
Statin-Induced Myopathy: Translational Studies from Preclinical to Clinical Evidence

 , , and
, , and 
Abstract
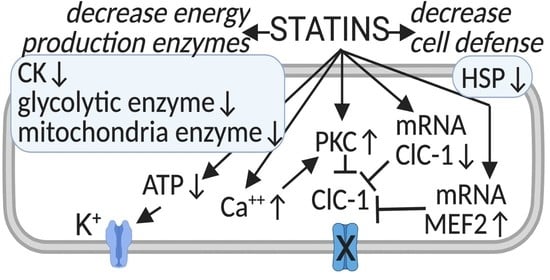
:
1. Introduction
2. The Causes of Statin-Induced Myopathy
3. Ion Channels as Biomarkers of Statin-Induced Muscle Symptoms of Myopathy
4. Translational Studies: Ion Channel Function and Statin-Induced Myopathy in Patients
5. Higher Risk with Statin Therapy: Role of Comorbidity, Genetic and Drug Interactions
6. Management of the Risk of Myopathy with Statin Therapy
7. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Endo, A.; Kuroda, M.; Tanzawa, K. Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme a reductase by ML-236A and ML-236B fungal metabolites, having hypocholesterolemic activity. FEBS Lett. 1976, 72, 323–326. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.; Chan, D.C.; Watts, G.F. The Knowns and Unknowns of Contemporary Statin Therapy for Familial Hypercholesterolemia. Curr. Atheroscler. Rep. 2020, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cignarella, A.; Bellosta, S.; Corsini, A.; Bolego, C. Hypolipidemic therapy for the metabolic syndrome. Pharmacol. Res. 2006, 53, 492–500. [Google Scholar] [CrossRef]
- Weitz-Schmidt, G. Statins as anti-inflammatory agents. Trends Pharmacol. Sci. 2002, 23, 482–487. [Google Scholar] [CrossRef]
- Jeong, G.H.; Lee, K.H.; Kim, J.Y.; Eisenhut, M.; Kronbichler, A.; Van Der Vliet, H.J.; Shin, J.I.; Gamerith, G. Statin and Cancer Mortality and Survival: An Umbrella Systematic Review and Meta-Analysis. J. Clin. Med. 2020, 9, 326. [Google Scholar] [CrossRef] [Green Version]
- Schönbeck, U.; Libby, P. Inflammation, immunity, and HMG-CoA reductase inhibitors: Statins as antiinflammatory agents? Circulation 2004, 109 (Suppl. 1), II18–II26. [Google Scholar] [CrossRef] [Green Version]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Urbach, D.; Awiszus, F.; Leiß, S.; Venton, T.; De Specht, A.V.; Apfelbacher, C. Associations of Medications With Lower Odds of Typical COVID-19 Symptoms: Cross-Sectional Symptom Surveillance Study. JMIR Public Health Surveill. 2020, 6, e22521. [Google Scholar] [CrossRef]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgözoğlu, L.; Nordestgaard, B.G.; et al. Statin-associated muscle symptoms: Impact on statin therapy—European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Rizzo, M.; Toth, P.P.; Farnier, M.; Davidson, M.H.; Al-Rasadi, K.; Aronow, W.S.; Athyros, V.; Djuric, D.M.; Ezhov, M.V.; et al. Statin intolerance-an attempt at a unified definition. Position paper from an international lipid expert panel. Expert Opin. Drug Saf. 2015, 14, 935–955. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.A.; Gomes, T.; Camacho, X.; Juurlink, D.N.; Shah, B.R.; Mamdani, M.M. Risk of incident diabetes among patients treated with statins: Population based study. BMJ 2013, 346, f2610. [Google Scholar] [CrossRef] [Green Version]
- Brault, M.; Ray, J.; Gomez, Y.-H.; Mantzoros, C.S.; Daskalopoulou, S.S. Statin treatment and new-onset diabetes: A review of proposed mechanisms. Metabolism 2014, 63, 735–745. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Pierno, S.; Camerino, D.C. Electrical properties of diaphragm and EDL muscles during the life of dystrophic mice. Am. J. Physiol. Physiol. 1997, 272, C333–C340. [Google Scholar] [CrossRef]
- Furberg, C.D.; Pitt, B. Withdrawal of cerivastatin from the world market. Curr. Control. Trials Cardiovasc. Med. 2001, 2, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Plutzky, J.; Skentzos, S.; Morrison, F.; Mar, P.; Shubina, M.M.; Turchin, A. Discontinuation of statins in routine care settings: A cohort study. Ann. Intern. Med. 2013, 158, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Newman, C.B.; Preiss, D.; Tobert, J.A.; Jacobson, T.A.; Page, R.L.; Goldstein, L.B.; Chin, C.; Tannock, L.R.; Miller, M.; Raghuveer, G.; et al. Statin Safety and Associated Adverse Events: A Scientific Statement From the American Heart Association. Arter. Thromb. Vasc. Biol. 2019, 39, e38–e81. [Google Scholar] [CrossRef] [Green Version]
