
A Cumulative Effect of Food and Viruses to Trigger Celiac Disease (CD): A Commentary on the Recent Literature


Abstract
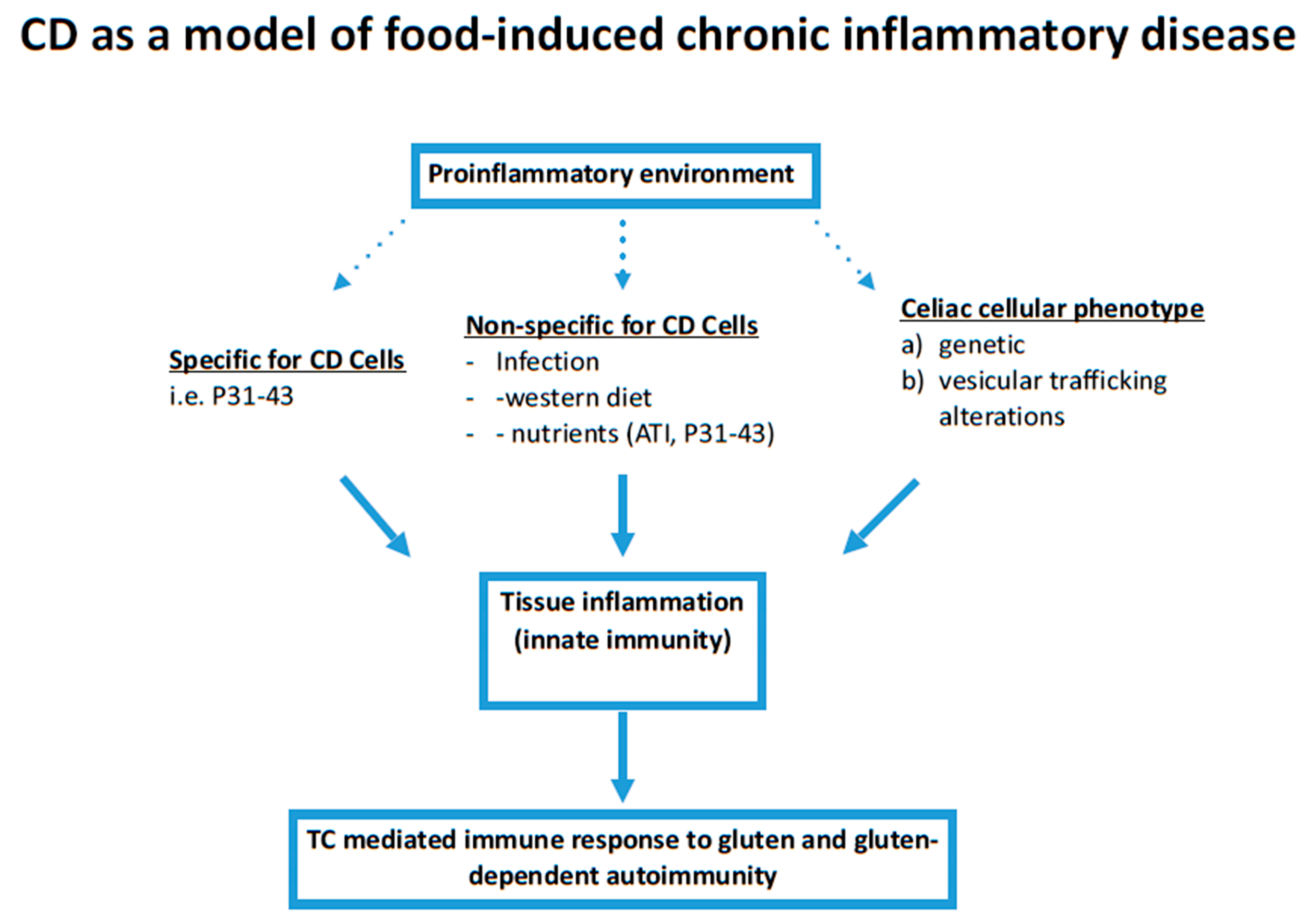
:1. Introduction
2. Results
2.1. Role of Infections in CD
2.2. Nutrients Are Able to Induce and Regulate Tissue Inflammation
2.3. Gliadin and Inflammation in CD
2.4. Cooperative Mechanisms between Gliadin and Infections Inducing Tissue Inflammation
2.5. Protective Role of the Mediterranean Diet in CD
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATI | Alpha-amylase/trypsin inhibitors |
| CD | Celiac Disease |
| GFD-CD | Gluten Free Diet-CD |
| HLA | Human Leukocyte Antigen |
| HRS | Hepatocyte growth factor-regulated tyrosine kinase substrate |
| MAPK | Mitogen-Activated Protein Kinase |
| NFkB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOD | Non-Obese-Diabetic |
| Poli I:C | Polyinosinic-polycytidylic acid |
| TC | T cells |
| TLR | Toll Like Receptor |
References
- Davidson, A.; Diamond, B. Autoimmune diseases. N. Engl. J. Med. 2001, 345, 340–350. [Google Scholar] [CrossRef]
- Amital, H.; Govoni, M.; Maya, R.; Meroni, P.L.; Ori, B.; Shoenfeld, Y.; Tincani, A.; Trotta, F.; Sarzi-Puttini, P.; Atzeni, F. Role of infectious agents in systemic rheumatic diseases. Clin. Exp. Rheumatol. 2008, 26 (Suppl. 48), S27–S32. [Google Scholar] [PubMed]
- Maya, R.; Gershwin, M.E.; Shoenfeld, Y. Hepatitis B virus (HBV) and autoimmune disease. Clin. Rev. Allergy Immunol. 2008, 34, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Tozzoli, R.; Barzilai, O.; Ram, M.; Villalta, D.; Bizzaro, N.; Sherer, Y.; Shoenfeld, Y. Infections and autoimmune thyroid diseases: Parallel detection of antibodies against pathogens with proteomic technology. Autoimmun. Rev. 2008, 8, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Vatanen, T.; Franzosa, E.A.; Schwager, R.; Tripathi, S.; Arthur, T.D.; Vehik, K.; Lernmark, A.; Hagopian, W.A.; Rewers, M.J.; She, J.X.; et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 2018, 562, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Marijon, E.; Mirabel, M.; Celermajer, D.S.; Jouven, X. Rheumatic heart disease. Lancet 2012, 379, 953–964. [Google Scholar] [CrossRef]
- Sollid, L.M. Molecular basis of celiac disease. Annu. Rev. Immunol. 2000, 18, 53–81. [Google Scholar] [CrossRef] [Green Version]
- Sollid, L.M.; Jabri, B. Triggers and drivers of autoimmunity: Lessons from coeliac disease. Nat. Rev. Immunol. 2013, 13, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemppainen, K.M.; Lynch, K.F.; Liu, E.; Lonnrot, M.; Simell, V.; Briese, T.; Koletzko, S.; Hagopian, W.; Rewers, M.; She, J.X.; et al. Factors That Increase Risk of Celiac Disease Autoimmunity After a Gastrointestinal Infection in Early Life. Clin. Gastroenterol. Hepatol. 2017, 15, 694–702 e5. [Google Scholar] [CrossRef] [Green Version]
- Marild, K.; Kahrs, C.R.; Tapia, G.; Stene, L.C.; Stordal, K. Infections and risk of celiac disease in childhood: A prospective nationwide cohort study. Am. J. Gastroenterol. 2015, 110, 1475–1484. [Google Scholar] [CrossRef]
