
Pre-Clinical Neuroprotective Evidences and Plausible Mechanisms of Sulforaphane in Alzheimer’s Disease


Abstract
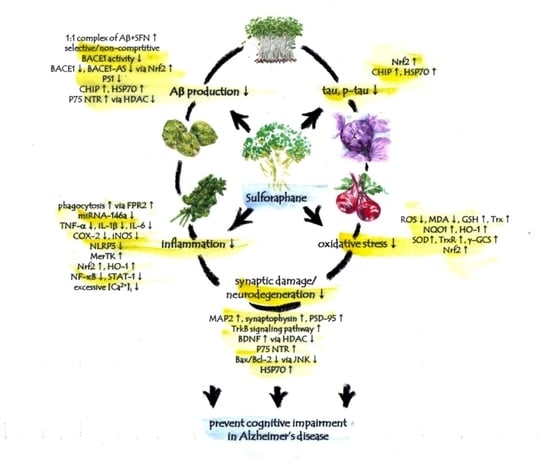
:
1. History of Sulforaphane Research in Brain Health
2. Evidence of Anti-AD Activity of Sulforaphane in Animals and Cells

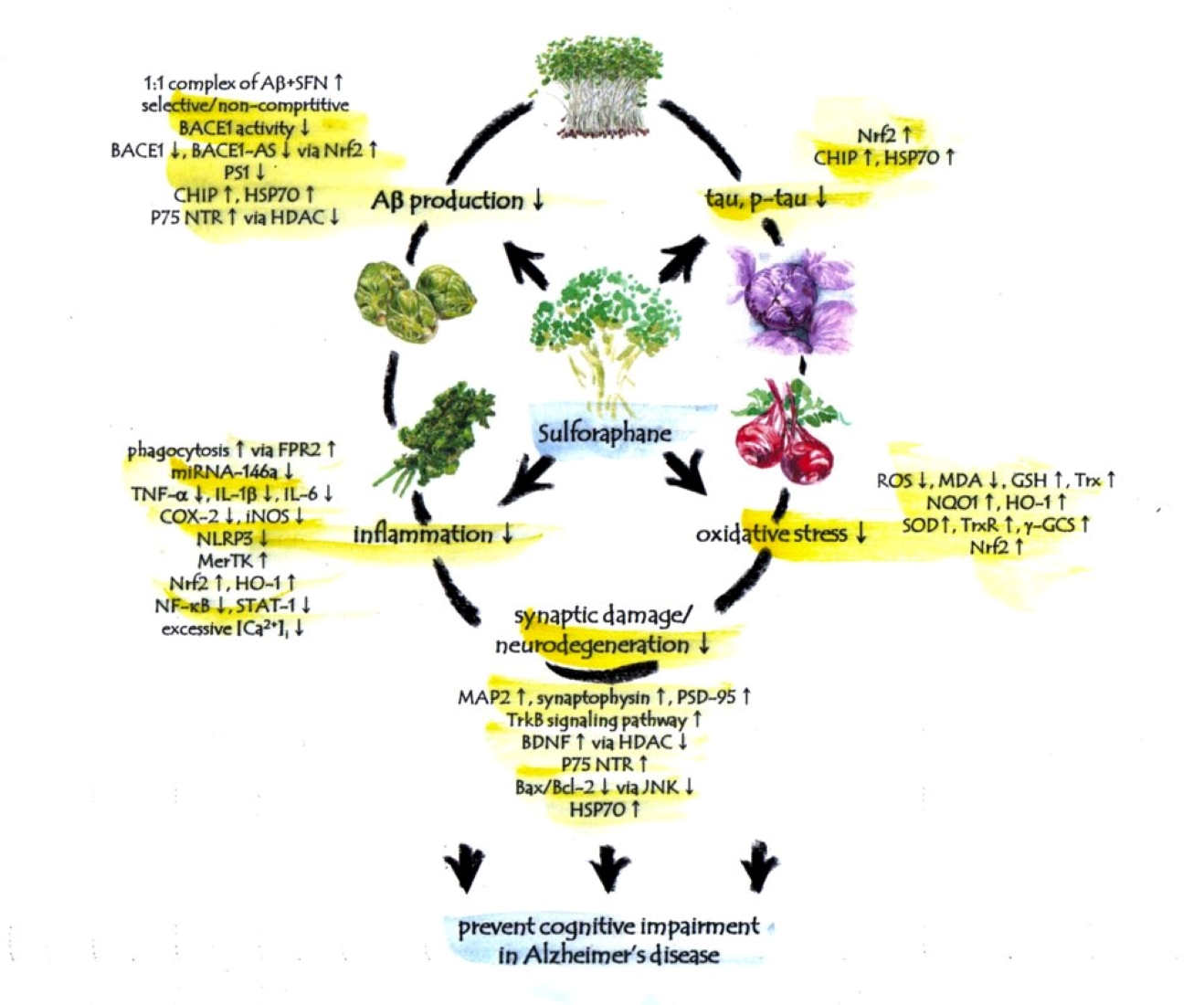{kind=link}
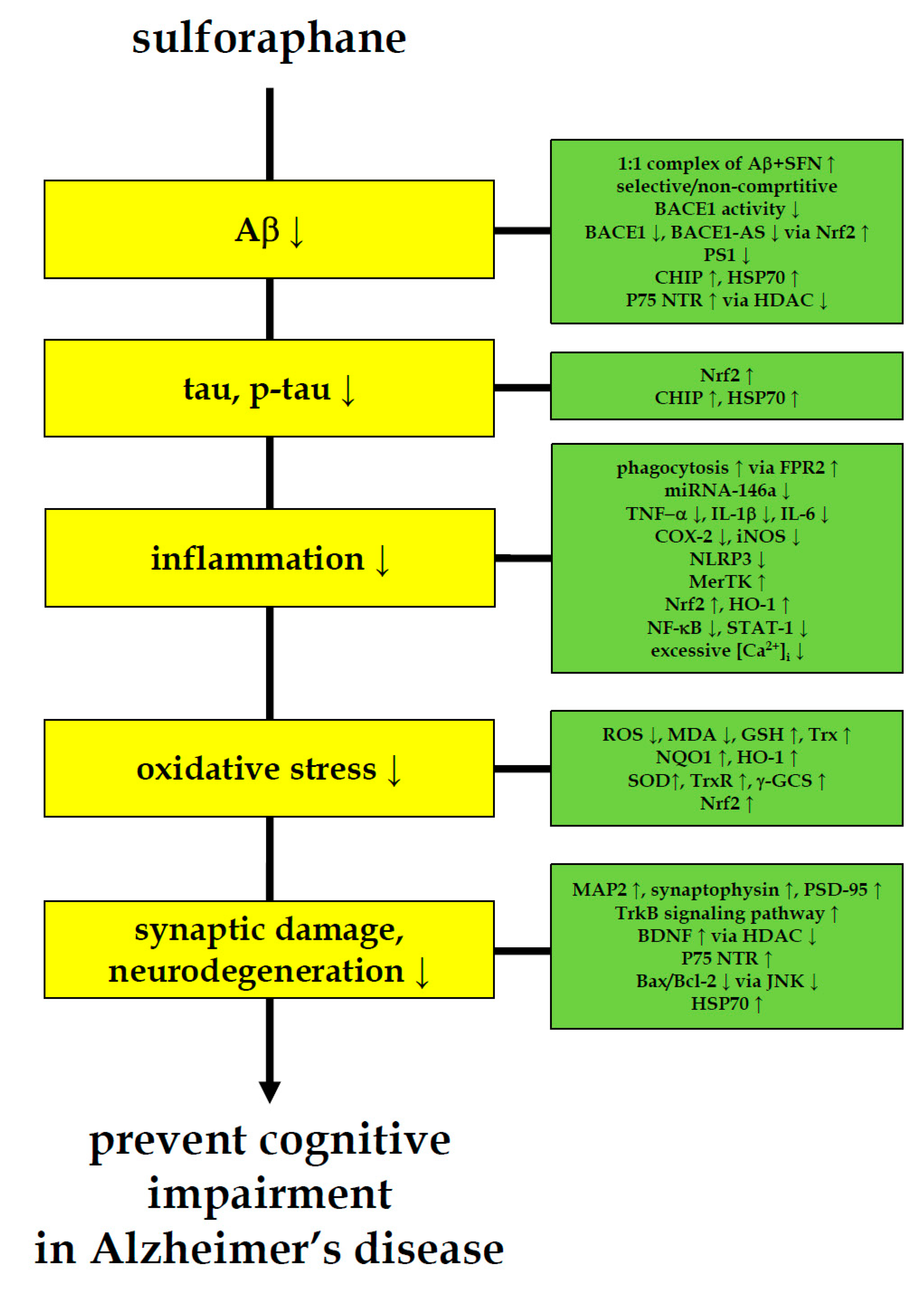{kind=link}
| Model | Description | Ref. |
|---|---|---|
| transgenic animal models carrying gene mutations associated with AD | ||
| 5×FAD mice | 5×FAD mice were made to harbor five-transgenes APPSwe, APPFlorida, APPLondon, PS1M146L, and PS1L286V. The resulting 5×FAD shows impaired memory and quickly constitute a major feature of AD amyloid pathology. 5×FAD has been proposed as a useful model for neurodegeneration and amyloid plaque formation induced by intraneuronal Aβ42. | [31,44] |
| 3×Tg-AD mice | 3×Tg-AD mice were designed to accommodate triple-transgenes APPSwe, PS1M146V, and tauP301L. 3×Tg-AD is the first model of the developed AD-like animal models to exhibit both plaque and tangle pathology. Consequently, 3×Tg-AD mice show synaptic damage and memory impairment. | [31,33,34,45] |
| APP/PS1 mice | APP/PS1 mice harbor double-transgenes APPSwe and PS1dE. APPSwe/PS1dE gene mutations are the Swedish-mutated APP gene combined with the exon-9-deleted PS1 gene. APP/PS1 mouse model exhibits amyloid plaque and memory impairment, recapitulating the onset and progression of early-onset familial AD. | [35,46] |
| PS1V97L mice | PS1V97L is a single-mutant transgenic mouse model harboring PS1V97L. It was generated by the report of a single missense mutation Val97Leu (V97L) of PS1 in a Chinese pedigree suffering from early-onset AD. Human Val97Leu mutant PS1 increases Aβ oligomers and tau phosphorylation level as well as AD-associated neuroinflammation and oxidative stress and finally causes spatial memory deficit in mice. | [32,47] |
| cell models carrying gene mutations associated with AD | ||
| primary cortical neurons derived from 3×Tg-AD mice | Mouse primary cortical cells derived from 3×Tg-AD mice stably express APPSwe, PS1M146V, and tauP301L and produce AD-associated high amount of Aβ, tau and p-tau. | [34] |
| mouse neuroblastoma N2a cells expressing APPswe | Mouse neuroblastoma Neuro2a cells stably expressing the human APPSwe produce AD-associated high amount of Aβ and exhibit neuroinflammation and oxidative stress. | [9] |
| AD-like animal models induced by Aβ | ||
| AD-like rat induced by Aβ42 | SD rat model administered Aβ42 by i.c.v. injection shows AD-associated neuroinflammation and oxidative stress in its brain. This model also exhibits impaired spatial learning. | [37] |
| AD-like mice induced by Aβ1–40 | This mouse model implemented the i.c.v. injection of Aβ1–40 shows impaired cognitive function. | [36] |
| AD-like cell models induced by Aβ | ||
| murine cortical neurons treated with Aβ42 | Rat primary cortical cells isolated from 18-day-old SD rat embryos exposed to Aβ42 show AD-associated tau hyperphosphorylation, damaged dendritic integrity and neuronal cell death. | [32] |
| human neuroblastoma SH-SY5Y cells treated with Aβ25–35 | Human neuroblastoma SH-SY5Y cells exposed to Aβ25–35 show AD-associated oxidative stress and neuronal cell death. | [40,43] |
| murine neuroblastoma N2a cells treated with Aβ1–42 | Mouse neuroblastoma Neuro2a cells exposed to Aβ1–42 show AD-associated neuronal cell death. | [41] |
| murine neuroblastoma N1E-115 cells treated with Aβ1–42 | Mouse neuroblastoma N1E-115 cells exposed to Aβ1–42 show AD-associated neuronal cell death. | [41] |