- Bruckert, E.; Hayem, G.; Dejager, S.; Yau, C.; Bégaud, B. Mild to Moderate Muscular Symptoms with High-Dosage Statin Therapy in Hyperlipidemic Patients—The PRIMO Study. Cardiovasc. Drugs Ther. 2005, 19, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Noyes, A.M.; Thompson, P.D. The effects of statins on exercise and physical activity. J. Clin. Lipidol. 2017, 11, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, D.; Banach, M.; Chianetta, R.; Luzzu, L.M.; Stoian, A.P.; Diaconu, C.C.; Citarrella, R.; Montalto, G.; Rizzo, M. An overview of statin-induced myopathy and perspectives for the future. Expert Opin. Drug Saf. 2020, 19, 601–615. [Google Scholar] [CrossRef]
- Rosenson, R.S. Current overview of statin-induced myopathy. Am. J. Med. 2004, 116, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.J.; Gotto, A.M., Jr. Update on statins. Circulation 2004, 110, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Reidenberg, M.M. Statins, lack of energy and ubiquinone. Br. J. Clin. Pharmacol. 2005, 59, 606–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, P.O.; Lerman, L.O.; Napoli, C.; Lerman, A. Statin effects beyond lipid lowering—Are they clinically relevant? Eur. Heart J. 2003, 24, 225–248. [Google Scholar] [CrossRef] [Green Version]
- Moosmann, B.; Behl, C. Selenoprotein synthesis and side-effects of statins. Lancet 2004, 363, 892–894. [Google Scholar] [CrossRef]
- Muntean, D.M.; Thompson, P.D.; Catapano, A.L.; Stasiolek, M.; Fabis, J.; Muntner, P.; Serban, M.-C.; Banach, M. Statin-associated myopathy and the quest for biomarkers: Can we effectively predict statin-associated muscle symptoms? Drug Discov. Today 2017, 22, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Chatzizisis, Y.S.; Koskinas, K.C.; Misirli, G.; Vaklavas, C.; Hatzitolios, A.; Giannoglou, G.D. Risk factors and drug interactions predisposing to statin-induced myopathy: Implications for risk assessment, prevention and treatment. Drug Saf. 2010, 33, 171–187. [Google Scholar] [CrossRef]
- Cao, P.; Hanai, J.; Tanksale, P.; Imamura, S.; Sukhatme, V.P.; Lecker, S.H. Statin-induced muscle damage and atrogin-1 in-duction is the result of a geranylgeranylation defect. FASEB J. 2009, 23, 2844–2854. [Google Scholar] [CrossRef] [Green Version]
- Mallinson, J.E.; Constantin-Teodosiu, D.; Sidaway, J.; Westwood, F.R.; Greenhaff, P.L. Blunted Akt/FOXO signalling and ac-tivation of genes controlling atrophy and fuel use in statin myopathy. J Physiol. 2009, 587, 219–230. [Google Scholar] [CrossRef]
- Copaja, M.; Venegas, D.; Aranguiz, P.; Canales, J.; Vivar, R.; Avalos, Y.; Garcia, L.; Chiong, M.; Olmedo, I.; Catalán, M.; et al. Simvastatin disrupts cytoskeleton and decreases cardiac fibroblast adhesion, migration and viability. Toxicology 2012, 294, 42–49. [Google Scholar] [CrossRef]
- Sakamoto, K.; Wada, I.; Kimura, J. Inhibition of Rab1 GTPase and Endoplasmic Reticulum-to-Golgi Trafficking Underlies Statin’s Toxicity in Rat Skeletal Myofibers. J. Pharmacol. Exp. Ther. 2011, 338, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, L.; Melli, L.; Segatto, M.; Trezza, V.; Campolongo, P.; Jozwiak, A.; Swiezewska, E.; Pucillo, L.P.; Moreno, S.; Fanelli, F.; et al. Effects of myosin heavy chain (MHC) plasticity induced by HMGCoA-reductase inhibition on skeletal muscle functions. FASEB J. 2011, 25, 4037–4047. [Google Scholar] [CrossRef] [Green Version]
- Christopher-Stine, L.; Casciola-Rosen, L.A.; Hong, G.; Chung, T.; Corse, A.M.; Mammen, A.L. A novel autoantibody recog-nizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010, 62, 2757–2766. [Google Scholar] [CrossRef] [Green Version]
- Selva-O’Callaghan, A.; Alvarado-Cardenas, M.; Pinal-Fernández, I.; Trallero-Araguás, E.; Milisenda, J.C.; Martínez, M.A.; Marín, A.; Labrador-Horrillo, M.; Juárez, C.; Grau-Junyent, J.M. Statin-induced myalgia and myositis: An update on patho-genesis and clinical recommendations. Expert Rev. Clin. Immunol. 2018, 14, 215–224. [Google Scholar] [CrossRef]
- Mohassel, P.; Mammen, A.L. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve 2013, 48, 477–483. [Google Scholar] [CrossRef]
- Needham, M.; Fabian, V.; Knezevic, W.; Panegyres, P.; Zilko, P.; Mastaglia, F.L. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul. Disord. 2007, 17, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.D.; Cooper, R.G.; McHugh, N.J.; Chinoy, H. Statin-induced necrotizing myositis-a discrete autoimmune entity within the “statin-induced myopathy spectrum”. Autoimmun. Rev. 2013, 12, 1177–1181. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, S.; Langguth, D.; Hardy, T.A.; Garg, N.; Bundell, C.; Rojana-Udomsart, A.; Dale, R.C.; Robertson, T.; Mammen, A.L.; Reddel, S.W. Clinical course and treatment of anti-HMGCR antibody-associated necrotizing autoimmune myopathy. Neurol. Neuroimmunol. Neuroinflammation 2015, 2, e96. [Google Scholar] [CrossRef] [Green Version]
