
Nc886, a Novel Suppressor of the Type I Interferon Response Upon Pathogen Intrusion


 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
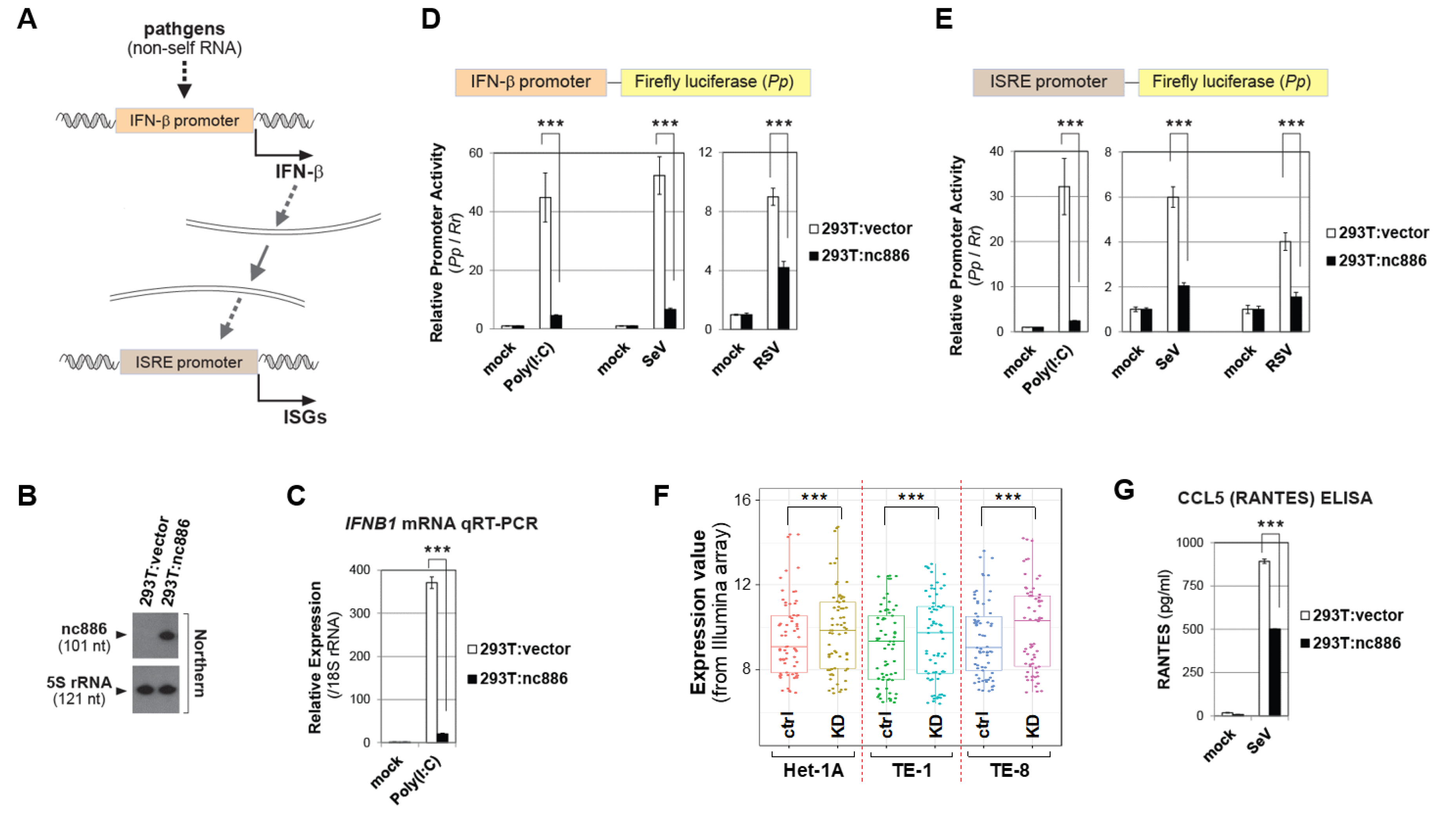
2.1. nc886 Antagonizes the IFN-β Signaling
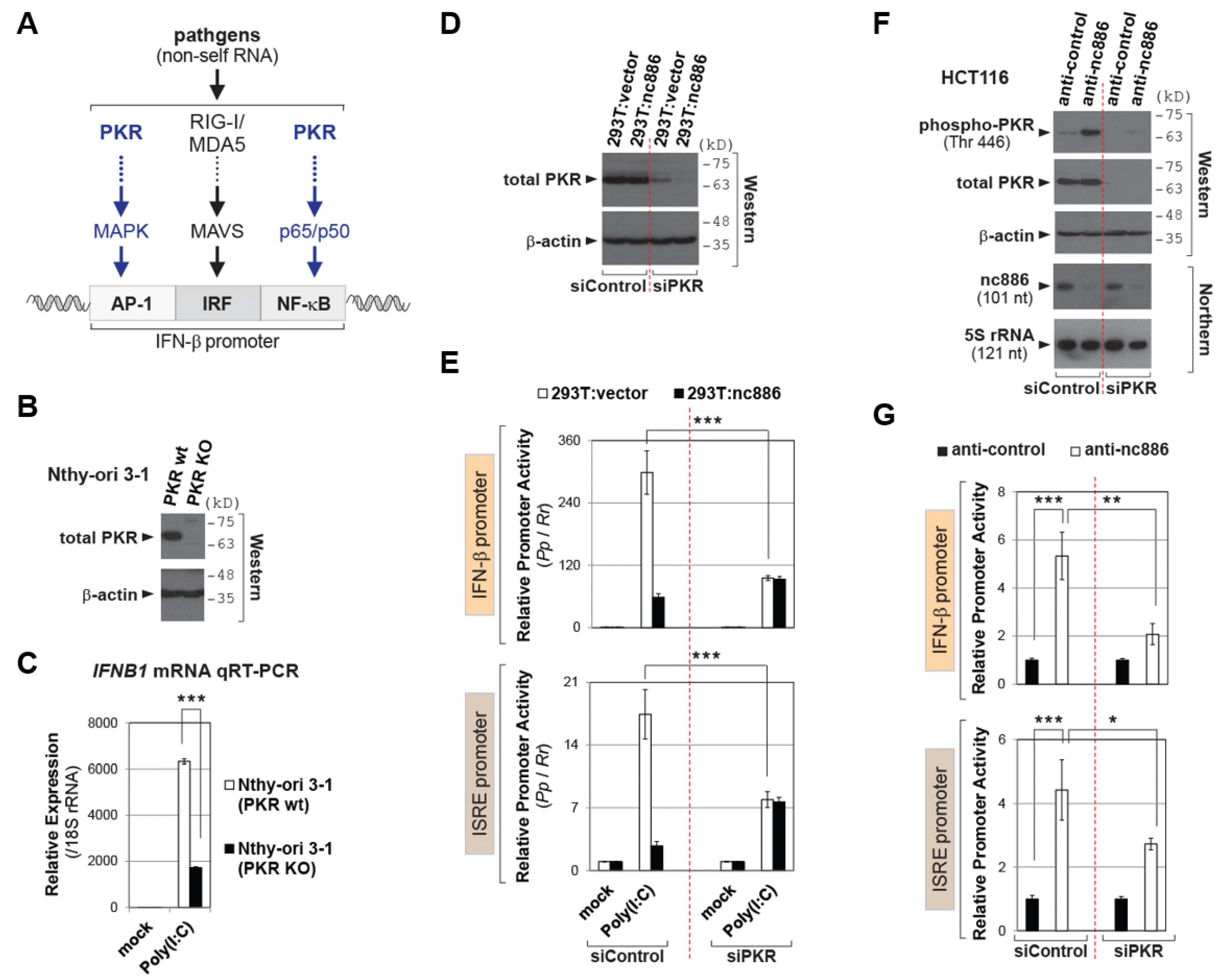
2.2. nc886 Suppresses the IFN-β Promoter Activity via Inhibiting PKR
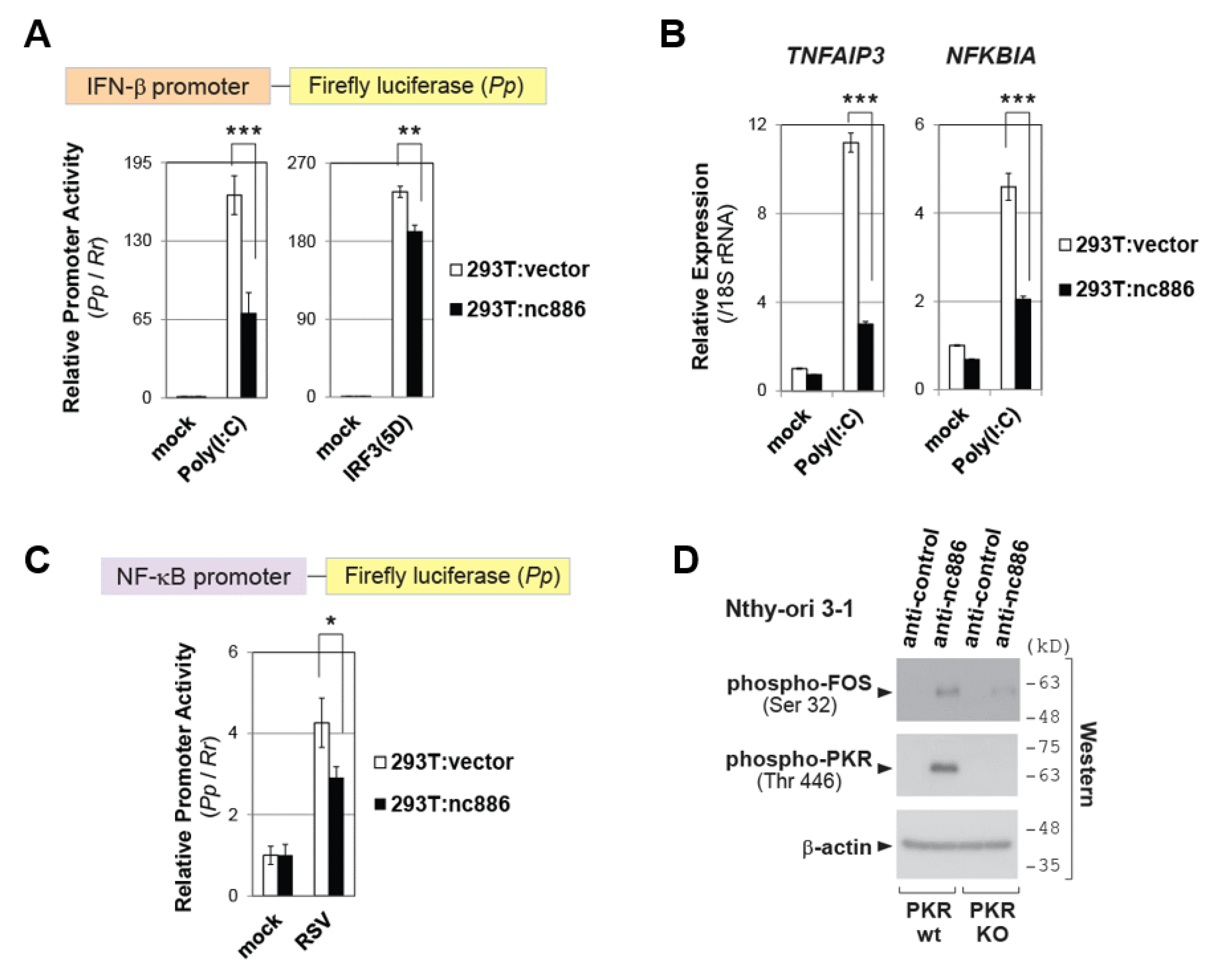
2.3. nc886 Restricts the IFN-β Promoter Activation by Inhibiting NF-κB and/or AP-1 Pathways
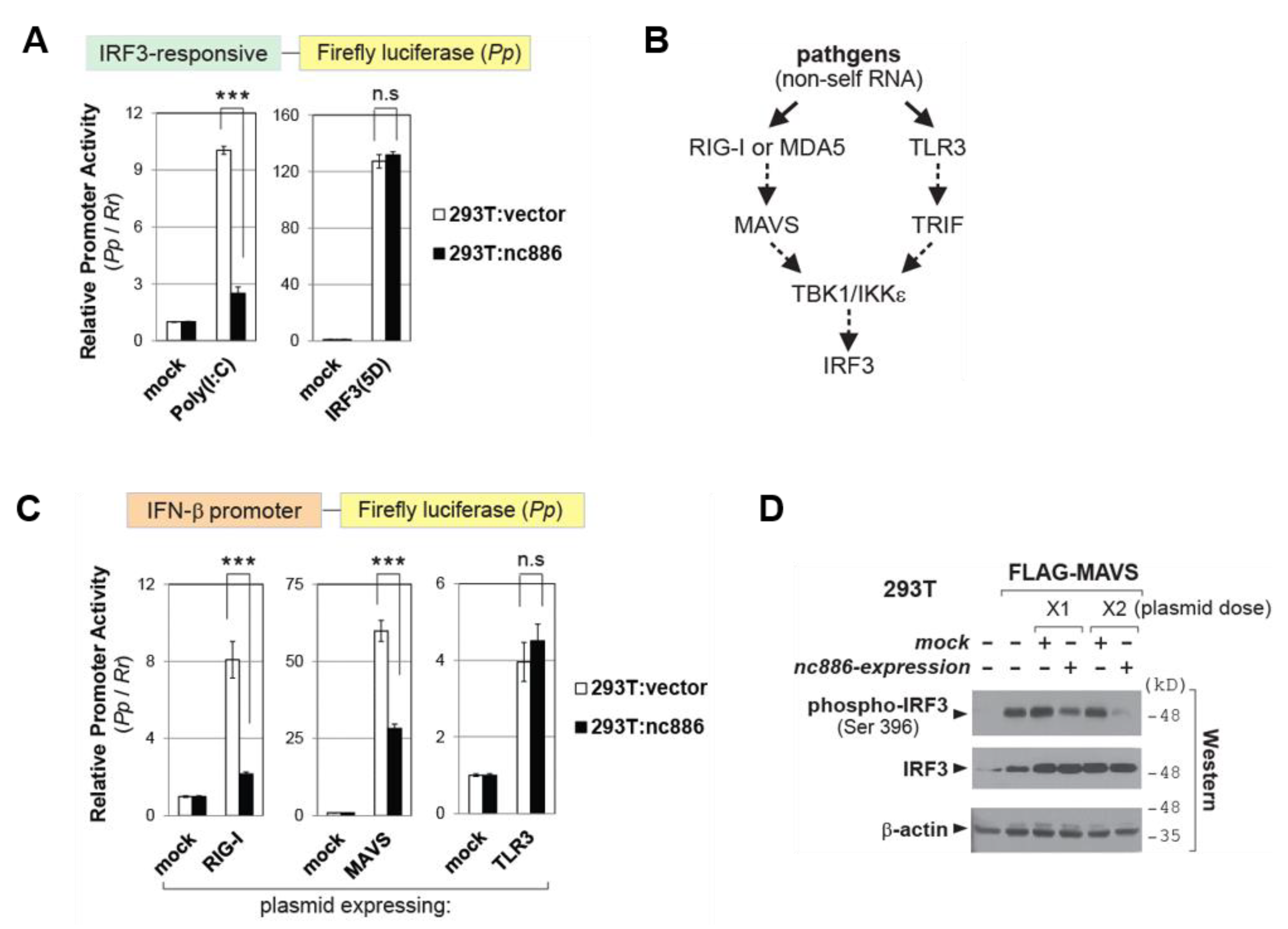
2.4. nc886 Inhibits IRF3 via the RIG-I/MAVS Pathway
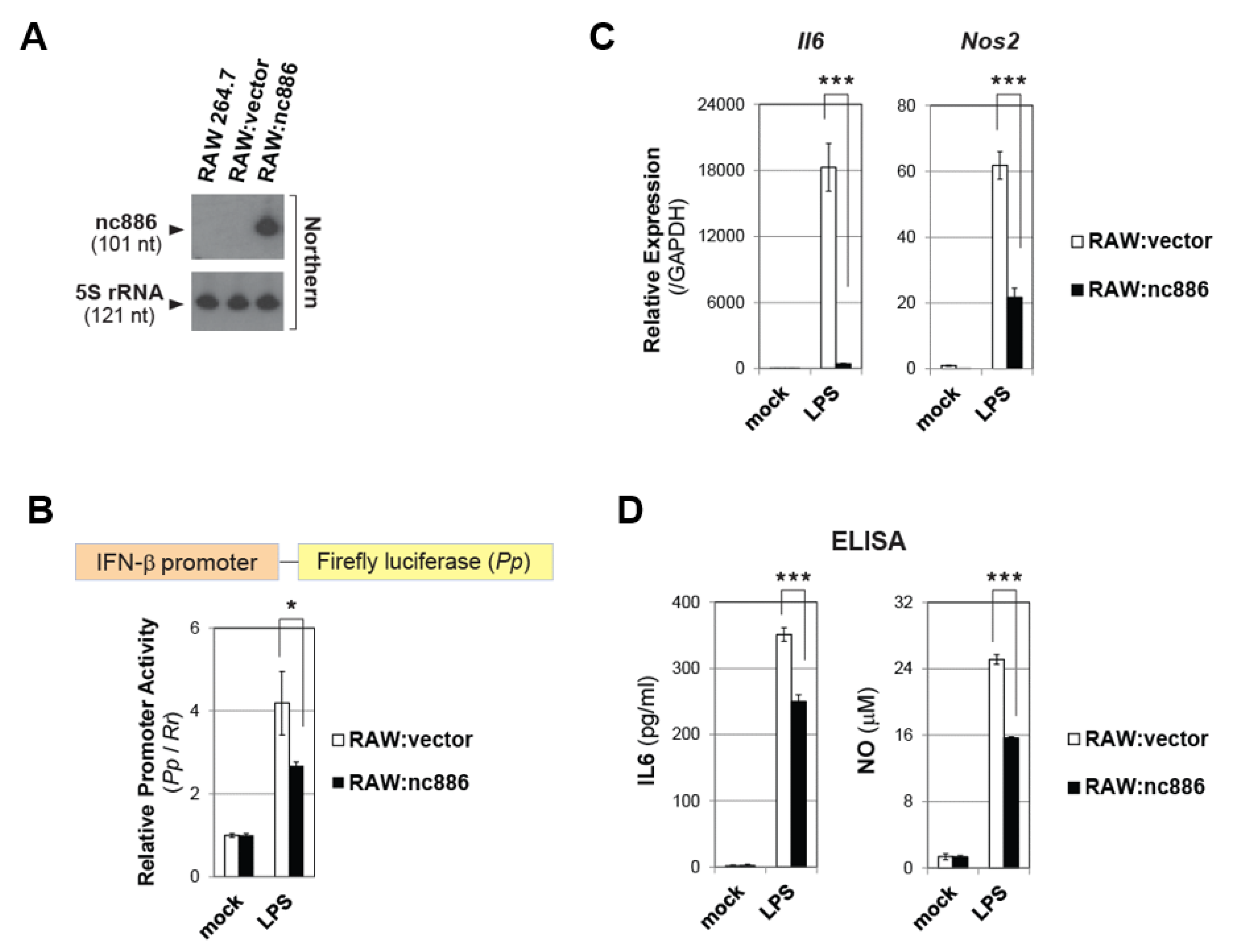
2.5. nc886 also Inhibits Inflammatory Genes in Macrophages
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Viruses, Antibodies, and Other Reagents
4.2. Luciferase Reporter Assays
4.3. Plasmids, Synthetic RNAs, and Their Transfection
4.4. RNA Isolation and Measurement
4.5. ELISA and NO Measurement
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossol, M.; Heine, H.; Meusch, U.; Quandt, D.; Klein, C.; Sweet, M.J.; Hauschildt, S. LPS-induced cytokine production in human monocytes and macrophages. Crit. Rev. Immunol. 2011, 31, 379–446. [Google Scholar] [CrossRef]