| human microglia-like THP-1 cells treated with Aβ1–42 | Differentiated human monocytic THP-1 cells, most closely resembling microglia, exposed to Aβ1–42 mimic AD-associated inflammatory microglial activation. | [38,39] |
| murine EOC-20 microglial cells treated with Aβ1–42 | Mouse EOC-20 microglial cells exposed to Aβ1–42 mimic AD-associated damaged microglial phagocytosis. | [42] |
2.1. Sulforaphane and Aβ
| Model | Sulforaphane Dose | Findings | Ref. |
|---|---|---|---|
| 5×FAD mice | every other day 10 mg/kg i.p. for 2 months | in cortex: (1) reduced the numbers of Aβ plaques/mm2 in cerebral cortex: (1) reduced Aβ1–40 and Aβ1–42 levels (2) reduced BACE1 protein expression (3) reduced BACE1 and BACE1-AS transcript (4) increased NQO1 transcript and protein expression (maybe through Nrf2 activation) in hippocampus: (1) reduced the numbers of Aβ plaques/mm2 | [31] |
| 3×Tg-AD mice | every other day 5 or 10 mg/kg i.p. for 2 months | in cortex: (1) reduced Aβ40 and Aβ42 in cerebral cortex: (1) reduced p-tau level (2) reduced BACE1 mRNA and protein expression (3) increased HO-1 mRNA and protein expression (maybe through Nrf2 activation) in hippocampus: (1) reduced p-tau pathology | [31] |
| daily 10 or 50 mg/kg p.o. 6 days/week for 2 months | in whole cortex and the fifth layer of the cortex: (1) decreased AβPP/Aβ level (2) decreased tau level (3) increased CHIP level in hippocampus: (1) decreased AβPP, polymeric Aβ, and monomeric Aβ (2) did not alter AβPP mRNA level (3) decreased tau and p-tau (4) did not alter tau mRNA level (5) increased CHIP and HSP70 | [34] | |
| APP/PS1 mice | daily 25 mg/kg p.o. for 5 months | in cerebral cortex: (1) protected against the increment of Aβ plaques (2) up-regulated p75 NTR (3) increased levels of Ace-H3K9 and Ace-H4K12 (4) reduced expression of HDAC1 and 3 suggested to contribute to up-regulation of p75 NTR | [35] |
| PS1V97L mice | daily 5 mg/kg i.p. for 4 months | in brain: (1) inhibited the generation of all types of Aβ oligomers (monomer, trimer, tetramer, hexamer, nonamer, and dodecamer) (2) alleviated tau hyperphosphorylation (3) decreased the expression levels of BACE1 and PS1 | [32] |
| primary cortical neurons derived from 3×Tg-AD mice | 10 μM for 6 h | (1) decreased Aβ (2) decreased tau (3) increased CHIP in the absence of CHIP expression: (1) failed to decrease Aβ (2) failed to decrease tau | [34] |
| mouse neuroblastoma N2a cells expressing APPswe | 1.25 or 2.5 μM for 48 h | in both cells and culture medium: (1) decreased levels of Aβ1–40 and Aβ1–42 | [9] |
| murine cortical neurons treated with Aβ42 | 0.01, 0.03 or 0.1 μM pre-treatment for 30 min followed by Aβ42 | (1) decreased hyperphosphorylation of tau | [32] |
| sulforaphane and Aβ | analyzed by mass spectrometry: (1) showed a 1:1 complex of [Aβ+sulforaphane] (2) formed three different [Aβ+sulforaphane] complexes due to covalent binding of sulforaphane to Aβ at three different sites (3) sulforaphane bound to free NH2 groups (N-terminal amino acid and lysines) in Aβ | [49] | |
| sulforaphane and BACE1 | analyzed by fluorescence resonance energy transfer: (1) sulforaphane shown selective and non-competitive BACE1 inhibitory activity | [48] |
2.2. Sulforaphane and Tau
2.3. Sulforaphane and AD-Associated Inflammatory Biomarkers
| Model | Sulforaphane Dose | Findings | Ref. |
|---|---|---|---|
| PS1V97L mice | daily 5 mg/kg i.p. for 4 months | in brain: (1) decreased IL-1β and TNF-α | [32] |
| AD-like rat induced by Aβ42 | daily 5 mg/kg i.p. for 7 days | in brain: (1) decreased IL-1β and TNF-α | [37] |
| mouse neuroblastoma N2a cells expressing APPswe | 1.25 or 2.5 μM for 48 h | (1) decreased IL-1β and IL-6 (2) decreased COX-2 and iNOS (3) reduced NF-κB p-p65 | [9] |
