
Ym155 Induces Oxidative Stress-Mediated DNA Damage and Cell Cycle Arrest, and Causes Programmed Cell Death in Anaplastic Thyroid Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
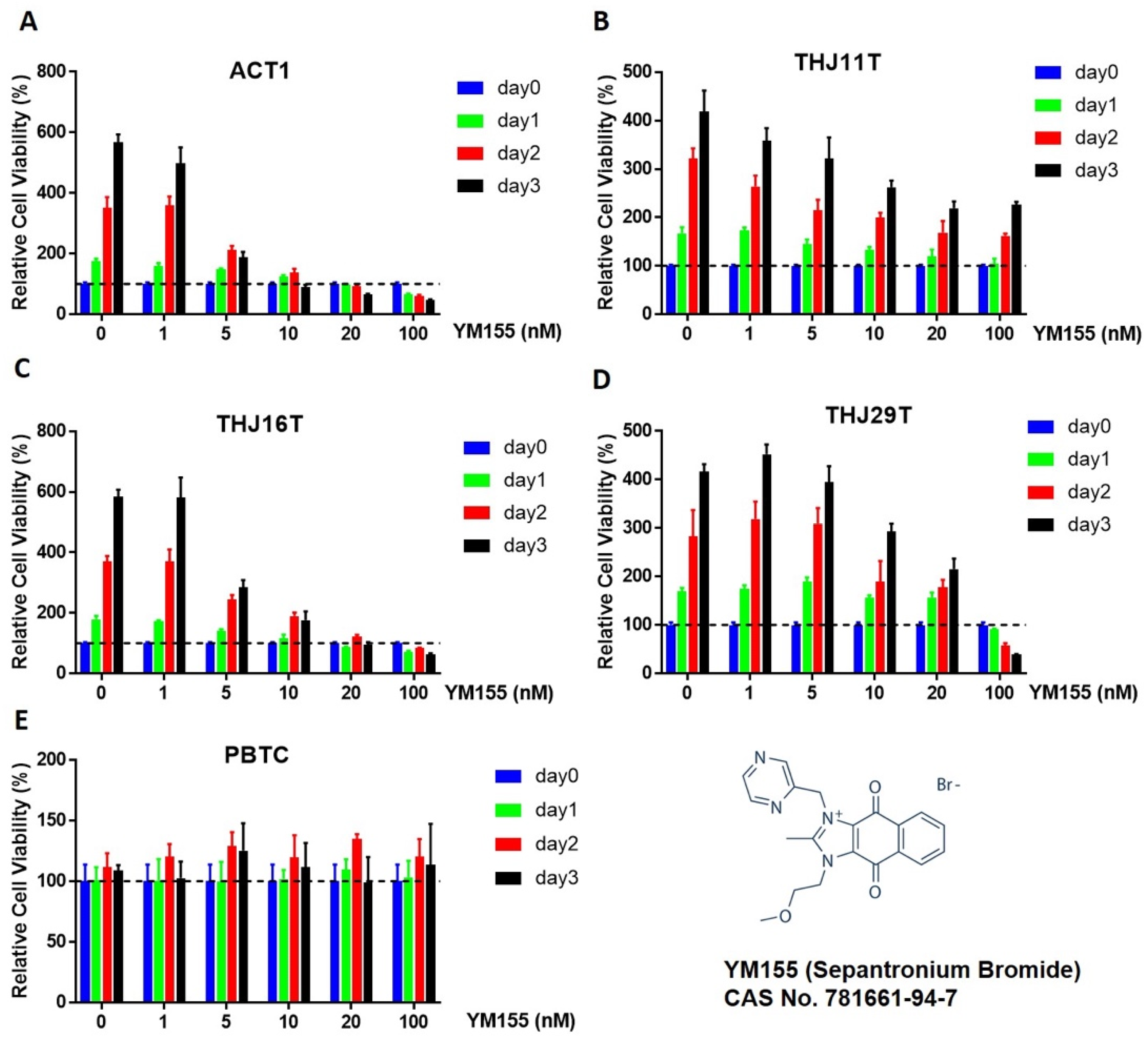
2.1. Effect of YM155 on ATC and Benign Thyroid Cell Growth
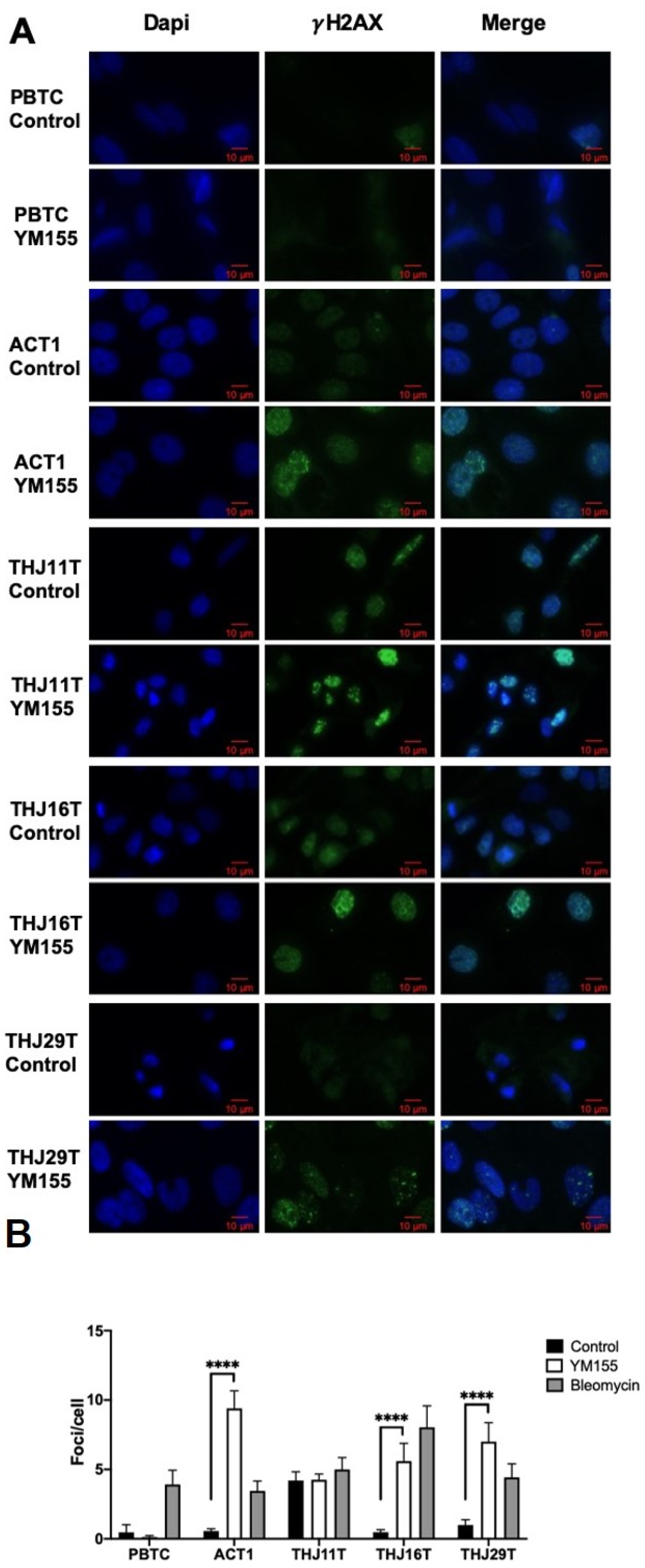
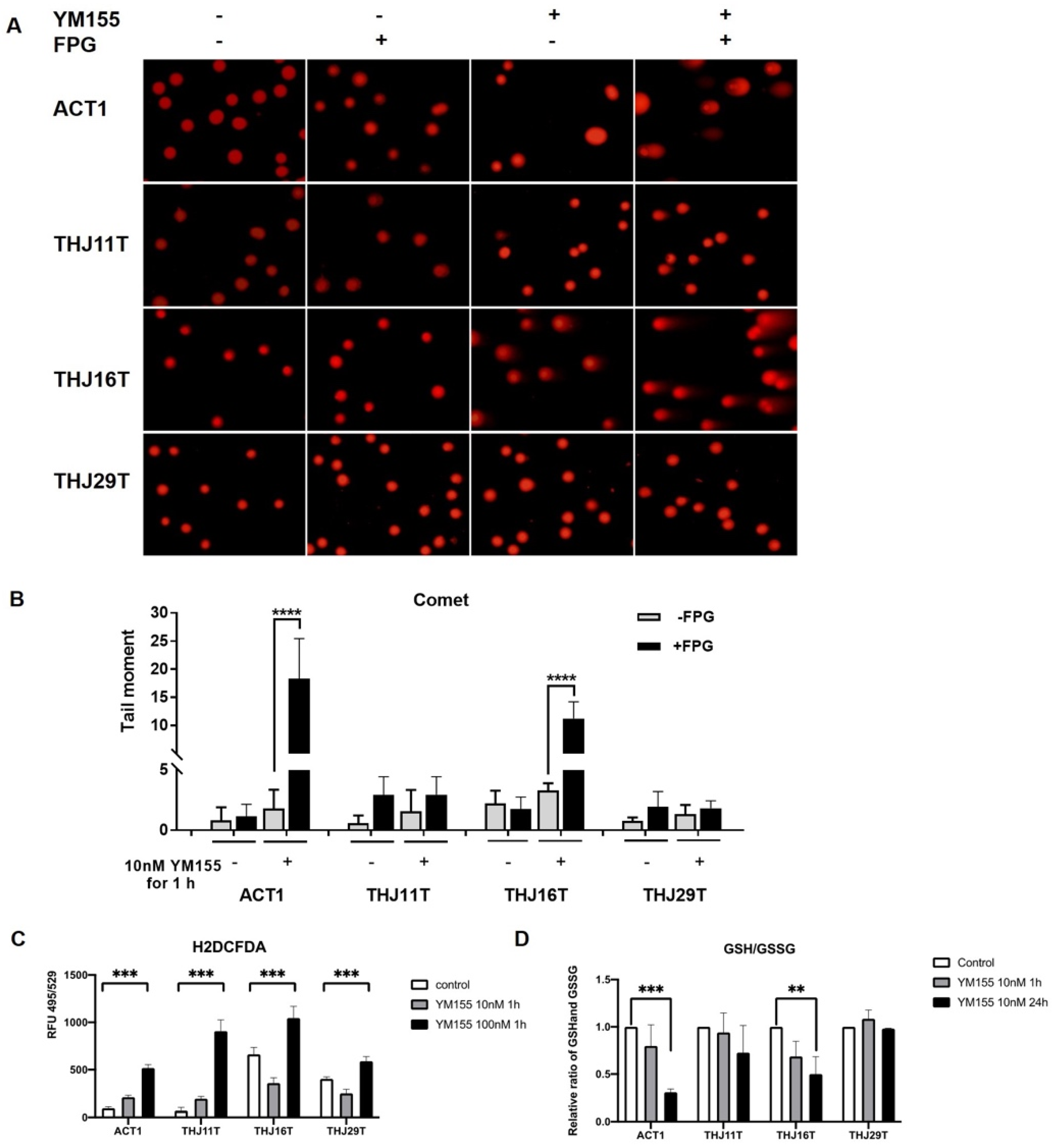
2.2. YM155 Selectively Induced DNA Damage in ATC Cells
2.3. YM155 Increased Oxidative Stress in ATC Cells
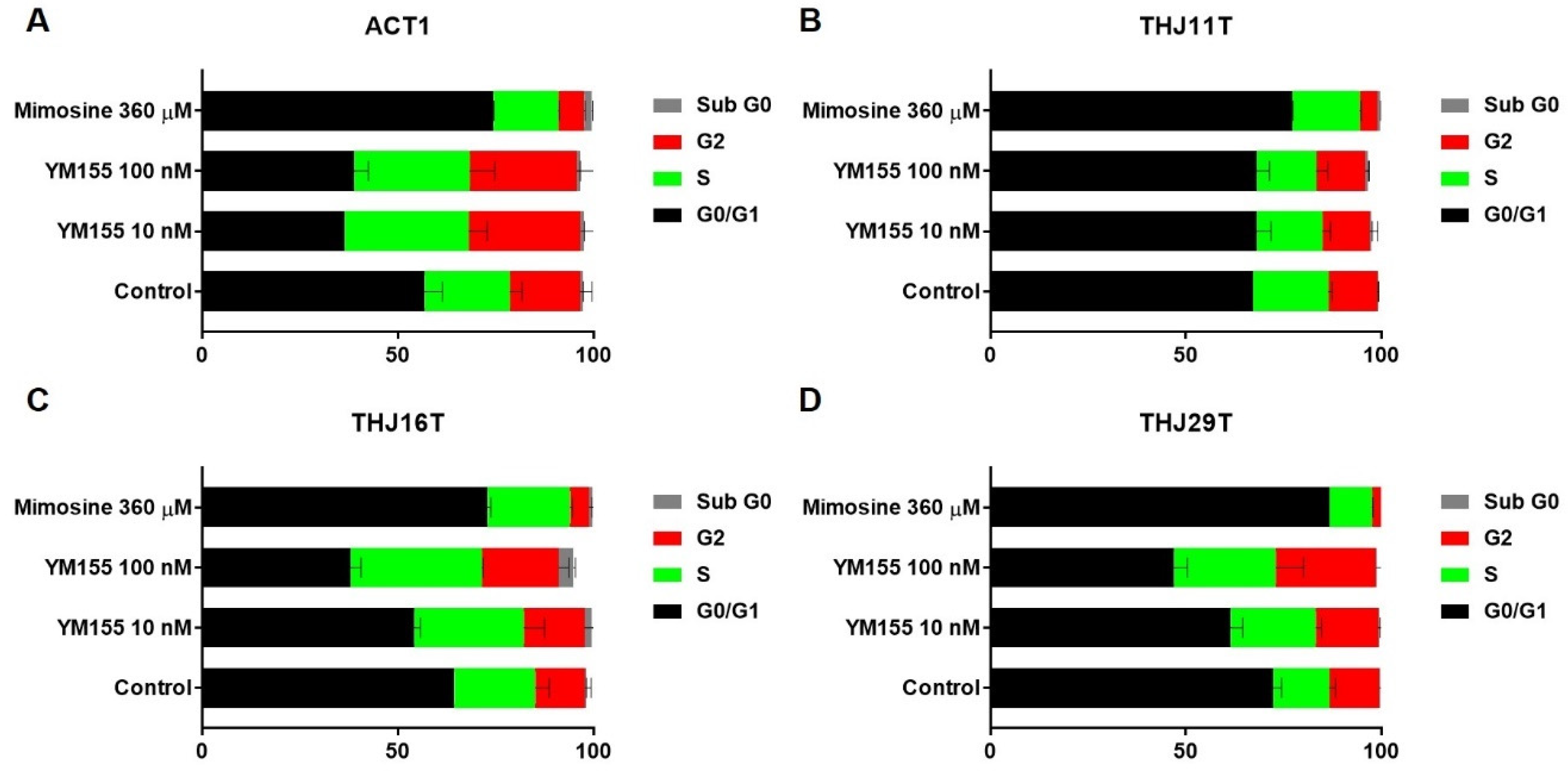
2.4. YM155 Induced S Phase or G2/M Arrest in ATC Cells
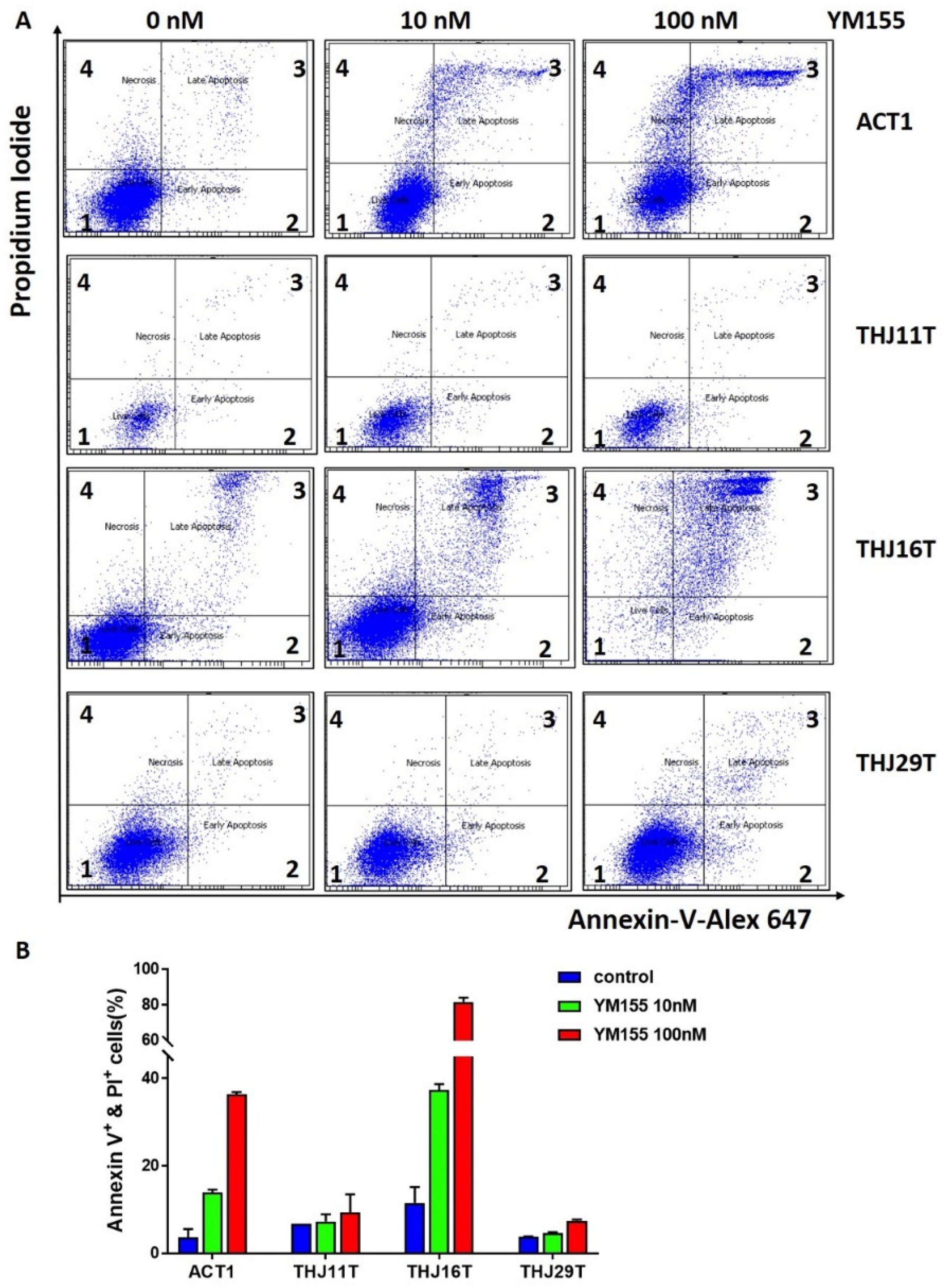
2.5. YM155 Induced Apoptosis in ATC Cells ACT1 and THJ16T
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. AlamarBlue Assay
4.3. Immunofluorescence
4.4. Fpg-Modified Alkaline Comet Assay
4.5. ROS Assay
4.6. Glutathione Fluorometric Assay
4.7. Cell Cycle Analysis
4.8. Apoptosis Analysis
4.9. Statistical Alanalysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saini, S.; Tulla, K.; Maker, A.V.; Burman, K.D.; Prabhakar, B.S. Therapeutic advances in anaplastic thyroid cancer: A current perspective. Mol. Cancer 2018, 17, 154. [Google Scholar] [CrossRef] [Green Version]