- Hemming-Harlo, M.; Lahdeaho, M.L.; Maki, M.; Vesikari, T. Rotavirus Vaccination Does Not Increase Type 1 Diabetes and May Decrease Celiac Disease in Children and Adolescents. Pediatr. Infect. Dis. J. 2019, 38, 539–541. [Google Scholar] [CrossRef]
- Kahrs, C.R.; Chuda, K.; Tapia, G.; Stene, L.C.; Mårild, K.; Rasmussen, T.; Rønningen, K.S.; Lundin, K.E.A.; Kramna, L.; Cinek, O.; et al. Enterovirus as trigger of coeliac disease: Nested case-control study within prospective birth cohort. BMJ 2019, 364, l231. [Google Scholar] [CrossRef] [Green Version]
- Caminero, A.; Verdu, E.F. Celiac disease: Should we care about microbes? Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G161–G170. [Google Scholar] [CrossRef]
- Kagnoff, M.F.; Paterson, Y.J.; Kumar, P.J.; Kasarda, D.D.; Carbone, F.R.; Unsworth, D.J.; Austin, R.K. Evidence for the role of a human intestinal adenovirus in the pathogenesis of coeliac disease. Gut 1987, 28, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Lahdeaho, M.L.; Lehtinen, M.; Rissa, H.R.; Hyoty, H.; Reunala, T.; Maki, M. Antipeptide antibodies to adenovirus E1b protein indicate enhanced risk of celiac disease and dermatitis herpetiformis. Int. Arch. Allergy Immunol. 1993, 101, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Bouziat, R.; Biering, S.B.; Kouame, E.; Sangani, K.A.; Kang, S.; Ernest, J.D.; Varma, M.; Brown, J.J.; Urbanek, K.; Dermody, T.S.; et al. Murine Norovirus Infection Induces TH1 Inflammatory Responses to Dietary Antigens. Cell Host Microbe 2018, 24, 677–688 e5. [Google Scholar] [CrossRef] [Green Version]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Azimi, T.; Nasser, A.; Shariati, A.; Shiadeh, S.M.J.; Safari, H.; Alizade-Sani, M.; Taghipour, A.; Dehghan, A. The Possible Role of Pathogenic and Non-Pathogenic Bacteria in Initiation and Exacerbation of Celiac Disease; A Comprehensive Review. Curr. Pharm. Biotechnol. 2020, 21, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Riddle, M.S.; Joseph, A. Murray, Brooks D Cash, Mark Pimentel, Chad K Porter Pathogen-specific risk of celiac disease following bacterial causes of foodborne illness: A retrospective cohort study. Dig. Dis. Sci. 2013, 58, 3242–3245. [Google Scholar] [CrossRef] [PubMed]
- Dore, M.P.; Salis, R.; Loria, M.F.; Villanacci, V.; Bassotti, G.; Pes, G.P. Helicobacter pylori infection and occurrence of celiac disease in subjects HLA-DQ2/DQ8 positive: A prospective study. Helicobacter 2018, 23, e12465. [Google Scholar] [CrossRef]
