
Regorafenib-Attenuated, Bleomycin-Induced Pulmonary Fibrosis by Inhibiting the TGF-β1 Signaling Pathway

 , and
, and
Abstract
:1. Introduction
2. Results
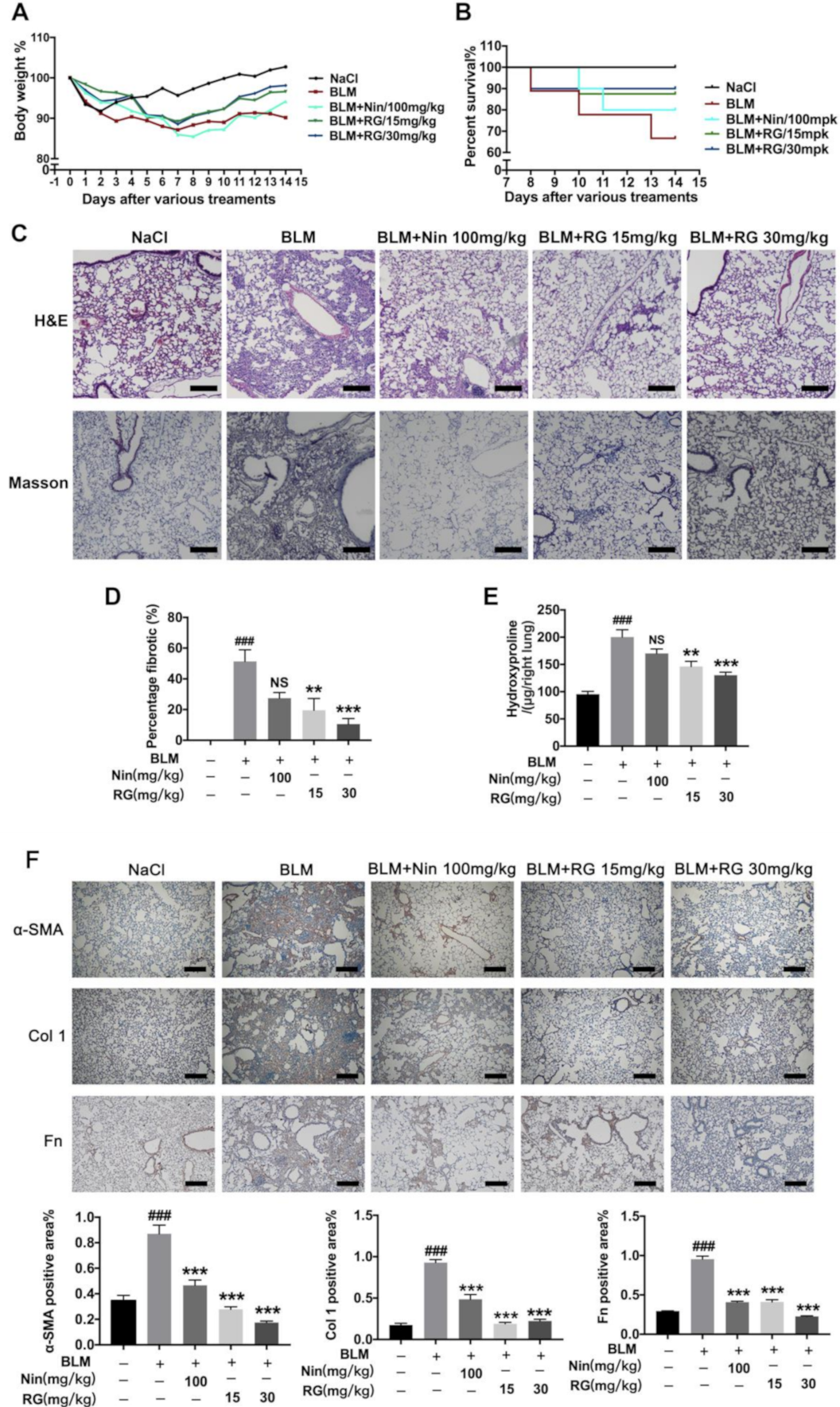
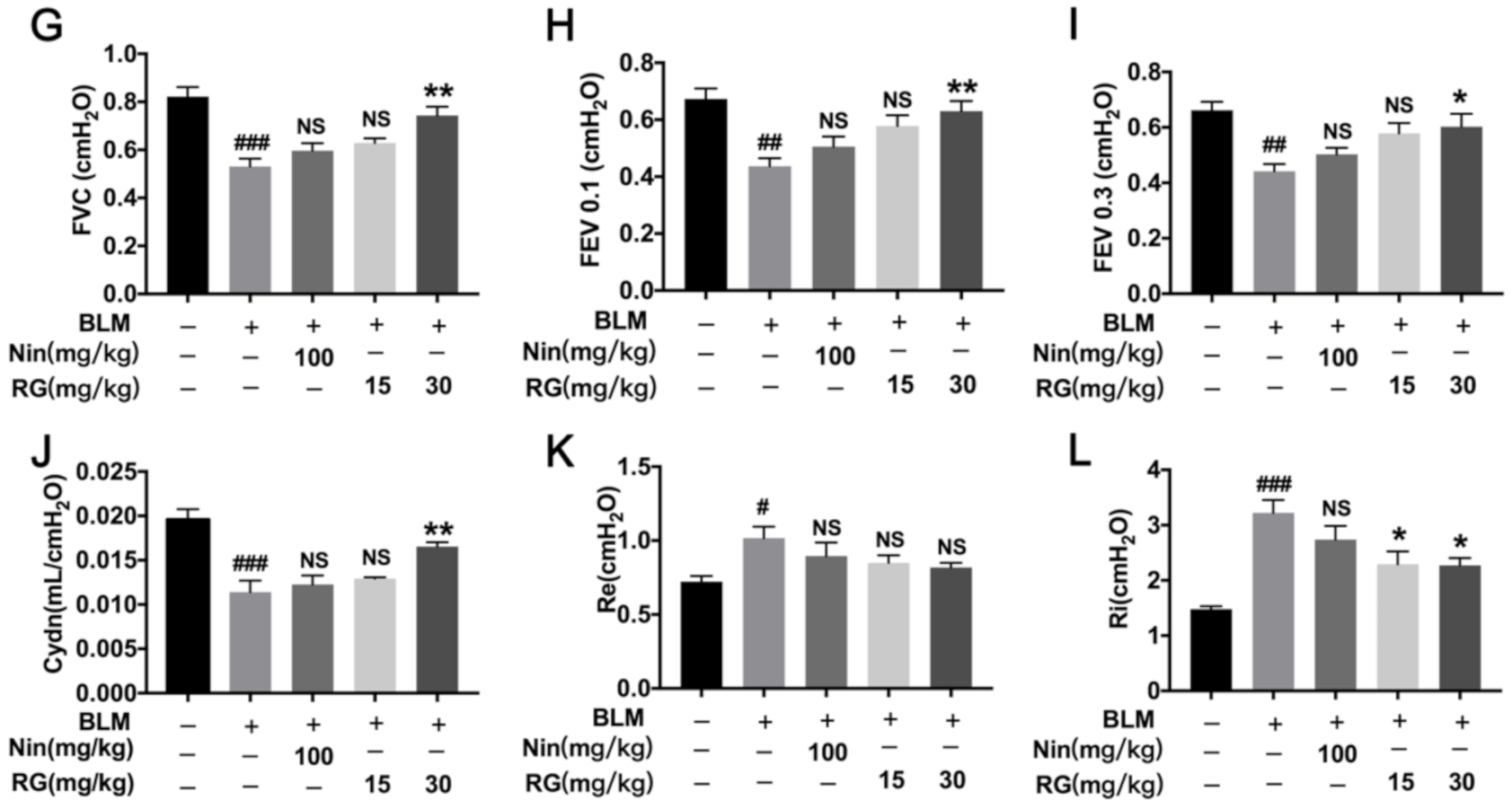
2.1. RG Attenuates BLM-Induced Pulmonary Fibrosis in Mice
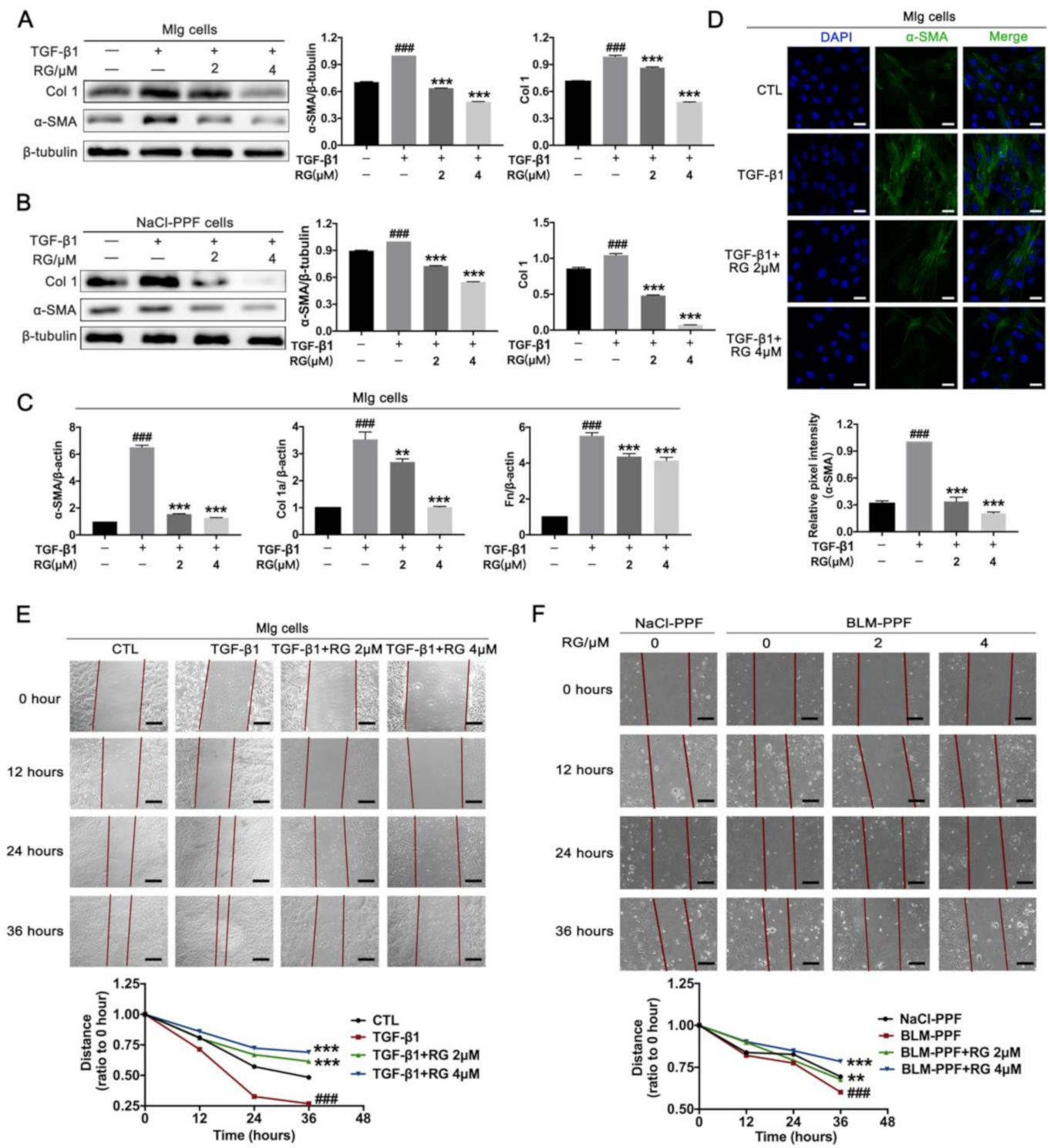
2.2. RG Suppresses TGF-β1-Induced Activation, ECM Accumulation, and Pulmonary Fibroblast Migration
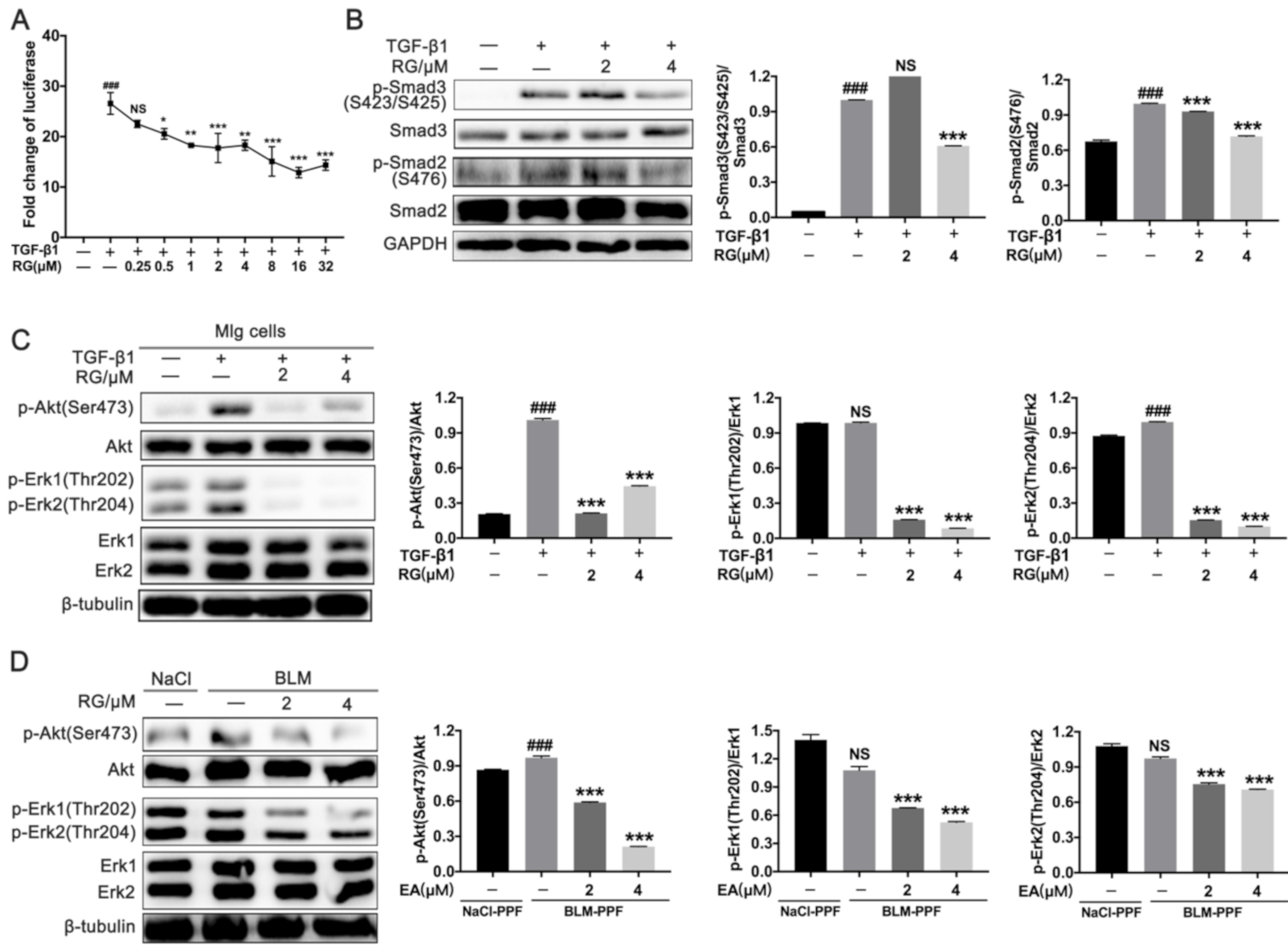
2.3. RG Down-Regulates TGF-β1/Smad and TGF-β1/Non-Smad Signals in Pulmonary Fibroblasts
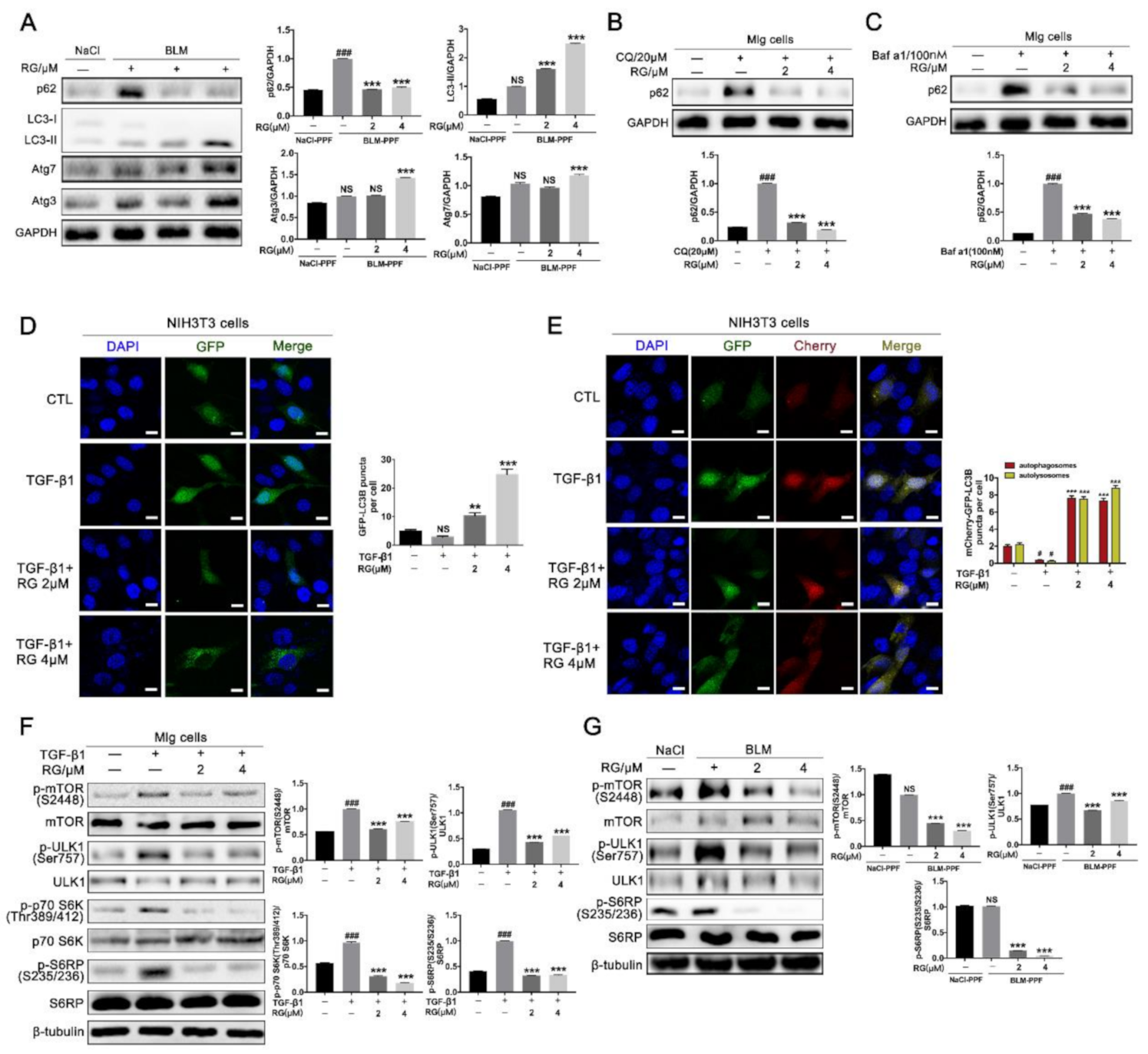
2.4. RG Induces Autophagy in Pulmonary Fibroblasts Mainly by Inhibiting the TGF-β1/mTOR Signaling Pathway
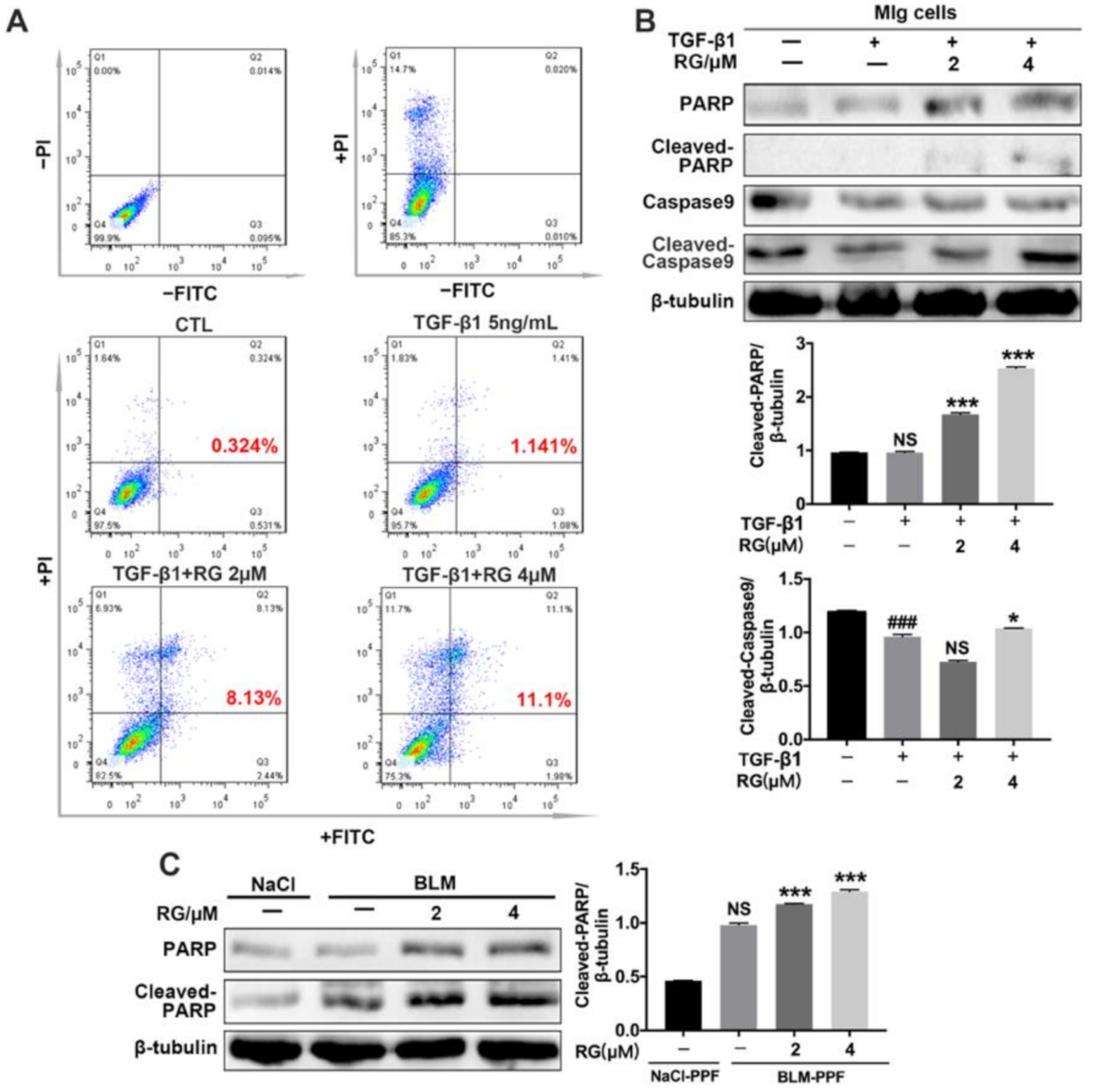
2.5. RG Promotes the Apoptosis of Myofibroblasts
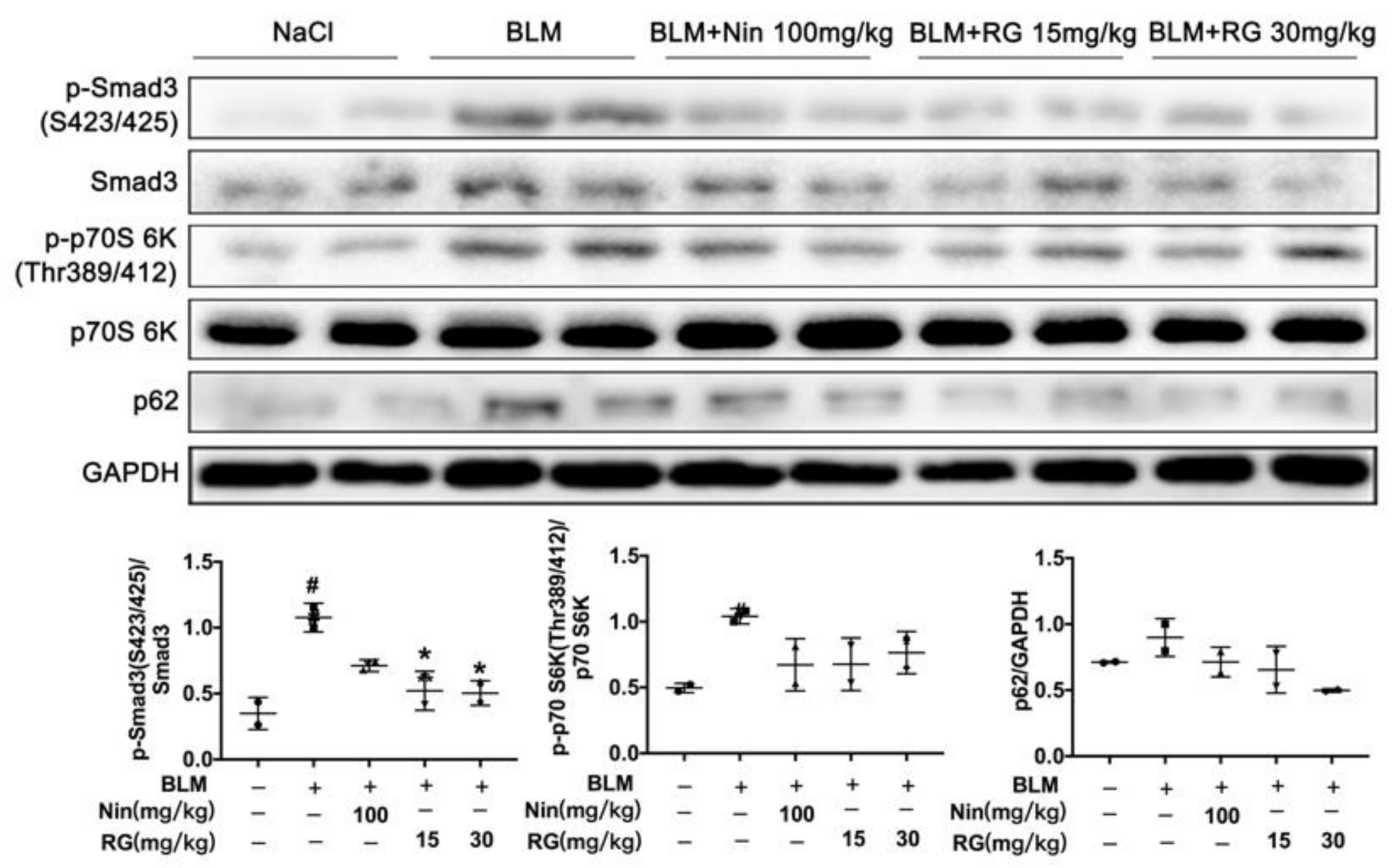
2.6. RG Promotes Autophagy Formation and Inhibits TGF-β1/mTOR Signaling In Vivo
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animals
4.3. Isolation Protocol of Primary Pulmonary Fibroblasts (PPFs)
4.4. Cell Culture
4.5. Western Blot
4.6. Real-Time Quantitative PCR
4.7. Luciferase Assay
4.8. Wound-Healing Assays
4.9. Hematoxylin–Eosin (H&E) Staining
4.10. Immunohistochemistry
4.11. Immunofluorescence
4.12. Flow Cytometric Analysis of Apoptosis
4.13. Hydroxyproline Assay
4.14. Evaluation of Pulmonary Function