- Neff, R.L.; Farrar, W.B.; Kloos, R.T.; Burman, K.D. Anaplastic thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2008, 37, 525–538, xi. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J.P.; Shaha, A.R. Anaplastic thyroid cancer. Oral Oncol. 2013, 49, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Perrier, N.D.; Brierley, J.D.; Tuttle, R.M. Differentiated and anaplastic thyroid carcinoma: Major changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2018, 68, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Ma, H.; Ma, M.; Zhang, Z.; Sun, Z.; Hsieh, I.Y.; Okenwa, O.; Guan, H.; Li, J.; Lv, W. The incidence and survival analysis for anaplastic thyroid cancer: A SEER database analysis. Am. J. Transl. Res. 2019, 11, 5888–5896. [Google Scholar] [PubMed]
- Mehta, A.; Zhang, L.; Boufraqech, M.; Liu-Chittenden, Y.; Zhang, Y.; Patel, D.; Davis, S.; Rosenberg, A.; Ylaya, K.; Aufforth, R.; et al. Inhibition of Survivin with YM155 Induces Durable Tumor Response in Anaplastic Thyroid Cancer. Clin. Cancer Res. 2015, 21, 4123–4132. [Google Scholar] [CrossRef] [Green Version]
- Clemens, M.R.; Gladkov, O.A.; Gartner, E.; Vladimirov, V.; Crown, J.; Steinberg, J.; Jie, F.; Keating, A. Phase II, multicenter, open-label, randomized study of YM155 plus docetaxel as first-line treatment in patients with HER2-negative metastatic breast cancer. Breast Cancer Res. Treat 2015, 149, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaccone, G.; Zatloukal, P.; Roubec, J.; Floor, K.; Musil, J.; Kuta, M.; van Klaveren, R.J.; Chaudhary, S.; Gunther, A.; Shamsili, S. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J. Clin. Oncol. 2009, 27, 4481–4486. [Google Scholar] [CrossRef]