- Petersen, I.; Ciacchi, L.; Tran, M.T.; Loh, K.L.; Kooy-Winkelaar, Y.; Croft, N.P.; Hardy, M.Y.; Chen, Z.; McCluskey, J.; Anderson, R.P.; et al. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat. Struct. Mol. Biol. 2020, 27, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Labruna, G.; Nanayakkara, M.; Pagliuca, C.; Nunziato, M.; Iaffaldano, L.; D’Argenio, V.; Colicchio, R.; Budelli, A.L.; Nigro, R.; Salvatore, P.; et al. Celiac disease-associated Neisseria flavescens decreases mitochondrial respiration in CaCo-2 epithelial cells: Impact of Lactobacillus paracasei CBA L74 on bacterial-induced cellular imbalance. Cell Microbiol. 2019, 21, e13035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R. Food, immunity, and the microbiome. Gastroenterology 2015, 148, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Moszak, M.; Szulińska, M.; Bogdański, P. You Are What You Eat-The Relationship between Diet, Microbiota, and Metabolic Disorders-A Review. Nutrients 2020, 12, 1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirdo, F.A.; Auricchio, S.; Troncone, R.; Barone, M.V. The gliadin p31–43 peptide: Inducer of multiple proinflammatory effects. Int. Rev. Cell Mol. Biol. 2020, in press. [Google Scholar]
- Shan, L.; Molberg, O.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef] [Green Version]
- Mamone, G.; Ferranti, P.; Rossi, M.; Roepstorff, P.; Fierro, O.; Malorni, A.; Addeo, F. Identification of a peptide from alpha-gliadin resistant to digestive enzymes: Implications for celiac disease. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 855, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Comino, I.; Real, A.; Vivas, S.; Siglez, M.A.; Caminero, A.; Nistal, E.; Casqueiro, J.; Rodriguez-Herrera, A.; Cebolla, A.; Sousa, C. Monitoring of gluten-free diet compliance in celiac patients by assessment of gliadin 33-mer equivalent epitopes in feces. Am. J. Clin. Nutr. 2012, 95, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, K.P.; Fischer, I.; Mothes, T.; Weissen-Plenz, G.; Schmitz, M.; Wieser, H.; Buning, J.; Lerch, M.M.; Ciclitira, P.C.; Weber, P.; et al. Endocytotic segregation of gliadin peptide 31-49 in enterocytes. Gut 2010, 59, 300–310. [Google Scholar] [CrossRef]
- Nanayakkara, M.; Lania, G.; Maglio, M.; Auricchio, R.; De Musis, C.; Discepolo, V.; Miele, E.; Jabri, B.; Troncone, R.; Auricchio, S.; et al. P31-43, an undigested gliadin peptide, mimics and enhances the innate immune response to viruses and interferes with endocytic trafficking: A role in celiac disease. Sci. Rep. 2018, 8, 10821. [Google Scholar] [CrossRef] [Green Version]
- Araya, R.E.; Gomez Castro, M.F.; Carasi, P.; McCarville, J.L.; Jury, J.; Mowat, A.M.; Verdu, E.F.; Chirdo, F.G. Mechanisms of innate immune activation by gluten peptide p31-43 in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G40–G49. [Google Scholar] [CrossRef]
- Dotsenko, V.; Oittinen, M.; Taavela, J.; Popp, A.; Peräaho, M.; Staff, S.; Sarin, J.; Leon, F.; Isola, J.; Mäki, M.; et al. Genome-Wide Transcriptomic Analysis of Intestinal Mucosa in Celiac Dis- ease Patients on a Gluten-Free Diet and Postgluten Challenge. Cell. Mol. Gastroenterol. Hepatol. 2020, 11, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Stamnaes, J.; Stray, D.; Stensland, M.; Sarna, V.S.; Nyman, T.A.; Lundin, K.E.A.; Sollid, M.L. Quantitative proteomics of coeliac gut during 14-day gluten challenge: Low-level baseline inflammation despite clinical and histological normality predicts subsequent response. medRxiv 2020. [Google Scholar] [CrossRef]