- The SEARCH Collaborative Group; Link, E.M.; Parish, S.; Armitage, J.M.; Bowman, L.H.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R.A. SLCO1B1Variants and Statin-Induced Myopathy—A Genomewide Study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef]
- Stenzel, W.; Goebel, H.-H.; Aronica, E. Review: Immune-mediated necrotizing myopathies-a heterogeneous group of diseases with specific myopathological features. Neuropathol. Appl. Neurobiol. 2012, 38, 632–646. [Google Scholar] [CrossRef] [Green Version]
- Bernatsky, S.; Joseph, L.; Pineau, C.A.; Bélisle, P.; Boivin, J.F.; Banerjee, D.; Clarke, A.E. Estimating the prevalence of poly-myositis and dermatomyositis from administrative data: Age, sex and regional differences. Ann. Rheum. Dis. 2009, 68, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Grable-Esposito, P.; Katzberg, H.D.; Greenberg, S.A.; Srinivasan, J.; Katz, J.; Amato, A.A. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2009, 41, 185–190. [Google Scholar] [CrossRef]
- Loganathan, P.; Oddis, C.V.; Aggarwal, R. Immune-mediated statin myopathy. Expert Rev. Clin. Immunol. 2015, 12, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Tiniakou, E. Statin-Associated Autoimmune Myopathy: Current Perspectives. Ther. Clin. Risk Manag. 2020, 16, 483–492. [Google Scholar] [CrossRef]
- Pierno, S.; De Luca, A.; Tricarico, D.; Ferrannini, E.; Conte, T.; D’Alò, G.; Camerino, D.C. Experimental Evaluation of the Effects of Pravastatin on Electrophysiological Parameters of Rat Skeletal Muscle. Pharmacol. Toxicol. 1992, 71, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Pierno, S.; Didonna, M.P.; Cippone, V.; De Luca, A.; Pisoni, M.; Frigeri, A.; Nicchia, G.P.; Svelto, M.; Chiesa, G.; Sirtori, C.; et al. Effects of chronic treatment with statins and fenofibrate on rat skeletal muscle: A biochemical, histological and electrophysiological study. Br. J. Pharmacol. 2006, 149, 909–919. [Google Scholar] [CrossRef] [Green Version]
- Pierno, S.; Camerino, G.; Cippone, V.; Rolland, J.-F.; Desaphy, J.-F.; De Luca, A.; Liantonio, A.; Bianco, G.; Kunic, J.; George, A.L.; et al. Statins and fenofibrate affect skeletal muscle chloride conductance in rats by differently impairing ClC-1 channel regulation and expression. Br. J. Pharmacol. 2009, 156, 1206–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camerino, G.M.; Pellegrino, M.A.; Brocca, L.; Digennaro, C.; Camerino, D.C.; Pierno, S.; Bottinelli, R. Statin or fibrate chronic treatment modifies the proteomic profile of rat skeletal muscle. Biochem. Pharmacol. 2011, 81, 1054–1064. [Google Scholar] [CrossRef] [Green Version]
- Camerino, G.M.; Bouchè, M.; De Bellis, M.; Cannone, M.; Liantonio, A.; Musaraj, K.; Romano, R.; Smeriglio, P.; Madaro, L.; Giustino, A.; et al. Protein kinase C theta (PKCθ) modulates the ClC-1 chloride channel activity and skeletal muscle phenotype: A biophysical and gene expression study in mouse models lacking the PKCθ. Pflügers Archiv-Eur. J. Physiol. 2014, 466, 2215–2228. [Google Scholar] [CrossRef]
- Camerino, G.M.; De Bellis, M.; Conte, E.; Liantonio, A.; Musaraj, K.; Cannone, M.; Fonzino, A.; Giustino, A.; De Luca, A.; Romano, R.; et al. Statin-induced myotoxicity is exacerbated by aging: A biophysical and molecular biology study in rats treated with atorvastatin. Toxicol. Appl. Pharmacol. 2016, 306, 36–46. [Google Scholar] [CrossRef]
- Camerino, G.M.; Musumeci, O.; Conte, E.; Musaraj, K.; Fonzino, A.; Barca, E.; Marino, M.; Rodolico, C.; Tricarico, D.; Camerino, C.; et al. Risk of Myopathy in Patients in Therapy with Statins: Identification of Biological Markers in a Pilot Study. Front. Pharmacol. 2017, 8, 500. [Google Scholar] [CrossRef]
- Desaphy, J.-F.; Pierno, S.; De Luca, A.; Didonna, P.; Camerino, D.C.; Conte, D. Different Ability of Clenbuterol and Salbutamol to Block Sodium Channels Predicts Their Therapeutic Use in Muscle Excitability Disorders. Mol. Pharmacol. 2003, 63, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desaphy, J.F.; Gramegna, G.; Altamura, C.; Dinardo, M.M.; Imbrici, P.; George, A.L., Jr.; Modoni, A.; Lomonaco, M.; Conte Camerino, D. Functional characterization of ClC-1 mutations from patients affected by recessive myotonia congenita pre-senting with different clinical phenotypes. Exp. Neurol. 2013, 248, 530–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altamura, C.; Desaphy, J.-F.; Conte, D.; De Luca, A.; Imbrici, P. Skeletal muscle ClC-1 chloride channels in health and diseases. Pflügers Archiv-Eur. J. Physiol. 2020, 472, 961–975. [Google Scholar] [CrossRef]