- Kelly, R.J.; Thomas, A.; Rajan, A.; Chun, G.; Lopez-Chavez, A.; Szabo, E.; Spencer, S.; Carter, C.A.; Guha, U.; Khozin, S.; et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2013, 24, 2601–2606. [Google Scholar] [CrossRef]
- Chang, B.H.; Johnson, K.; LaTocha, D.; Rowley, J.S.; Bryant, J.; Burke, R.; Smith, R.L.; Loriaux, M.; Müschen, M.; Mullighan, C.; et al. YM155 potently kills acute lymphoblastic leukemia cells through activation of the DNA damage pathway. J. Hematol. Oncol. 2015, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.M.; Chang, Y.C.; Liu, C.Y.; Lee, J.Y.; Chan, H.H.; Kuo, C.W.; Lin, K.Y.; Tsai, S.L.; Chen, S.H.; Li, C.F.; et al. YM155 down-regulates survivin and XIAP, modulates autophagy and induces autophagy-dependent DNA damage in breast cancer cells. Br. J. Pharmacol. 2015, 172, 214–234. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, N.; Yamauchi, T.; Hiramoto, M.; Yuri, M.; Naito, M.; Takeuchi, M.; Yamanaka, K.; Kita, A.; Nakahara, T.; Kinoyama, I.; et al. Interleukin enhancer-binding factor 3/NF110 is a target of YM155, a suppressant of survivin. Mol. Cell Proteomics 2012, 11, M111.013243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.; Ren, M.Q.; Silva, J.; Paul, A.; Wilson, W.D.; Schroeder, C.; Weinberger, P.; Janik, J.; Hao, Z. YM155 