- Junker, Y.; Zeissig, S.; Kim, S.J.; Barisani, D.; Wieser, H.; Leffler, D.A.; Zevallos, V.; Libermann, T.A.; Dillon, S.; Freitag, T.L.; et al. Wheat amylase trypsin inhibitors drive intestinal inflammation via activation of toll-like receptor 4. J. Exp. Med. 2012, 209, 2395–2408. [Google Scholar] [CrossRef]
- Caminero, A.; McCarville, J.L.; Zevallos, V.F.; Pigrau, M.; Yu, X.B.; Jury, J.; Galipeau, H.J.; Clarizio, A.V.; Casqueiro, J.; Murray, J.A.; et al. Lactobacilli Degrade Wheat Amylase Trypsin Inhibitors to Reduce Intestinal Dysfunction Induced by Immunogenic Wheat Proteins. Gastroenterology 2019, 156, 2266–2280. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.F.; Lebwohl, B. Three papers indicate that amount of gluten play a role for celiac disease—But only a minor role. Acta Paediatr. 2020, 109, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Lindfors, K.; Lin, J.; Lee, H.S.; Hyoty, H.; Nykter, M.; Kurppa, K.; Liu, E.; Koletzko, S.; Rewers, M.; Hagopian, W.; et al. Metagenomics of the faecal virome indicate a cumulative effect of enterovirus and gluten amount on the risk of coeliac disease autoimmunity in genetically at risk children: The TEDDY study. Gut 2020, 69, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
- Araya, R.E.; Jury, J.; Bondar, C.; Verdu, E.F.; Chirdo, F.G. Intraluminal administration of poly I:C causes an enteropathy that is exacerbated by administration of oral dietary antigen. PLoS ONE 2014, 9, e99236. [Google Scholar] [CrossRef]
- Barroso, M.; Beth, S.A.; Voortman, T.; Jaddoe, V.W.V.; van Zelm, M.C.; Moll, H.A.; Kiefte-de Jong, J.C. Dietary Patterns After the Weaning and Lactation Period Are Associated With Celiac Disease Autoimmunity in Children. Gastroenterology 2018, 154, 2087–2096 e7. [Google Scholar] [CrossRef] [Green Version]
- Vrdoljak, J.; Vilovic, M.; Živkovic, P.M.; Hadjina, I.T.; Rušic, D.; Bukic, J.; Borovac, J.A.; Božić, J. Mediterranean Diet Adherence and Dietary Attitudes in Patients with Inflammatory Bowel Disease. Nutrients 2020, 12, 3429. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Arora, A.; Strand, T.A.; Leffler, D.A.; Catassi, C.; Green, P.H.; Kelly, C.P.; Ahuja, V.; Makharia, G.K. Global Prevalence of Celiac Disease: Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 823–836 e2. [Google Scholar] [CrossRef] [Green Version]
- King, J.A.; Jeong, J.; Underwood, F.E.; Quan, J.; Panaccione, N.; Windsor, J.W.; Coward, S.; deBruyn, J.; Ronksley, P.E.; Shaheen, A.A.; et al. Incidence of Celiac Disease Is Increasing Over Time: A Systematic Review and Meta-analysis. Am. J. Gastroenterol. 2020, 115, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Lania, G.; Nanayakkara, M.; Maglio, M.; Auricchio, R.; Porpora, M.; Conte, M.; De Matteis, M.A.; Rizzo, R.; Luini, A.; Discepolo, V.; et al. Constitutive alterations in vesicular trafficking increase the sensitivity of cells from celiac disease patients to gliadin. Commun. Biol. 2019, 2, 190. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, W.; Neurath, M.F.; Zopf, Y. Intestinal ex vivo organoid culture reveals altered programmed crypt stem cells in patients with celiac disease. Sci. Rep. 2020, 10, 3535. [Google Scholar] [CrossRef] [Green Version]