- Edwards, M.R.; Slater, L.; Johnston, S.L. Signalling pathways mediating type I interferon gene expression. Microbes Infect. Inst. Pasteur 2007, 9, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.M. The Jak-STAT pathway stimulated by interferon alpha or interferon beta. Sci. STKE 2004, 2004, tr10. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, K.; Jang, H.J.; Lee, G.K.; Park, J.L.; Kim, S.Y.; Kim, S.B.; Johnson, B.H.; Zo, J.I.; Lee, J.S.; et al. Epigenetic silencing of the non-coding RNA nc886 provokes oncogenes during human esophageal tumorigenesis. Oncotarget 2014, 5, 3472–3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [Green Version]
- McAllister, C.S.; Samuel, C.E. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J. Biol. Chem. 2009, 284, 1644–1651. [Google Scholar] [CrossRef] [Green Version]
- McAllister, C.S.; Toth, A.M.; Zhang, P.; Devaux, P.; Cattaneo, R.; Samuel, C.E. Mechanisms of protein kinase PKR-mediated amplification of beta interferon induction by C protein-deficient measles virus. J. Virol. 2010, 84, 380–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Kunkeaw, N.; Jeon, S.H.; Lee, I.; Johnson, B.H.; Kang, G.Y.; Bang, J.Y.; Park, H.S.; Leelayuwat, C.; Lee, Y.S. Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA 2011, 17, 1076–1089. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.H.; Lee, K.; Lee, K.S.; Kunkeaw, N.; Johnson, B.H.; Holthauzen, L.M.; Gong, B.; Leelayuwat, C.; Lee, Y.S. Characterization of the direct physical interaction of nc886, a cellular non-coding RNA, and PKR. FEBS Lett. 2012, 586, 3477–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Magee, W.E.; Griffith, M.J. The liver as a site for interferon production in response to poly I:poly C. Life Sci. II 1972, 11, 1081–1086. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Genin, P.; Algarte, M.; Roof, P.; Lin, R.; Hiscott, J. Regulation of RANTES chemokine gene expression requires cooperativity between NF-kappa B and IFN-regulatory factor transcription factors. J. Immunol. 2000, 164, 5352–5361. [Google Scholar] [CrossRef] [Green Version]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Kim, S.; Matsuo, K.; Suzuki, H.; Suzuki, T.; Sato, K.; Yokochi, T.; Oda, H.; Nakamura, K.; Ida, N.; et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature 2002, 416, 744–749. [Google Scholar] [CrossRef]
- Chow, K.T.; Gale, M., Jr.; Loo, Y.M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- Lee, Y.S. A Novel Type of Non-coding RNA, nc886, Implicated in Tumor Sensing and Suppression. Genom. Inform. 2015, 13, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Lee, H.S.; Lee, J.S.; Lee, Y.S.; Park, J.L.; Kim, S.Y.; Hwang, J.A.; Kunkeaw, N.; Jung, S.Y.; Kim, T.J.; et al. nc886 is induced by TGF-beta and suppresses the microRNA pathway in ovarian cancer. Nat. Commun. 2018, 9, 1166. [Google Scholar] [CrossRef]
- Nandy, C.; Mrazek, J.; Stoiber, H.; Grasser, F.A.; Huttenhofer, A.; Polacek, N. Epstein-barr virus-induced expression of a novel human vault RNA. J. Mol. Biol. 2009, 388, 776–784. [Google Scholar] [CrossRef]
- Park, J.L.; Lee, Y.S.; Kunkeaw, N.; Kim, S.Y.; Kim, I.H.; Lee, Y.S. Epigenetic regulation of noncoding RNA transcription by mammalian RNA polymerase III. Epigenomics 2017, 9, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Silver, M.J.; Kessler, N.J.; Hennig, B.J.; Dominguez-Salas, P.; Laritsky, E.; Baker, M.S.; Coarfa, C.; Hernandez-Vargas, H.; Castelino, J.M.; Routledge, M.N.; et al. Independent genomewide screens identify the tumor suppressor VTRNA2-1 as a human epiallele responsive to periconceptional environment. Genome Biol. 2015, 16, 118. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kunkeaw, N.; Lee, Y.S. Protein kinase R and its cellular regulators in cancer: An active player or a surveillant? Wiley Interdiscip Rev RNA 2020, 11, e1558. [Google Scholar] [CrossRef]