inhibits topoisomerase function. Anticancer Drugs 2017, 28, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- AN, I.; OA, M.; AJ, S.; CE, R.; WM, B.; RF, M.; PN, L. γH2AX Foci as a Measure of DNA Damage: A Computational Approach to Automatic Analysis. Mutat. Res. 2011, 711. [Google Scholar] [CrossRef] [Green Version]
- Jakl, L.; Lobachevsky, P.; Vokálová, L.; Durdík, M.; Marková, E.; Belyaev, I. Validation of JCountPro software for efficient assessment of ionizing radiation-induced foci in human lymphocytes. Int. J. Radiat. Biol. 2016, 92, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. Investigating oxidative DNA damage and its repair using the comet assay. Mutat. Res. 2009, 681, 24–32. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [Green Version]
- Krude, T. Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp. Cell Res. 1999, 247, 148–159. [Google Scholar] [CrossRef]
- Lai, W.A.; Liu, C.Y.; Lin, S.Y.; Chen, C.C.; Hang, J.F. Characterization of Driver Mutations in Anaplastic Thyroid Carcinoma Identifies. Cancers 2020, 12, 1973. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Chintakuntlawar, A.V.; Rumilla, K.M.; Smith, C.Y.; Jenkins, S.M.; Foote, R.L.; Kasperbauer, J.L.; Morris, J.C.; Ryder, M.; Alsidawi, S.; Hilger, C.; et al. Expression of PD-1 and PD-L1 in Anaplastic Thyroid Cancer Patients Treated With Multimodal Therapy: Results From a Retrospective Study. J. Clin. Endocrinol. Metab. 2017, 102, 1943–1950. [Google Scholar] [CrossRef]