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BLM | Bleomycin |
| IPF | Idiopathic pulmonary fibrosis |
| ECM | Extracellular matrix |
| RG | Regorafenib |
| mTOR | mammalian target of rapamycin |
| BCL2 | B-cell lymphoma-2 |
| TFEB | Transcription factor EB |
| PPF | Primary pulmonary fibroblasts |
| Mlg | Mouse lung fibroblasts cell |
| HLF-1 | Human lung fibroblasts |
| DMEM | Dulbecco’s modified eagle medium |
| NIH-3T3 | Mouse embryonic fibroblasts cell line |
| FBS | Fetal bovine serum |
| CQ | Chloroquine |
| Baf a1 | Bafilomycin a1 |
| FVC | Forced vital capacity |
| Cydn | Dynamic compliance |
| Ri | Inspiratory resistance |
| Re | Expiratory resistance |
| Pf | Fibrosis area pixel |
| Pw | Total lung pixel |
| H&E | Hematoxylin–eosin |
| TGF-β | Transforming growth factor β |
| Fn | Fibronectin |
| Col 1 | Collagen I |
| α-SMA | α-smooth muscle actin |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| P38 | P38 mitogen-activated protein kinase |
| ERK | Extracellular regulated protein kinases |
| JNK | c-Jun N-terminal kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| Src | Steroid receptor coactivator |
| VEGFR | Vascular endothelial growth factor receptor |
| TIE2 | Endothelial tyrosine kinase Tie2 |
| c-KIT | c-kit proto-oncogene |
| RET | RET proto-oncogene |
| CSF1R | Colony-stimulating factor 1 receptor |