- Arico, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers (Basel) 2019, 11, 1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.K.; Hong, S.H.; Shin, S.; Lee, H.S.; Lee, J.S.; Park, E.J.; Choi, S.S.; Min, J.W.; Park, D.; Hwang, J.A.; et al. nc886, a non-coding RNA and suppressor of PKR, exerts an oncogenic function in thyroid cancer. Oncotarget 2016, 7, 75000–75012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueba, O. Respiratory syncytial virus. I. Concentration and purification of the infectious virus. Acta Med. Okayama 1978, 32, 265–272. [Google Scholar]
- Wang, Q.; Lee, I.; Ren, J.; Ajay, S.S.; Lee, Y.S.; Bao, X. Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol. Ther. 2013, 21, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Contursi, C.; Wang, I.M.; Gabriele, L.; Gadina, M.; O’Shea, J.; Morse, H.C.; Ozato, K. IFN consensus sequence binding protein potentiates STAT1-dependent activation of IFNgamma-responsive promoters in macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.J.; Ren, Y.; Chen, Y.; Liu, S.; Wu, W.; Ren, J.; Wang, P.; Garofalo, R.P.; Zhou, J.; Bao, X. Exchange Proteins Directly Activated by cAMP and Their Roles in Respiratory Syncytial Virus Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Wang, Q.; Kolli, D.; Prusak, D.J.; Tseng, C.T.; Chen, Z.J.; Li, K.; Wood, T.G.; Bao, X. Human metapneumovirus M2-2 protein inhibits innate cellular signaling by targeting MAVS. J. Virol. 2012, 86, 13049–13061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkeaw, N.; Lee, Y.S.; Im, W.R.; Jang, J.J.; Song, M.J.; Yang, B.; Park, J.L.; Kim, S.Y.; Ku, Y.; Kim, Y.; et al. Mechanism mediated by a noncoding RNA, nc886, in the cytotoxicity of a DNA-reactive compound. Proc. Natl. Acad. Sci. USA 2019, 116, 8289–8294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casola, A.; Burger, N.; Liu, T.; Jamaluddin, M.; Brasier, A.R.; Garofalo, R.P. Oxidant tone regulates RANTES gene expression in airway epithelial cells infected with respiratory syncytial virus. Role in viral-induced interferon regulatory factor activation. J. Biol. Chem. 2001, 276, 19715–19722. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-S.; Bao, X.; Lee, H.-H.; Jang, J.J.; Saruuldalai, E.; Park, G.; Im, W.R.; Park, J.-L.; Kim, S.-Y.; Shin, S.; et al. Nc886, a Novel Suppressor of the Type I Interferon Response Upon Pathogen Intrusion. Int. J. Mol. Sci. 2021, 22, 2003. https://doi.org/10.3390/ijms22042003