- Feng, W.; Yoshida, A.; Ueda, T. YM155 induces caspase-8 dependent apoptosis through downregulation of survivin and Mcl-1 in human leukemia cells. Biochem. Biophys. Res. Commun. 2013, 435, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Jane, E.P.; Premkumar, D.R.; Sutera, P.A.; Cavaleri, J.M.; Pollack, I.F. Survivin inhibitor YM155 induces mitochondrial dysfunction, autophagy, DNA damage and apoptosis in Bcl-xL silenced glioma cell lines. Mol. Carcinog. 2017, 56, 1251–1265. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Discovery of survivin inhibitors and beyond: FL118 as a proof of concept. Int. Rev. Cell Mol. Biol. 2013, 305, 217–252. [Google Scholar] [CrossRef]
- Danielpour, D.; Gao, Z.; Zmina, P.M.; Shankar, E.; Shultes, B.C.; Jobava, R.; Welford, S.M.; Hatzoglou, M. Early Cellular Responses of Prostate Carcinoma Cells to Sepantronium Bromide (YM155) Involve Suppression of mTORC1 by AMPK. Sci. Rep. 2019, 9, 11541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.H.; Onoda, N.; Ishikawa, T.; Ogisawa, K.; Takenaka, C.; Yano, Y.; Hato, F.; Hirakawa, K. Peroxisome proliferator-activated receptor gamma activation induces cell cycle arrest via the p53-independent pathway in human anaplastic thyroid cancer cells. Jpn. J. Cancer Res. 2002, 93, 1358–1365. [Google Scholar] [CrossRef]