| GFP-LC3B | Green fluorescent protein-microtubule-associated protein 1 light chain 3B |
| mCherry-GFP-LC3B | mCherry fluorescent protein-Green fluorescent protein-microtubule-associated protein 1 light chain 3B |
| ULK-1 | Protein kinase ULK1/Autophagy-related protein 1 |
| p70 S6K | p70 S6 Kinase |
| S6RP | S6 ribosomal protein |
References
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Biffi, A.; Richeldi, L. Idiopathic pulmonary fibrosis: Diagnosis, epidemiology and natural history. Respirology 2016, 21, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Kolb, M.; Vasakova, M. The natural history of progressive fibrosing interstitial lung diseases. Respir. Res. 2019, 20, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goss, C.H.; Newsom, S.A.; Schildcrout, J.S.; Sheppard, L.; Kaufman, J.D. Effect of ambient air pollution on pulmonary exacerbations and lung function in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2004, 169, 816–821. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Shah, B.A.; Chowdhury, B.A. Forced vital capacity in idiopathic pulmonary fibrosis—FDA review of pirfenidone and nintedanib. N. Engl. J. Med. 2015, 372, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Xaubet, A.; Marin-Arguedas, A.; Lario, S.; Ancochea, J.; Morell, F.; Ruiz-Manzano, J.; Rodriguez-Becerra, E.; Rodriguez-Arias, J.M.; Inigo, P.; Sanz, S.; et