- Altamura, C.; Mangiatordi, G.F.; Nicolotti, O.; Sahbani, D.; Farinato, A.; Leonetti, F.; Carratù, M.R.; Conte, D.; Desaphy, J.-F.; Imbrici, P. Mapping ligand binding pockets in chloride ClC-1 channels through an integrated in silico and experimental ap-proach using anthracene-9-carboxylic acid and niflumic acid. Br. J. Pharmacol. 2018, 175, 1770–1780. [Google Scholar] [CrossRef] [Green Version]
- Cozzoli, A.; Liantonio, A.; Conte, E.; Cannone, M.; Massari, A.M.; Giustino, A.; Scaramuzzi, A.; Pierno, S.; Mantuano, P.; Capogrosso, R.F.; et al. Angiotensin II modulates mouse skeletal muscle resting conductance to chloride and potassium ions and calcium homeostasis via the AT1 receptor and NADPH oxidase. Am. J. Physiol. Physiol. 2014, 307, C634–C647. [Google Scholar] [CrossRef] [Green Version]
- Pierno, S.; Camerino, G.M.; Cannone, M.; Liantonio, A.; De Bellis, M.; Digennaro, C.; Gramegna, G.; De Luca, A.; Germinario, E.; Danieli-Betto, D.; et al. Paracrine Effects of IGF-1 Overexpression on the Functional Decline Due to Skeletal Muscle Disuse: Molecular and Functional Evaluation in Hindlimb Unloaded MLC/mIgf-1 Transgenic Mice. PLoS ONE 2013, 8, e65167. [Google Scholar] [CrossRef]
- Pedersen, T.H.; Macdonald, W.A.; de Paoli, F.V.; Gurung, I.S.; Nielsen, O.B. Comparison of regulated passive membrane conductance in action potential-firing fast- and slow-twitch muscle. J. Gen. Physiol. 2009, 134, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Sehra, D.; Sehra, S.; Sehra, S.T. Cardiovascular pleiotropic effects of statins and new onset diabetes: Is there a common link: Do we need to evaluate the role of KATP channels? Expert. Opin. Drug. Saf. 2017, 16, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Liantonio, A.; Giannuzzi, V.; Cippone, V.; Camerino, G.M.; Pierno, S.; Camerino, D.C. Fluvastatin and Atorvastatin Affect Calcium Homeostasis of Rat Skeletal Muscle Fibers in Vivo and in Vitro by Impairing the Sarcoplasmic Reticulum/Mitochondria Ca2+-Release System. J. Pharmacol. Exp. Ther. 2007, 321, 626–634. [Google Scholar] [CrossRef] [Green Version]
- Galtier, F.; Mura, T.; De Mauverger, E.R.; Chevassus, H.; Farret, A.; Gagnol, J.-P.; Costa, F.; Dupuy, A.; Petit, P.; Cristol, J.; et al. Effect of a high dose of simvastatin on muscle mitochondrial metabolism and calcium signaling in healthy volunteers. Toxicol. Appl. Pharmacol. 2012, 263, 281–286. [Google Scholar] [CrossRef]
- Conte, E.; Camerino, G.M.; Mele, A.; De Bellis, M.; Pierno, S.; Rana, F.; Fonzino, A.; Caloiero, R.; Rizzi, L.; Bresciani, E.; et al. Growth hormone secretagogues prevent dysregulation of skeletal muscle calcium homeostasis in a rat model of cisplatin-induced cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 386–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirvent, P.; Mercier, J.; Vassort, G.; Lacampagne, A. Simvastatin triggers mitochondria-induced Ca2+ signaling alteration in skeletal muscle. Biochem. Biophys. Res. Commun. 2005, 329, 1067–1075. [Google Scholar] [CrossRef]
- Sirvent, P.; Fabre, O.; Bordenave, S.; Hillaire-Buys, D.; Raynaud De Mauverger, E.; Lacampagne, A.; Mercier, J. Muscle mi-tochondrial metabolism and calcium signaling impairment in patients treated with statins. Toxicol. Appl. Pharmacol. 2012, 259, 263–268. [Google Scholar] [CrossRef]
- Venturi, E.; Lindsay, C.; Lotteau, S.; Yang, Z.; Steer, E.; Witschas, K.; Wilson, A.D.; Wickens, J.R.; Russell, A.J.; Steele, D.; et al. Simvastatin activates single skeletal RyR1 channels but exerts more complex regulation of the cardiac RyR2 isoform. Br. J. Pharmacol. 2018, 175, 938–952. [Google Scholar] [CrossRef] [Green Version]
- Malerba, A.; Roth, F.; Harish, P.; Dhiab, J.; Lu-Nguyen, N.; Cappellari, O.; Jarmin, S.; Mahoudeau, A.; Ythier, V.; Lainé, J.; et al. Pharmacological modulation of the ER stress response ameliorates oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2019, 28, 1694–1708. [Google Scholar] [CrossRef]