- Marlow, L.A.; D’Innocenzi, J.; Zhang, Y.; Rohl, S.D.; Cooper, S.J.; Sebo, T.; Grant, C.; McIver, B.; Kasperbauer, J.L.; Wadsworth, J.T.; et al. Detailed molecular fingerprinting of four new anaplastic thyroid carcinoma cell lines and their use for verification of RhoB as a molecular therapeutic target. J. Clin. Endocrinol. Metab. 2010, 95, 5338–5347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaros, T.G.; Stockwin, L.H.; Mullendore, M.E.; Smith, B.; Morrison, B.L.; Newton, D.L. The “survivin suppressants” NSC 80467 and YM155 induce a DNA damage response. Cancer Chemother. Pharmacol. 2012, 70, 207–212. [Google Scholar] [CrossRef]
- Hu, S.; Fu, S.; Xu, X.; Chen, L.; Xu, J.; Li, B.; Qu, Y.; Yu, H.; Lu, S.; Li, W. The mechanism of radiosensitization by YM155, a novel small molecule inhibitor of survivin expression, is associated with DNA damage repair. Cell Physiol. Biochem. 2015, 37, 1219–1230. [Google Scholar] [CrossRef]
- Wani, T.H.; Surendran, S.; Mishra, V.S.; Chaturvedi, J.; Chowdhury, G.; Chakrabarty, A. Adaptation to chronic exposure to sepantronium bromide (YM155), a prototypical survivin suppressant is due to persistent DNA damage-response in breast cancer cells. Oncotarget 2018, 9, 33589–33600. [Google Scholar] [CrossRef] [PubMed]