al. Transforming growth factor-beta1 gene polymorphisms are associated with disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 431–435. [Google Scholar] [CrossRef]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Miyazono, K.; ten Dijke, P.; Heldin, C.H. TGF-beta signaling by Smad proteins. Adv. Immunol. 2000, 75, 115–157. [Google Scholar]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [Green Version]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Ghavami, S.; Cunnington, R.H.; Gupta, S.; Yeganeh, B.; Filomeno, K.L.; Freed, D.H.; Chen, S.; Klonisch, T.; Halayko, A.J.; Ambrose, E.; et al. Autophagy is a regulator of TGF-beta1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis. 2015, 6, e1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.S.; Zeglinski, M.R.; Rattan, S.G.; Landry, N.M.; Ghavami, S.; Wigle, J.T.; Klonisch, T.; Halayko, A.J.; Dixon, I.M. Inhibition of autophagy inhibits the conversion of cardiac fibroblasts to cardiac myofibroblasts. Oncotarget 2016, 7, 78516–78531. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, H.V.; Eley, J.D.; Guillotin, D.; Plate, M.; Nanthakumar, C.B.; Martufi, M.; Peace, S.; Joberty, G.; Poeckel, D.; Good, R.B.; et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nat. Commun. 2019, 10, 6. [Google Scholar]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schutz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Arai, H.; Battaglin, F.; Wang, J.; Lo, J.H.; Soni, S.; Zhang, W.; Lenz, H.J. Molecular insight of regorafenib treatment for colorectal cancer. Cancer Treat. Rev. 2019, 81, 101912. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Seufferlein, T. Regorafenib. Recent Results Cancer Res. 2018, 211, 45–56. [Google Scholar] [PubMed]
- Kuhn, C.; McDonald, J.A. The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am. J. Pathol. 1991, 138, 1257–1265. [Google Scholar]
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-beta: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Park, J.S.; Park, H.J.; Park, Y.S.; Lee, S.M.; Yim, J.J.; Yoo, C.G.; Han, S.K.; Kim, Y.W. Clinical significance of mTOR, ZEB1, ROCK1 expression in lung tissues of pulmonary fibrosis patients. BMC Pulm. Med. 2014, 14, 168. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, T.; Lei, Y.; Li, X.; Jiang, J.; Lan, J.; Liu, Y.; Chen, H.; Gao, W.; Xie, N.; et al. Identification of ANXA2 (annexin A2) as a specific bleomycin target to induce pulmonary fibrosis by impeding TFEB-mediated autophagic flux. Autophagy 2018, 14, 269–282. [Google Scholar] [CrossRef]