Lee Y-S, Bao X, Lee H-H, Jang JJ, Saruuldalai E, Park G, Im WR, Park J-L, Kim S-Y, Shin S, et al. Nc886, a Novel Suppressor of the Type I Interferon Response Upon Pathogen Intrusion. International Journal of Molecular Sciences. 2021; 22(4):2003. https://doi.org/10.3390/ijms22042003
Chicago/Turabian StyleLee, Yeon-Su, Xiaoyong Bao, Hwi-Ho Lee, Jiyoung Joan Jang, Enkhjin Saruuldalai, Gaeul Park, Wonkyun Ronny Im, Jong-Lyul Park, Seon-Young Kim, Sooyong Shin, and et al. 2021. "Nc886, a Novel Suppressor of the Type I Interferon Response Upon Pathogen Intrusion" International Journal of Molecular Sciences 22, no. 4: 2003. https://doi.org/10.3390/ijms22042003
APA StyleLee, Y. -S., Bao, X., Lee, H. -H., Jang, J. J., Saruuldalai, E., Park, G., Im, W. R., Park, J. -L., Kim, S. -Y., Shin, S., Jeon, S. H., Kang, S., Lee, H. -S., Lee, J. -S., Zhang, K., Park, E. J., Kim, I. -H., & Lee, Y. S. (2021). Nc886, a Novel Suppressor of the Type I Interferon Response Upon Pathogen Intrusion. International Journal of Molecular Sciences, 22(4), 2003. https://doi.org/10.3390/ijms22042003