- Barone, M.V.; Troncone, R.; Auricchio, S. Gliadin peptides as triggers of the proliferative and stress/innate immune response of the celiac small intestinal mucosa. Int. J. Mol. Sci. 2014, 15, 20518–20537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Jimenez, N.; Castellanos-Rubio, A.; Plaza-Izurieta, L.; Irastorza, I.; Elcoroaristizabal, X.; Jauregi-Miguel, A.; Lopez-Euba, T.; Tutau, C.; de Pancorbo, M.M.; Vitoria, J.C.; et al. Coregulation and modulation of NFkappaB-related genes in celiac disease: Uncovered aspects of gut mucosal inflammation. Hum. Mol. Genet. 2014, 23, 1298–1310. [Google Scholar] [CrossRef]
- Trynka, G.; Zhernakova, A.; Romanos, J.; Franke, L.; Hunt, K.A.; Turner, G.; Bruinenberg, M.; Heap, G.A.; Platteel, M.; Ryan, A.W.; et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut 2009, 58, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-kappaB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef] [Green Version]
- Majer, O.; Liu, B.; Barton, G.M. Nucleic acid-sensing TLRs: Trafficking and regulation. Curr. Opin. Immunol. 2017, 44, 26–33. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Pathways | HRS Silencing * | P31–43 * and ** | Loxorubine * | ATIs | Poly I:C *** |
|---|---|---|---|---|---|
| Delay of vesicular trafficking (EEA1, LAMP2, EGFR) | YES | YES | YES | Not tested | Not tested |
| Activation of innate immunity (IL15, IL15Ralpha) | YES | YES | YES | Yes | YES |
| Activation of inflammatory markers (NFkB and MAPK) | YES | YES | YES | Yes | YES |
| TLRs activation (MXA, Mydd 88, INF-alpha) | YES | YES | YES | Yes | YES |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barone, M.V.; Auricchio, S. A Cumulative Effect of Food and Viruses to Trigger Celiac Disease (CD): A Commentary on the Recent Literature. Int. J. Mol. Sci. 2021, 22, 2027. https://doi.org/10.3390/ijms22042027
Barone MV, Auricchio S. A Cumulative Effect of Food and Viruses to Trigger Celiac Disease (CD): A Commentary on the Recent Literature. International Journal of Molecular Sciences. 2021; 22(4):2027. https://doi.org/10.3390/ijms22042027
Chicago/Turabian StyleBarone, Maria Vittoria, and Salvatore Auricchio. 2021. "A Cumulative Effect of Food and Viruses to Trigger Celiac Disease (CD): A Commentary on the Recent Literature" International Journal of Molecular Sciences 22, no. 4: 2027. https://doi.org/10.3390/ijms22042027
APA StyleBarone, M. V., & Auricchio, S. (2021). A Cumulative Effect of Food and Viruses to Trigger Celiac Disease (CD): A Commentary on the Recent Literature. International Journal of Molecular Sciences, 22(4), 2027. https://doi.org/10.3390/ijms22042027