| human microglia-like THP-1 cells treated with Aβ1–42 | 5 μM pre-treatment for 30 min followed by Aβ1–42 | (1) inhibited IL-1β secretion (2) inhibited miRNA-146a production (3) reduced NLRP3 inflammasome (4) reduced STAT-1 activation (5) induced HO-1 gene expression 6) increased nuclear Nrf2 levels | [38] |
| 5 μM pre-treatment for 30 min followed by Aβ1–42 | (1) decreased IL-1β and TNF-α (2) attenuated MerTK reduction (3) inhibited NF-κB signaling (4) decreased intracellular Ca2+ levels | [39] | |
| murine EOC-20 microglial cells treated with Aβ1–42 | 5 μM co-treatment with Aβ1–42 for 24 h | (1) induced the phagocytic activity (2) induced FPR2 expression | [42] |
2.4. Sulforaphane and AD-Associated Oxidative Stress Biomarkers
| Model | Sulforaphane Dose | Findings | Ref. |
|---|---|---|---|
| PS1V97L mice | daily 5 mg/kg i.p. for 4 months | in brain: (1) increased GSH (2) decreased MDA | [32] |
| AD-like rat induced by Aβ42 | daily 5 mg/kg i.p. for 7 days | in brain: (1) increased GSH (2) decreased MDA | [37] |
| mouse neuroblastoma N2a cells expressing APPswe | 1.25 or 2.5 μM for 48 h | (1) decreased ROS and MDA (2) increased SOD activity (3) upregulated Nrf2 expression and promoted Nrf2 nuclear translocation via decreasing DNA methylation levels of the Nrf2 promoter | [9] |
| human neuroblastoma SH-SY5Y cells treated with Aβ25–35 | 1–5 μM pre-treatment for 30 min followed by Aβ25–35 | (1) inhibited ROS production and subsequent oxidative damages (2) increased NQO1, HO-1 and g-GCS (3) activated Nrf2 | [40] |
| 1 μM co-treatment with Aβ25–35 | (1) increased GSH (2) increased Trx expression (3) increased HO-1 and TrxR expression 4) increased NQO1 activity (5) activated Nrf2 | [43] |
2.5. Sulforaphane and AD-Associated Biomarkers of Synaptic Damage and Neurodegeneration
| Model | Sulforaphane Dose | Findings | Ref. |
|---|---|---|---|
| 3×Tg-AD mice | 10 or 50 mg/kg p.o., 6 days/week for 2 months | in the frontal cortex: (1) increased MAP2, synaptophysin, and PSD-95 (2) activated TrkB signaling pathway in the cortex and hippocampal CA1: (1) increased BDNF levels | [33] |
| murine cortical neurons treated with Aβ42 | 0.01, 0.03 or 0.1 μM pre-treatment for 30 min followed by Aβ42 | (1) protected against cell death (2) rescued dendritic integrity | [32] |
| human neuroblastoma SH-SY5Y cells treated with Aβ25–35 | 2 μM pre-treatment for 3 h followed by Aβ25–35 | (1) protected against cell death (2) up-regulated p75 NTR (3) increased levels of Ace-H3K9 and Ace-H4K12 (4) reduced expression of HDAC1 and 3 suggested to contribute to up-regulation of p75 NTR | [35] |
| 1–5 μM pre-treatment for 30 min followed by Aβ25–35 | (1) protected against cell death (2) reduced Bax/Bcl-2 (3) reduced activation of JNK | [40] | |
| 1 μM co-treatment with Aβ25–35 | (1) protected against cell death (2) increased HSP70 | [43] | |
| murine neuroblastoma N2A cells treated with Aβ1–42 | 2.5 μM pre-treatment for 18 h followed by Aβ1–42 | (1) protected against cell death (2) sulforaphane effect dependent on proteasome activity | [41] |
| murine neuroblastoma N1E-115 treated with Aβ1–42 | 5 μM pre-treatment for 18 h followed by Aβ1–42 | (1) protected against cell death |
2.6. Sulforaphane and Cognitive Impairment in AD-Like Animal Models
| Model | Sulforaphane Dose | Findings | Ref. |
|---|---|---|---|
| 5×FAD mice | every other day 10 mg/kg i.p. for 2 months | ameliorated cognitive deficits (Morris water maze tests and passive avoidance tests) | [31] |
| 3×Tg-AD mice | every other day 5 or 10 mg/kg i.p. for 2 months | ameliorated cognitive deficits (Morris water maze tests) | [31] |