- Mi, S.; Li, Z.; Yang, H.Z.; Liu, H.; Wang, J.P.; Ma, Y.G.; Wang, X.X.; Liu, H.Z.; Sun, W.; Hu, Z.W. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J. Immunol. 2011, 187, 3003–3014. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, L.; Chen, H.; Lei, Y.; Zhang, T.; Wang, Y.; Jin, P.; Lan, J.; Zhou, L.; Huang, Z.; et al. Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma. Autophagy 2020, 16, 106–122. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, J.C.; Thannickal, V.J. Idiopathic pulmonary fibrosis: New concepts in pathogenesis and implications for drug therapy. Treat. Respir. Med. 2006, 5, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Phan, S.H. Inhibition of myofibroblast apoptosis by transforming growth factor beta(1). Am. J. Respir. Cell Mol. Biol 1999, 21, 658–665. [Google Scholar] [CrossRef]
- Ning, W.; Li, C.J.; Kaminski, N.; Feghali-Bostwick, C.A.; Alber, S.M.; Di, Y.P.; Otterbein, S.L.; Song, R.; Hayashi, S.; Zhou, Z.; et al. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. USA 2004, 101, 14895–14900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Liang, J.; Hodge, J.; Lu, B.; Zhu, Z.; Yu, S.; Fan, J.; Gao, Y.; Yin, Z.; Homer, R.; et al. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. J. Clin. Investig. 2004, 114, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Gilhodes, J.C.; Jule, Y.; Kreuz, S.; Stierstorfer, B.; Stiller, D.; Wollin, L. Quantification of Pulmonary Fibrosis in a Bleomycin Mouse Model Using Automated Histological Image Analysis. PLoS ONE 2017, 12, e0170561. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company and Item No. | Antibody | Company and Item No. |