- Conte, E.; Bresciani, E.; Rizzi, L.; Cappellari, O.; De Luca, A.; Torsello, A.; Liantonio, A. Cisplatin-Induced Skeletal Muscle Dysfunction: Mechanisms and Counteracting Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 1242. [Google Scholar] [CrossRef] [Green Version]
- Isackson, P.J.; Wang, J.; Zia, M.; Spurgeon, P.; Levesque, A.; Bard, J.; James, S.; Nowak, N.; Lee, T.K.; Vladutiu, G.D. RYR1 and CACNA1S genetic variants identified with statin-associated muscle symptoms. Pharmacogenomics 2018, 19, 1235–1249. [Google Scholar] [CrossRef]
- D’Andrea, M.; Pisaniello, A.; Serra, C.; Senni, M.; Castaldi, L.; Molinaro, M.; Bouche, M. Protein kinase C theta co-operates with calcineurin in the activation of slow muscle genes in cultured myogenic cells. J. Cell. Physiol. 2006, 207, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Olson, E.N. Activation of the MEF2 transcription factor in skeletal muscles from myotonic mice. J. Clin. Investig. 2002, 109, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Crowe, A.; Wang, X.; Zhang, P.; Ding, K.; Li, L.; Yue, W. Regulation of Organic Anion Transporting Polypeptides (OATP) 1B1- and OATP1B3-Mediated Transport: An Updated Review in the Context of OATP-Mediated Drug-Drug Inter-actions. Int. J. Mol. Sci. 2018, 19, 855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierno, S.; De Luca, A.; Beck, C.L.; George, A.L., Jr.; Conte Camerino, D. Aging-associated down-regulation of ClC-1 expression in skeletal muscle: Phenotypic-independent relation to the decrease of chloride conductance. FEBS Lett. 1999, 449, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Zacharek, A.; Chen, J.; Cui, X.; Yang, Y.; Chopp, M. Simvastatin Increases Notch Signaling Activity and Promotes Arteriogenesis After Stroke. Stroke 2009, 40, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Mele, A.; Mantuano, P.; De Bellis, M.; Rana, F.; Sanarica, F.; Conte, E.; Morgese, M.G.; Bove, M.; Rolland, J.-F.; Capogrosso, R.F.; et al. A long-term treatment with taurine prevents cardiac dysfunction in mdx mice. Transl. Res. 2019, 204, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Draeger, A.; Monastyrskaya, K.; Mohaupt, M.; Hoppeler, H.; Savolainen, H.; Allemann, C.; Babiychuk, E.B. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J. Pathol. 2006, 210, 94–102. [Google Scholar] [CrossRef]
- Mohaupt, M.G.; Karas, R.H.; Babiychuk, E.B.; Sanchez-Freire, V.; Monastyrskaya, K.; Iyer, L.; Hoppeler, H.; Breil, F.; Draeger, A. Association between statin-associated myopathy and skeletal muscle damage. Can. Med. Assoc. J. 2009, 181, E11–E18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, G.J.; Baker, S.; Bergeron, J.; Fitchett, D.; Frohlich, J.; Genest, J.; Gupta, M.; Hegele, R.A.; Ng, D.; Pearson, G.J.; et al. Diagnosis, Prevention, and Management of Statin Adverse Effects and Intolerance: Canadian Consensus Working Group Update (2016). Can. J. Cardiol. 2016, 32, S35–S65. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Barberi, L.; Bijlsma, A.Y.; Blaauw, B.; Dyar, K.A.; Milan, G.; Mammucari, C.; Meskers, C.G.; Pallafacchina, G.; Paoli, A.; et al. Signalling pathways regulating muscle mass in ageing skeletal muscle: The role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 2013, 14, 303–323. [Google Scholar] [CrossRef]
- Visser, M.; Simonsick, E.M.; Colbert, L.H.; Brach, J.S.; Rubin, S.M.; Kritchevsky, S.B.; Newman, A.B.; Harris, T.B. For the Health ABC study Type and Intensity of Activity and Risk of Mobility Limitation: The Mediating Role of Muscle Parameters. J. Am. Geriatr. Soc. 2005, 53, 762–770. [Google Scholar] [CrossRef]
- Fraysse, B.; Desaphy, J.F.; Rolland, J.F.; Pierno, S.; Liantonio, A.; Giannuzzi, V.; Camerino, C.; Didonna, M.P.; Cocchi, D.; De Luca, A.; et al. Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and resto-ration by growth hormone. Neurobiol. Dis. 2006, 21, 372–380. [Google Scholar] [CrossRef]