| daily 10 or 50 mg/kg p.o. 6 days/week for 2 months | ameliorated memory deficit (novel object/location recognition tests and contextual fear conditioning tests) | [34] | |
| APP/PS1 mice | daily 25 mg/kg p.o. for 5 months | ameliorated cognitive dysfunction (open field and Morris water maze tests) | [35] |
| PS1V97L mice | daily 5 mg/kg i.p. for 4 months | alleviated cognitive deficit (Morris water maze tests) | [32] |
| AD-like mice induced by Aβ1–40 | daily 30 mg/kg i.p. for 6 days | ameliorated cognitive function (Y-maze and passive avoidance behavior tests) | [36] |
| AD-like rat induced by Aβ42 | daily 5 mg/kg i.p. for 7 days | improved spatial learning | [37] |
3. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palliyaguru, D.L.; Yuan, J.M.; Kensler, T.W.; Fahey, J.W. Isothiocyanates: Translating the power of plants to people. Mol. Nutr. Food Res. 2018, 62, e1700965. [Google Scholar] [CrossRef]
- Thangstad, O.P.; Winge, P.; Husebye, H.; Bones, A. The myrosinase (thioglucoside glucohydrolase) gene family in Brassicaceae. Plant Mol. Biol. 1993, 23, 511–524. [Google Scholar] [CrossRef]
- Bones, A.M.; Rossiter, J.T. The enzymic and chemically induced decomposition of glucosinolates. Phytochemistry 2006, 67, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Wehage, S.L.; Holtzclaw, W.D.; Kensler, T.W.; Egner, P.A.; Shapiro, T.A.; Talalay, P. Protection of humans by plant glucosinolates: efficiency of conversion of glucosinolates to isothiocyanates by the gastrointestinal microflora. Cancer Prev. Res. 2012, 5, 603–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [Green Version]
- Posner, G.H.; Cho, C.G.; Green, J.V.; Zhang, Y.; Talalay, P. Design and synthesis of bifunctional isothiocyanate analogs of sulforaphane: correlation between structure and potency as inducers of anticarcinogenic detoxication enzymes. J. Med. Chem. 1994, 37, 170–176. [Google Scholar] [CrossRef]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Konwinski, R.R.; Haddad, R.; Chun, J.A.; Klenow, S.; Larson, S.C.; Haab, B.B.; Furge, L.L. Oltipraz, 3H-1,2-dithiole-3-thione, and sulforaphane induce overlapping and protective antioxidant responses in murine microglial cells. Toxicol. Lett. 2004, 153, 343–355. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, J.; Chang, N. Epigenetic modification of Nrf2 by sulforaphane increases the antioxidative and anti-inflammatory capacity in a cellular model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 824, 1–10. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, B.; Wang, X.; Wu, L.; Yang, Y.; Cheng, X.; Hu, Z.; Cai, X.; Yang, J.; Sun, X.; et al. Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2 and autophagy pathways. Sci. Rep. 2016, 6, 32206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef]
- Li, B.; Cui, W.; Liu, J.; Li, R.; Liu, Q.; Xie, X.H.; Ge, X.L.; Zhang, J.; Song, X.J.; Wang, Y.; et al. Sulforaphane ameliorates the development of experimental autoimmune encephalomyelitis by antagonizing oxidative stress and Th17-related inflammation in mice. Exp. Neurol. 2013, 250, 239–249. [Google Scholar] [CrossRef]
- Nadeem, A.; Ahmad, S.F.; Al-Ayadhi, L.Y.; Attia, S.M.; Al-Harbi, N.O.; Alzahrani, K.S.; Bakheet, S.A. Differential regulation of Nrf2 is linked to elevated inflammation and nitrative stress in monocytes of children with autism. Psychoneuroendocrinology 2020, 113, 104554. [Google Scholar] [CrossRef]
- Shirai, Y.; Fujita, Y.; Hashimoto, K. Effects of the antioxidant sulforaphane on hyperlocomotion and prepulse inhibition deficits in mice after phencyclidine administration. Clin. Psychopharmacol. Neurosci. 2012, 10, 94–98. [Google Scholar] [CrossRef]