- Majera, D.; Mistrik, M. Effect of Sepatronium Bromide (YM-155) on DNA Double-Strand Breaks Repair in Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9431. [Google Scholar] [CrossRef]
- Wani, T.H.; Surendran, S.; Jana, A.; Chakrabarty, A.; Chowdhury, G. Quinone-Based Antitumor Agent Sepantronium Bromide (YM155) Causes Oxygen-Independent Redox-Activated Oxidative DNA Damage. Chem. Res. Toxicol. 2018, 31, 612–618. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Dasika, G.K.; Lin, S.C.; Zhao, S.; Sung, P.; Tomkinson, A.; Lee, E.Y. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene 1999, 18, 7883–7899. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Löbrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Mjelle, R.; Hegre, S.A.; Aas, P.A.; Slupphaug, G.; Drabløs, F.; Saetrom, P.; Krokan, H.E. Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair 2015, 30, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, W.; Phay, J.E.; Shen, R.; Pellegata, N.S.; Saji, M.; Ringel, M.D.; de la Chapelle, A.; He, H. Primary Cell Culture Systems for Human Thyroid Studies. Thyroid 2016, 26, 1131–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, A.Y.; Maynard, M.R.; Piett, C.G.; Nagel, Z.D.; Alexander, J.S.; Kevil, C.G.; Berridge, M.V.; Pattillo, C.B.; Rosen, L.R.; Miriyala, S.; et al. Sodium sulfide selectively induces oxidative stress, DNA damage, and mitochondrial dysfunction and radiosensitizes glioblastoma (GBM) cells. Redox Biol. 2019, 26, 101220. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Q.; Mackay, R.P.; Xiao, A.Y.; Copland, J.A.; Weinberger, P.M. Ym155 Induces Oxidative Stress-Mediated DNA Damage and Cell Cycle Arrest, and Causes Programmed Cell Death in Anaplastic Thyroid Cancer Cells. Int. J. Mol. Sci. 2021, 22, 1961. https://doi.org/10.3390/ijms22041961
Xu Q, Mackay RP, Xiao AY, Copland JA, Weinberger PM. Ym155 Induces Oxidative Stress-Mediated DNA Damage and Cell Cycle Arrest, and Causes Programmed Cell Death in Anaplastic Thyroid Cancer Cells. International Journal of Molecular Sciences. 2021; 22(4):1961. https://doi.org/10.3390/ijms22041961
Chicago/Turabian StyleXu, Qinqin, Ryan P. Mackay, Adam Y. Xiao, John A. Copland, and Paul M. Weinberger. 2021. "Ym155 Induces Oxidative Stress-Mediated DNA Damage and Cell Cycle Arrest, and Causes Programmed Cell Death in Anaplastic Thyroid Cancer Cells" International Journal of Molecular Sciences 22, no. 4: 1961. https://doi.org/10.3390/ijms22041961
APA StyleXu, Q., Mackay, R. P., Xiao, A. Y., Copland, J. A., & Weinberger, P. M. (2021). Ym155 Induces Oxidative Stress-Mediated DNA Damage and Cell Cycle Arrest, and Causes Programmed Cell Death in Anaplastic Thyroid Cancer Cells. International Journal of Molecular Sciences, 22(4), 1961. https://doi.org/10.3390/ijms22041961