- Conte, E.; Fonzino, A.; Cibelli, A.; De Benedictis, V.; Imbrici, P.; Nicchia, G.P.; Pierno, S.; Camerino, G.M. Changes in Expression and Cellular Localization of Rat Skeletal Muscle ClC-1 Chloride Channel in Relation to Age, Myofiber Phenotype and PKC Modulation. Front. Pharmacol. 2020, 11, 714. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Ouchi, N.; Izumiya, Y.; Bernardo, B.L.; Lebrasseur, N.K.; Walsh, K. Glycolytic fast-twitch muscle fiber restoration counters adverse age-related changes in body composition and metabolism. Aging Cell 2013, 13, 80–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.; Superko, H.R.; Martin, S.S.; Blumenthal, R.S.; Christopher-Stine, L. Genetic and immunologic susceptibility to statin-related myopathy. Atherosclerosis 2015, 240, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A. Statin safety: Lessons from new drug applications for marketed statins. Am. J. Cardiol. 2006, 97(8A), 44C–51C. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, E.; Del Re, M.; Fidilio, L.; Fogli, S.; Danesi, R.; Di Paolo, A. Pharmacogenetic Foundations of Therapeutic Efficacy and Adverse Events of Statins. Int. J. Mol. Sci. 2017, 18, 104. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Bogsrud, M.P.; Molden, E.; Asberg, A.; Mohebi, B.U.; Ose, L.; Retterstøl, K. Exposure of atorvastatin is un-changed but lactone and acid metabolites are increased several-fold in patients with atorvastatin-induced myopathy. Clin. Pharmacol. Ther. 2006, 79, 532–539. [Google Scholar] [CrossRef]
- Pasanen, M.K.; Fredrikson, H.; Neuvonen, P.J.; Niemi, M. Different Effects of SLCO1B1 Polymorphism on the Pharmacokinetics of Atorvastatin and Rosuvastatin. Clin. Pharmacol. Ther. 2007, 82, 726–733. [Google Scholar] [CrossRef]
- Donnelly, L.A.; Doney, A.S.F.; Tavendale, R.; Lang, C.C.; Pearson, E.R.; Colhoun, H.M.; McCarthy, M.I.; Hattersley, A.T.; Morris, A.D.; Palmer, C.N.A. Common Nonsynonymous Substitutions in SLCO1B1 Predispose to Statin Intolerance in Routinely Treated Individuals With Type 2 Diabetes: A Go-DARTS Study. Clin. Pharmacol. Ther. 2010, 89, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Bellosta, S.; Paoletti, R.; Corsini, A. Safety of statins: Focus on clinical pharmacokinetics and drug interactions. Circulation 2004, 109 (Suppl. 1), III50–III57. [Google Scholar] [CrossRef] [Green Version]
- Fallah, A.; Deep, M.; Smallwood, D.; Hughes, P. Life-threatening rhabdomyolysis following the interaction of two commonly prescribed medications. Australas. Med. J. 2013, 6, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A. With your genes? Take one of these, three times a day. Nat. Cell Biol. 2003, 425, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Frudakis, T.N.; Thomas, M.J.; Ginjupalli, S.N.; Handelin, B.; Gabriel, R.; Gomez, H.J. CYP2D6*4 polymorphism is associated with statin-induced muscle effects. Pharmacogenetics Genom. 2007, 17, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Mulder, A.B.; Van Lijf, H.J.; Bon, M.A.; Bergh, F.A.V.D.; Touw, D.J.; Neef, C.; Vermes, I. Association of polymorphism in the cytochrome CYP2D6 and the efficacy and tolerability of simvastatin. Clin. Pharmacol. Ther. 2001, 70, 546–551. [Google Scholar] [CrossRef]
- Ruaño, G.; Windemuth, A.; Wu, A.H.; Kane, J.P.; Malloy, M.J.; Pullinger, C.R.; Kocherla, M.; Bogaard, K.; Gordon, B.R.; Holford, T.R.; et al. Mechanisms of statin-induced myalgia assessed by physiogenomic asso-ciations. Atherosclerosis 2011, 218, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Mangravite, L.M.; Engelhardt, B.E.; Medina, M.W.; Smith, J.D.; Brown, C.D.; Chasman, D.I.; Mecham, B.H.; Howie, B.; Shim, H.; Naidoo, D.; et al. A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nat. Cell Biol. 2013, 502, 377–380. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.D.; Clarkson, P.; Karas, R.H. Statin-Associated Myopathy. JAMA 2003, 289, 1681–1690. [Google Scholar] [CrossRef]
- Marcoff, L.; Thompson, P.D. The role of coenzyme Q10 in statin-associated myopathy: A systematic review. J. Am. Coll. Cardiol. 2007, 49, 2231–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Laghezza, A.; Montanari, R.; Lavecchia, A.; Piemontese, L.; Pochetti, G.; Iacobazzi, V.; Infantino, V.; Capelli, D.; De Bellis, M.; Liantonio, A.; et al. On the metabolically active form of metaglidasen: Improved synthesis and investigation of its peculiar activity on peroxisome proliferator-activated receptors and skeletal muscles. ChemMedChem. 2015, 10, 555–565. [Google Scholar] [CrossRef]
- Fracchiolla, G.; Laghezza, A.; Piemontese, L.; Tortorella, P.; Mazza, F.; Montanari, R.; Pochetti, G.; Lavecchia, A.; Novellino, E.; Pierno, S.; et al. New 2-Aryloxy-3-phenyl-propanoic Acids As Peroxisome Proliferator-Activated Receptors α/γ Dual Agonists with Improved Potency and Reduced Adverse Effects on Skeletal Muscle Function. J. Med. Chem. 2009, 52, 6382–6393. [Google Scholar] [CrossRef]