- Lynch, R.; Diggins, E.L.; Connors, S.L.; Zimmerman, A.W.; Singh, K.; Liu, H.; Talalay, P.; Fahey, J.W. Sulforaphane from broccoli reduces symptoms of autism: A follow-up case series from a randomized double-blind Study. Glob. Adv. Health Med. 2017, 6, 2164957x17735826. [Google Scholar] [CrossRef] [PubMed]
- Shiina, A.; Kanahara, N.; Sasaki, T.; Oda, Y.; Hashimoto, T.; Hasegawa, T.; Yoshida, T.; Iyo, M.; Hashimoto, K. An open study of sulforaphane-rich broccoli sprout extract in patients with schizophrenia. Clin. Psychopharmacol Neurosci. 2015, 13, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Connors, S.L.; Macklin, E.A.; Smith, K.D.; Fahey, J.W.; Talalay, P.; Zimmerman, A.W. Sulforaphane treatment of autism spectrum disorder (ASD). Proc. Natl. Acad. Sci. USA 2014, 111, 15550–15555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bent, S.; Lawton, B.; Warren, T.; Widjaja, F.; Dang, K.; Fahey, J.W.; Cornblatt, B.; Kinchen, J.M.; Delucchi, K.; Hendren, R.L. Identification of urinary metabolites that correlate with clinical improvements in children with autism treated with sulforaphane from broccoli. Mol. Autism. 2018, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Zimmerman, A.W. Sulforaphane treatment of young men with autism spectrum disorder. CNS Neurol. Disord. Drug Targets 2016, 15, 597–601. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Amirimoghaddam-Yazdi, Z.; Moghaddam, H.S.; Mohammadi, M.R.; Akhondzadeh, S. Sulforaphane as an adjunctive treatment for irritability in children with autism spectrum disorder: A randomized, double-blind, placebo-controlled clinical trial. Psychiatry Clin. Neurosci. 2020, 74, 398–405. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmqvist, S.; Insel, P.S.; Stomrud, E.; Janelidze, S.; Zetterberg, H.; Brix, B.; Eichenlaub, U.; Dage, J.L.; Chai, X.; Blennow, K.; et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer’s disease. EMBO Mol. Med. 2019, 11, e11170. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid-β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Barcikowska, M. Guideline on the Clinical Investigation of Medicines for the Treatment of Alzheimer’s Disease; CPMP/EWP/553/95 Rev.2; European Medicines Agency: London, UK, 2018. [Google Scholar]
- Shaw, L.M.; Korecka, M.; Clark, C.M.; Lee, V.M.; Trojanowski, J.Q. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat. Rev. Drug Discov. 2007, 6, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Bendlin, B.B. Biomarkers for Alzheimer’s disease—preparing for a new era of disease-modifying therapies. Mol. Psychiatry 2021, 26, 296–308. [Google Scholar] [CrossRef]
- Collin, F.; Cheignon, C.; Hureau, C. Oxidative stress as a biomarker for Alzheimer’s disease. Biomark. Med. 2018, 12, 201–203. [Google Scholar] [CrossRef]
- Khoury, R.; Ghossoub, E. Diagnostic biomarkers of Alzheimer’s disease: A state-of-the-art review. Biomark. Neuropsychiatry 2019, 1, 100005. [Google Scholar] [CrossRef]
- Marcourakis, T.; Camarini, R.; Kawamoto, E.M.; Scorsi, L.R.; Scavone, C. Peripheral biomarkers of oxidative stress in aging and Alzheimer’s disease. Dement. Neuropsychol. 2008, 2, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Bahn, G.; Park, J.-S.; Yun, U.J.; Lee, Y.J.; Choi, Y.; Park, J.S.; Baek, S.H.; Choi, B.Y.; Cho, Y.S.; Kim, H.K.; et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. USA 2019, 116, 12516–12523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.T.; Yang, H.Y.; Wang, W.; Wu, Q.Q.; Tian, Y.R.; Jia, J.P. Sulforaphane inhibits the generation of amyloid-β oligomer and promotes spatial learning and memory in Alzheimer’s disease (PS1V97L) transgenic mice. J. Alzheimers Dis. 2018, 62, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Choi, B.R.; Yang, H.; Hwang, Y.; Park, J.H.; LaFerla, F.M.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane epigenetically enhances neuronal BDNF expression and TrkB signaling pathways. Mol. Nutr. Food Res. 2017, 61, 2. [Google Scholar] [CrossRef]