|---|---|---|---|
| GAPDH | Affinity, AF7021 | Beclin1 | CST, 3495S |
| β-tubulin | Affinity, T0023 | LC3A/B | CST, 12741S |
| α-SMA | Affinity, BF9212 | mTOR | Abcam, ab32028 |
| Collagen I | Affinity, AF7001 | p-mTOR (S2448) | Abcam, ab109268 |
| p-Smad3 (S423/425) | Affinity, AF8315 | S6 ribosomal protein (S6RP) | Affinity, AF7831 |
| Smad3 | Affinity, AF6362 | p-S6 ribosomal protein (S235/S236) | CST, 4858T |
| p-Smad2(S467) | Affinity, AF3449 | p-ULK1( Ser757) | CST, 14202T |
| Smad2 | Affinity, AF6449 | ULK1 | CST, 8054T |
| Akt | SANTA CRUZ, sc-56878 | p-p70 S6K (Thr289/412) | Affinity, AF3228 |
| p-Akt (Ser473) | SANTA CRUZ, sc-514032 | p70 S6K | Affinity, AF6226 |
| Erk1/2 | SANTA CRUZ, sc-514302 | PARP | CST, 9532T |
| p-Erk1/2 (Thr202/Thy204) | SANTA CRUZ, sc-81492 | Cleaved-PARP | CST, 5625T |
| Atg7 | CST, 8558T | Caspase9 | CST, 9009T |
| P62 | Proteintech, 8420-1-AP | Cleaved-Caspase9 | CST, 9504T |
| Atg3 | CST, 8089T |
| Gene (Mouse) | Forward Primer Sequence (5′-3′) | Reverse Primer Sequence (5′-3′) |
|---|---|---|
| α-SMA | GCTGGTGATGATGCTCCCA | GCCCATTCCAACCATTACTCC |
| Fn | GTGTAGCACAACTTCCAATTACGAA | GGAATTTCCGCCTCGAGTCT |
| Col1 A1 | CCAAGAAGACATCCCTGAAGTCA | TGCACGTCATCGCACACA |