- Hilton-Jones, D. Statin-related myopathies. Pract. Neurol. 2018, 18, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, B.; Drouot, S.; Barrail-Tran, A.; Taburet, A.M. Drug-drug interactions between HMG-CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin. Pharmacokinet. 2013, 52, 815–831. [Google Scholar] [CrossRef]
- Shitara, Y.; Sugiyama, Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: Drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol. Ther. 2006, 112, 71–105. [Google Scholar] [CrossRef] [PubMed]
- Neřoldová, M.; Stránecký, V.; Hodaňová, K.; Hartmannová, H.; Piherová, L.; Přistoupilová, A.; Mrázová, L.; Vrablík, M.; Adámková, V.; Hubáček, J.A.; et al. Rare variants in known and novel candidate genes predisposing to statin-associated myopathy. Pharmacogenomics 2016, 17, 1405–1414. [Google Scholar] [CrossRef]
- Camerino, G.M.; Fonzino, A.; Conte, E.; De Bellis, M.; Mele, A.; Liantonio, A.; Tricarico, D.; Tarantino, N.; Dobrowolny, G.; Musarò, A.; et al. Elucidating the Contribution of Skeletal Muscle Ion Channels to Amyotrophic Lateral Sclerosis in search of new therapeutic options. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, L.; Pradat, P.-F.; Ludolph, A.C.; Loeffler, J.-P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Waters, C.W.; Varuzhanyan, G.; Talmadge, R.J.; Voss, A.A. Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc. Natl. Acad. Sci. USA 2013, 110, 9160–9165. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, O.; Brady, S.; Rodolico, C.; Ciranni, A.; Montagnese, F.; Aguennouz, M.; Kirk, R.; Allen, E.; Godfrey, R.; Romeo, S.; et al. Recurrent rhabdomyolysis due to muscle β-enolase deficiency: Very rare or underestimated? J. Neurol. 2014, 261, 2424–2428. [Google Scholar] [CrossRef] [PubMed]
- Vladutiu, G.D.; Simmons, Z.; Isackson, P.J.; Tarnopolsky, M.; Peltier, W.L.; Barboi, A.C.; Sripathi, N.; Wortmann, R.L.; Phillips, P.S. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve 2006, 34, 153–162. [Google Scholar] [CrossRef]
- Similä, M.E.; Päivi, M.A.; Piirilä, L. Beneficial Effects of Ketogenic Diet on Phosphofructokinase Deficiency (Glycogen Storage Disease Type VII). Front. Neurol. 2020, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musumeci, O.; Bruno, C.; Mongini, T.; Rodolico, C.; Aguennouz, M.; Barca, E.; Amati, A.; Cassandrini, D.; Serlenga, L.; Vita, G.; et al. Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). Neuromuscul. Disord. 2012, 22, 325–330. [Google Scholar] [CrossRef]
- Comi, G.P.; Fortunato, F.; Lucchiari, S.; Bordoni, A.; Prelle, A.; Jann, S.; Keller, A.; Ciscato, P.; Galbiati, S.; Chiveri, L.; et al. Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Ann. Neurol. 2001, 50, 202–207. [Google Scholar] [CrossRef]
- Filosto, M.; Piccinelli, S.C.; Pichiecchio, A.; Musumeci, O.; Galvagni, A.; Caria, F.; Cassarino, S.G.; Baldelli, E.; Vitale, R.; Padovani, A.; et al. Late and Severe Myopathy in a Patient With Glycogenosis VII Worsened by Cyclosporine and Amiodarone. Front. Neurol. 2019, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Saxena, A.; Singh, V. Guillain-Barre syndrome complicated by acute fatal rhabdomyolysis. Indian J. Crit. Care Med. 2014, 18, 241–243. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, H.; Yang, Y.J.; Dong, Q.T.; Wang, T.J.; Qian, H.Y.; Li, N.; Wang, X.M.; Jin, C. Atorvastatin treatment im-proves the effects of mesenchymal stem cell transplantation on acute myocardial infarction: The role of the RhoA/ROCK/ERK pathway. Int. J. Cardiol. 2014, 176, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Lee, T.-S.; Zhu, M.; Gu, C.; Wang, Y.; Zhu, Y.; Shyy, J.Y.-J. Statins Activate AMP-Activated Protein Kinase In Vitro and In Vivo. Circulation 2006, 114, 2655–2662. [Google Scholar] [CrossRef] [Green Version]
- Sattar, N.; Taskinen, M.R. Statins are diabetogenic—Myth or reality? Atherosclerosis 2012, 13, 1–10. [Google Scholar] [CrossRef]
- Thakker, D.; Nair, S.; Pagada, A.; Jamdade, V.; Malik, A. Statin use and the risk of developing diabetes: A network me-ta-analysis. Pharmacoepidemiol. Drug Saf. 2016, 25, 1131–1149. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Pierno, S.; De Luca, A.; Tricarico, D.; Roselli, A.; Natuzzi, F.; Ferrannini, E.; Laico, M.; Camerino, D.C. Potential risk of myo-pathy by HMG-CoA reductase inhibitors: A comparison of pravastatin and simvastatin effects on membrane electrical prop-erties of rat skeletal muscle fibers. J. Pharmacol. Exp. Ther. 1995, 275, 1490–1496. [Google Scholar] [PubMed]