- Lee, S.; Choi, B.R.; Kim, J.; LaFerla, F.M.; Park, J.H.Y.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane upregulates the heat shock protein co-chaperone CHIP and clears amyloid-β and tau in a mouse model of Alzheimer’s disease. Mol. Nutr. Food Res. 2018, 62, e1800240. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, R.; Zhan, Z.; Li, X.; Zhou, F.; Xing, A.; Jiang, C.; Chen, Y.; An, L. Beneficial effects of sulforaphane treatment in Alzheimer’s disease may be mediated through reduced HDAC1/3 and increased P75NTR expression. Front Aging Neurosci. 2017, 9, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y. Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef]
- Wang, W.; Wei, C.; Quan, M.; Li, T.; Jia, J. Sulforaphane reverses the amyloid-β oligomers induced depressive-like behavior. J Alzheimers Dis. 2020, 78, 127–137. [Google Scholar] [CrossRef]
- An, Y.W.; Jhang, K.A.; Woo, S.Y.; Kang, J.L.; Chong, Y.H. Sulforaphane exerts its anti-inflammatory effect against amyloid-β peptide via STAT-1 dephosphorylation and activation of Nrf2/HO-1 cascade in human THP-1 macrophages. Neurobiol. Aging 2016, 38, 1–10. [Google Scholar] [CrossRef]
- Jhang, K.A.; Park, J.S.; Kim, H.S.; Chong, Y.H. Sulforaphane rescues amyloid-β peptide-mediated decrease in MerTK expression through its anti-inflammatory effect in human THP-1 macrophages. J. Neuroinflammation 2018, 15, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Park, G.H.; Lee, S.R.; Jang, J.H. Attenuation of β-amyloid-induced oxidative cell death by sulforaphane via activation of NF-E2-related factor 2. Oxid. Med. Cell Longev. 2013, 2013, 313510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.M.; Kim, J.A.; Kwak, M.K. Protection against amyloid-β cytotoxicity by sulforaphane: role of the proteasome. Arch Pharm. Res. 2009, 32, 109–115. [Google Scholar] [CrossRef]
- Chilakala, R.R.; Manchikalapudi, A.L.; Kumar, A.; Sunkaria, A. Sulforaphane attenuates Aβ oligomers mediated decrease in phagocytic activity of microglial cells. Neuroscience 2020, 429, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Masci, A.; Mattioli, R.; Costantino, P.; Baima, S.; Morelli, G.; Punzi, P.; Giordano, C.; Pinto, A.; Donini, L.M.; d’Erme, M.; et al. Neuroprotective effect of Brassica oleracea sprouts crude juice in a cellular model of Alzheimer’s disease. Oxid. Med. Cell Longev. 2015, 2015, 781938. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Jankowsky, J.L.; Slunt, H.H.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. APP processing and amyloid deposition in mice haplo-insufficient for presenilin 1. Neurobiol Aging 2004, 25, 885–892. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, Z.; Qin, W.; Jia, J. Val97Leu mutant presenilin-1 induces tau hyperphosphorylation and spatial memory deficit in mice and the underlying mechanisms. J. Neurochem. 2012, 121, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Youn, K.; Yoon, J.H.; Lee, N.; Lim, G.; Lee, J.; Sang, S.; Ho, C.T.; Jun, M. Discovery of sulforaphane as a potent BACE1 inhibitor based on kinetics and computational studies. Nutrients 2020, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Nagaveni, V.; Lakshmi, V.V.; Prabhakar, S. Sulforaphane interaction with amyloid-β 1-40 peptide studied by electrospray ionization mass spectrometry. Rapid. Commun. Mass Spectrom. 2014, 28, 2171–2180. [Google Scholar] [CrossRef]
