Type Hereditary Cerebral Amyloid Angiopathy Pedigree: A Cross-Sectional and Longitudinal Investigation. International Journal of Molecular Sciences. 2021; 22(6):2931. https://doi.org/10.3390/ijms22062931
Chicago/Turabian StyleChatterjee, Pratishtha, Michelle Tegg, Steve Pedrini, Anne M. Fagan, Chengjie Xiong, Abhay K. Singh, Kevin Taddei, Samantha Gardener, Colin L. Masters, Peter R. Schofield, and et al. 2021. "Plasma Amyloid-Beta Levels in a Pre-Symptomatic Dutch-Type Hereditary Cerebral Amyloid Angiopathy Pedigree: A Cross-Sectional and Longitudinal Investigation" International Journal of Molecular Sciences 22, no. 6: 2931. https://doi.org/10.3390/ijms22062931
APA StyleChatterjee, P., Tegg, M., Pedrini, S., Fagan, A. M., Xiong, C., Singh, A. K., Taddei, K., Gardener, S., Masters, C. L., Schofield, P. R., Multhaup, G., Benzinger, T. L. S., Morris, J. C., Bateman, R. J., Greenberg, S. M., van Buchem, M. A., Stoops, E., Vanderstichele, H., Teunissen, C. E., ... the Dominantly Inherited Alzheimer Network. (2021). Plasma Amyloid-Beta Levels in a Pre-Symptomatic Dutch-Type Hereditary Cerebral Amyloid Angiopathy Pedigree: A Cross-Sectional and Longitudinal Investigation. International Journal of Molecular Sciences, 22(6), 2931. https://doi.org/10.3390/ijms22062931