| β-actin | AGGCCAACCGTGAAAAGATG | AGAGCATAGCCCTCGTAGATGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Ma, L.; Huang, K.; Wei, Y.; Long, S.; Liu, Q.; Zhang, D.; Wu, S.; Wang, W.; Yang, G.; et al. Regorafenib-Attenuated, Bleomycin-Induced Pulmonary Fibrosis by Inhibiting the TGF-β1 Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 1985. https://doi.org/10.3390/ijms22041985
Li X, Ma L, Huang K, Wei Y, Long S, Liu Q, Zhang D, Wu S, Wang W, Yang G, et al. Regorafenib-Attenuated, Bleomycin-Induced Pulmonary Fibrosis by Inhibiting the TGF-β1 Signaling Pathway. International Journal of Molecular Sciences. 2021; 22(4):1985. https://doi.org/10.3390/ijms22041985
Chicago/Turabian StyleLi, Xiaohe, Ling Ma, Kai Huang, Yuli Wei, Shida Long, Qinyi Liu, Deqiang Zhang, Shuyang Wu, Wenrui Wang, Guang Yang, and et al. 2021. "Regorafenib-Attenuated, Bleomycin-Induced Pulmonary Fibrosis by Inhibiting the TGF-β1 Signaling Pathway" International Journal of Molecular Sciences 22, no. 4: 1985. https://doi.org/10.3390/ijms22041985
APA StyleLi, X., Ma, L., Huang, K., Wei, Y., Long, S., Liu, Q., Zhang, D., Wu, S., Wang, W., Yang, G., Zhou, H., & Yang, C. (2021). Regorafenib-Attenuated, Bleomycin-Induced Pulmonary Fibrosis by Inhibiting the TGF-β1 Signaling Pathway. International Journal of Molecular Sciences, 22(4), 1985. https://doi.org/10.3390/ijms22041985