- Kordower, J.H.; Gash, D.M.; Bothwell, M.; Hersh, L.; Mufson, E.J. Nerve growth factor receptor and choline acetyltransferase remain colocalized in the nucleus basalis (Ch4) of Alzheimer’s patients. Neurobiol Aging 1989, 10, 67–74. [Google Scholar] [CrossRef]
- Salehi, A.; Ocampo, M.; Verhaagen, J.; Swaab, D.F. P75 neurotrophin receptor in the nucleus basalis of meynert in relation to age, sex, and Alzheimer’s disease. Exp. Neurol. 2000, 161, 245–258. [Google Scholar] [CrossRef]
- Wang, Y.J.; Wang, X.; Lu, J.J.; Li, Q.X.; Gao, C.Y.; Liu, X.H.; Sun, Y.; Yang, M.; Lim, Y.; Evin, G.; et al. p75NTR regulates Aβ deposition by increasing Aβ production but inhibiting Aβ aggregation with its extracellular domain. J. Neurosci. 2011, 31, 2292–2304. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Ambasta, R.K.; Veereshwarayya, V.; Rosen, K.M.; Kosik, K.S.; Band, H.; Mestril, R.; Patterson, C.; Querfurth, H.W. CHIP and HSPs interact with β-APP in a proteasome-dependent manner and influence Aβ metabolism. Hum. Mol. Genet. 2007, 16, 848–864. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl Neurodegener 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelke, M.W.; Attia, P.; Palenchar, D.J.; Kaplan, B.; Mureb, M.; Ganzer, C.A.; Scheyer, O.; Rahman, A.; Kachko, R.; Krikorian, R.; et al. Mechanisms of risk reduction in the clinical practice of Alzheimer’s disease prevention. Front Aging Neurosci. 2018, 10, 96. [Google Scholar] [CrossRef] [Green Version]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Escudero-Lourdes, C. Toxicity mechanisms of arsenic that are shared with neurodegenerative diseases and cognitive impairment: Role of oxidative stress and inflammatory responses. Neurotoxicology 2016, 53, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Viola, K.L.; Klein, W.L. Amyloid-β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V.W. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef]
- Zhang, R.; Miao, Q.W.; Zhu, C.X.; Zhao, Y.; Liu, L.; Yang, J.; An, L. Sulforaphane ameliorates neurobehavioral deficits and protects the brain from amyloid-β deposits and peroxidation in mice with Alzheimer-like lesions. Am. J. Alzheimers Dis. Other Demen 2015, 30, 183–191. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Kuiperij, H.B.; Claassen, J.A.; Kusters, B.; Verbeek, M.M. MicroRNAs in Alzheimer’s disease: differential expression in hippocampus and cell-free cerebrospinal fluid. Neurobiol. Aging 2014, 35, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Sethi, P.; Lukiw, W.J. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009, 459, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, H.; Fahey, J.W.; Bryan, K.E.; Healy, Z.R.; Talalay, P. Stimulation of phagocytosis by sulforaphane. Biochem. Biophys. Res. Commun. 2011, 405, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Bewley, M.A.; Budd, R.C.; Ryan, E.; Cole, J.; Collini, P.; Marshall, J.; Kolsum, U.; Beech, G.; Emes, R.D.; Tcherniaeva, I.; et al. Opsonic phagocytosis in chronic obstructive pulmonary disease is enhanced by Nrf2 agonists. Am. J. Respir. Crit. Care Med. 2018, 198, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cha, Y.N.; Surh, Y.J. A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat. Res. 2010, 690, 12–23. [Google Scholar] [CrossRef]
- Koh, K.; Kim, J.; Jang, Y.J.; Yoon, K.; Cha, Y.; Lee, H.J.; Kim, J. Transcription factor Nrf2 suppresses LPS-induced hyperactivation of BV-2 microglial cells. J. Neuroimmunol. 2011, 233, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. clinicaltrials.gov. Available online: https://www.clinicaltrials.gov/ (accessed on 13 March 2021).
- Shirai, Y.; Fujita, Y.; Hashimoto, R.; Ohi, K.; Yamamori, H.; Yasuda, Y.; Ishima, T.; Suganuma, H.; Ushida, Y.; Takeda, M.; et al. Dietary intake of sulforaphane-rich broccoli sprout extracts during juvenile and adolescence can prevent phencyclidine-induced cognitive deficits at adulthood. PLoS ONE 2015, 10, e0127244. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, A.; Fahey, J.W.; Fukumoto, A.; Nakayama, M.; Inoue, S.; Zhang, S.; Tauchi, M.; Suzuki, H.; Hyodo, I.; Yamamoto, M. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori-infected mice and humans. Cancer Prev. Res. 2009, 2, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Ng, D.; Carmella, S.G.; Chen, M.; Jacobson, L.P.; Muñoz, A.; Egner, P.A.; Chen, J.G.; Qian, G.S.; Chen, T.Y.; et al. Modulation of the metabolism of airborne pollutants by glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages in Qidong, China. Carcinogenesis 2012, 33, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, A. Daily intake of broccoli sprouts normalizes bowel habits in human healthy subjects. J. Clin. Biochem. Nutr. 2018, 62, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigliotti, J.C.; Tin, A.; Pourafshar, S.; Cechova, S.; Wang, Y.T.; Sung, S.J.; Bodonyi-Kovacs, G.; Cross, J.V.; Yang, G.; Nguyen, N.; et al. GSTM1 deletion exaggerates kidney injury in experimental mouse models and confers the protective effect of cruciferous vegetables in mice and humans. J. Am. Soc. Nephrol. 2020, 31, 102–116. [Google Scholar] [CrossRef]
- Tian, S.; Liu, X.; Lei, P.; Zhang, X.; Shan, Y. Microbiota: a mediator to transform glucosinolate precursors in cruciferous vegetables to the active isothiocyanates. J. Sci. Food Agric. 2018, 98, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Nugon-Baudon, L.; Rabot, S.; Wal, J.M.; Szylit, O. Interactions of the intestinal microflora with glucosinolates in rapeseed meal toxicity: First evidence of an intestinal lactobacillus possessing a myrosinase-like activity in vivo. J. Sci. Food Agric. 1990, 52, 547–559. [Google Scholar] [CrossRef]
- Elfoul, L.; Rabot, S.; Khelifa, N.; Quinsac, A.; Duguay, A.; Rimbault, A. Formation of allyl isothiocyanate from sinigrin in the digestive tract of rats monoassociated with a human colonic strain of Bacteroides thetaiotaomicron. FEMS Microbiol. Lett. 2001, 197, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.L.; Hashimoto, K.; Uda, Y. In vitro digestion of sinigrin and glucotropaeolin by single strains of Bifidobacterium and identification of the digestive products. Food Chem Toxicol. 2004, 42, 351–357. [Google Scholar] [CrossRef]
- Luang-In, V.; Narbad, A.; Nueno-Palop, C.; Mithen, R.; Bennett, M.; Rossiter, J.T. The metabolism of methylsulfinylalkyl- and methylthioalkyl-glucosinolates by a selection of human gut bacteria. Mol. Nutr. Food Res. 2014, 58, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Tani, N.; Ohtsuru, M.; Hata, T. Purification and general characteristics of bacterial myrosinase produced by enterobacter cloacae. Agr. Biol. Chem. 1974, 38, 1623–1630. [Google Scholar]
- Charron, C.S.; Vinyard, B.T.; Ross, S.A.; Seifried, H.E.; Jeffery, E.H.; Novotny, J.A. Absorption and metabolism of isothiocyanates formed from broccoli glucosinolates: effects of BMI and daily consumption in a randomised clinical trial. Br. J. Nutr. 2018, 120, 1370–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J. Pre-Clinical Neuroprotective Evidences and Plausible Mechanisms of Sulforaphane in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2929. https://doi.org/10.3390/ijms22062929
Kim J. Pre-Clinical Neuroprotective Evidences and Plausible Mechanisms of Sulforaphane in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(6):2929. https://doi.org/10.3390/ijms22062929
Chicago/Turabian StyleKim, Jiyoung. 2021. "Pre-Clinical Neuroprotective Evidences and Plausible Mechanisms of Sulforaphane in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 6: 2929. https://doi.org/10.3390/ijms22062929
APA StyleKim, J. (2021). Pre-Clinical Neuroprotective Evidences and Plausible Mechanisms of Sulforaphane in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(6), 2929. https://doi.org/10.3390/ijms22062929