- Sinzinger, H.; O’Grady, J. Professional athletes suffering from familial hypercholesterolaemia rarely tolerate statin treatment because of muscular problems. Br. J. Clin. Pharmacol. 2004, 57, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, G.; Spatz, E.S.; Jablecki, C.; Phillips, P.S. Statin myopathy: A common dilemma not reflected in clinical trials. Clevel. Clin. J. Med. 2011, 78, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, B.A.; Augeri, A.L.; Capizzi, J.A.; Ballard, K.D.; Troyanos, C.; Baggish, A.L.; D’Hemecourt, P.A.; Thompson, P.D. Effect of Statins on Creatine Kinase Levels Before and After a Marathon Run. Am. J. Cardiol. 2012, 109, 282–287. [Google Scholar] [CrossRef]
- De Luca, A.; Nico, B.; Liantonio, A.; Didonna, M.P.; Fraysse, B.; Pierno, S.; Burdi, R.; Mangieri, M.; Rolland, J.-F.; Camerino, C.; et al. A Multidisciplinary Evaluation of the Effectiveness of Cyclosporine A in Dystrophic Mdx Mice. Am. J. Pathol. 2005, 166, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, N.P.; Kim, M.J.; Bible, K.L.; Adams, M.E.; Froehner, S.C. A new therapeutic effect of simvastatin revealed by functional improvement in muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 12864–12869. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Reisman, J.; Morris-Eppolito, B.; Qian, S.X.; Kazis, L.E.; Wolozin, B.; Goldstein, L.E.; Xia, W. Beneficial association of angiotensin-converting enzyme inhibitors and statins on the occurrence of possible Alzheimer’s disease after traumatic brain injury. Alzheimer’s Res. Ther. 2020, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pasta, A.; Cremonini, A.L.; Formisano, E.; Fresa, R.; Bertolini, S.; Pisciotta, L. Long term follow-up of genetically confirmed patients with familial hypercholesterolemia treated with first and second-generation statins and then with PCSK9 monoclonal antibodies. Atherosclerosis 2020, 308, 6–14. [Google Scholar] [CrossRef]
- Norata, G.D.; Ballantyne, C.M.; Catapano, A.L. New therapeutic principles in dyslipidaemia: Focus on LDL and Lp(a) lowering drugs. Eur. Heart J. 2013, 34, 1783–1789. [Google Scholar] [CrossRef] [Green Version]
- Toth, P.P.; Farnier, M.E.; Tomassini, J.; Foody, J.M.; Tershakovec, A.M. Statin combination therapy and cardiovascular risk reduction. Futur. Cardiol. 2016, 12, 289–315. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.; Sheth, S.; Jacoby, D. A Clinical Guide to Combination Lipid-Lowering Therapy. Curr. Atheroscler. Rep. 2018, 20, 19. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P. Low-Density Lipoprotein Cholesterol Treatment in the Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitor Era: Getting Back on Target. JAMA Cardiol. 2017, 2, 935–936. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardio-vascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Ruscica, M.; Banach, M.; Sahebkar, A.; Corsini, A.; Sirtori, C.R. ETC-1002 (Bempedoic acid) for the management of hyper-lipidemia: From preclinical studies to phase 3 trials. Expert Opin. Pharmacother. 2019, 20, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Macchi, C.; Banach, M.; Corsini, A.; Sirtori, C.R.; Ferri, N.; Ruscica, M. Changes in circulating pro-protein convertase subtil-isin/kexin type 9 levels-experimental and clinical approaches with lipid-lowering agents. Eur. J. Prev. Cardiol. 2019, 26, 930–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raal, F.J.; Hovingh, G.K.; Catapano, A.L. Familial hypercholesterolemia treatments: Guidelines and new therapies. Atherosclerosis 2018, 277, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S.; Baker, S.; Banach, M.; Borow, K.M.; Braun, L.T.; Bruckert, E.; Brunham, L.R.; Catapano, A.L.; Elam, M.B.; Mancini, G.B.J.; et al. Optimizing Cholesterol Treatment in Patients With Muscle Complaints. J. Am. Coll. Cardiol. 2017, 70, 1290–1301. [Google Scholar] [CrossRef]
- Ward, N.; Sahebkar, A.; Banach, M.; Watts, G. Recent perspectives on the role of nutraceuticals as cholesterol-lowering agents. Curr. Opin. Lipidol. 2017, 28, 495–501. [Google Scholar] [CrossRef]



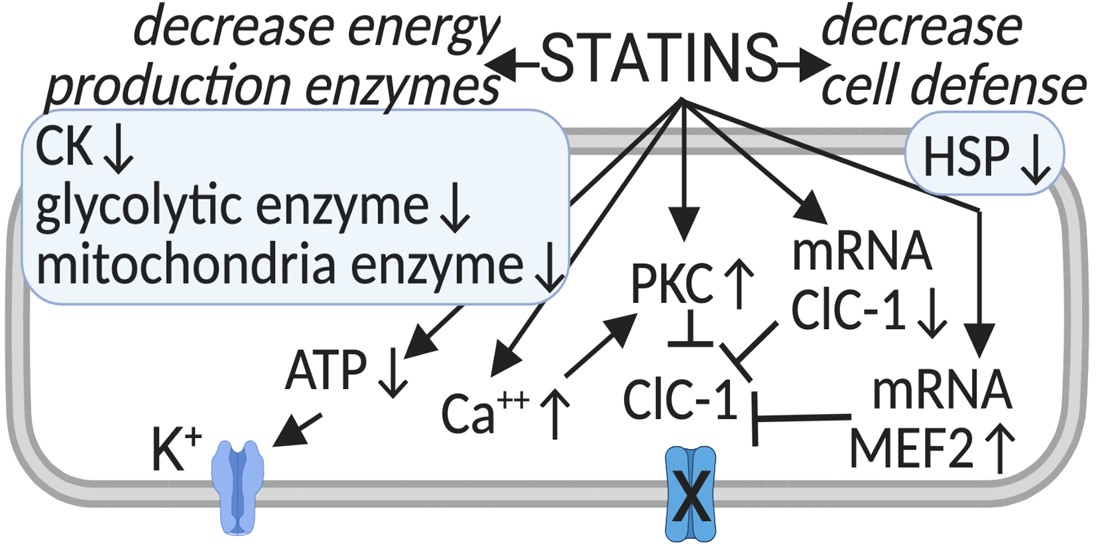{kind=link}
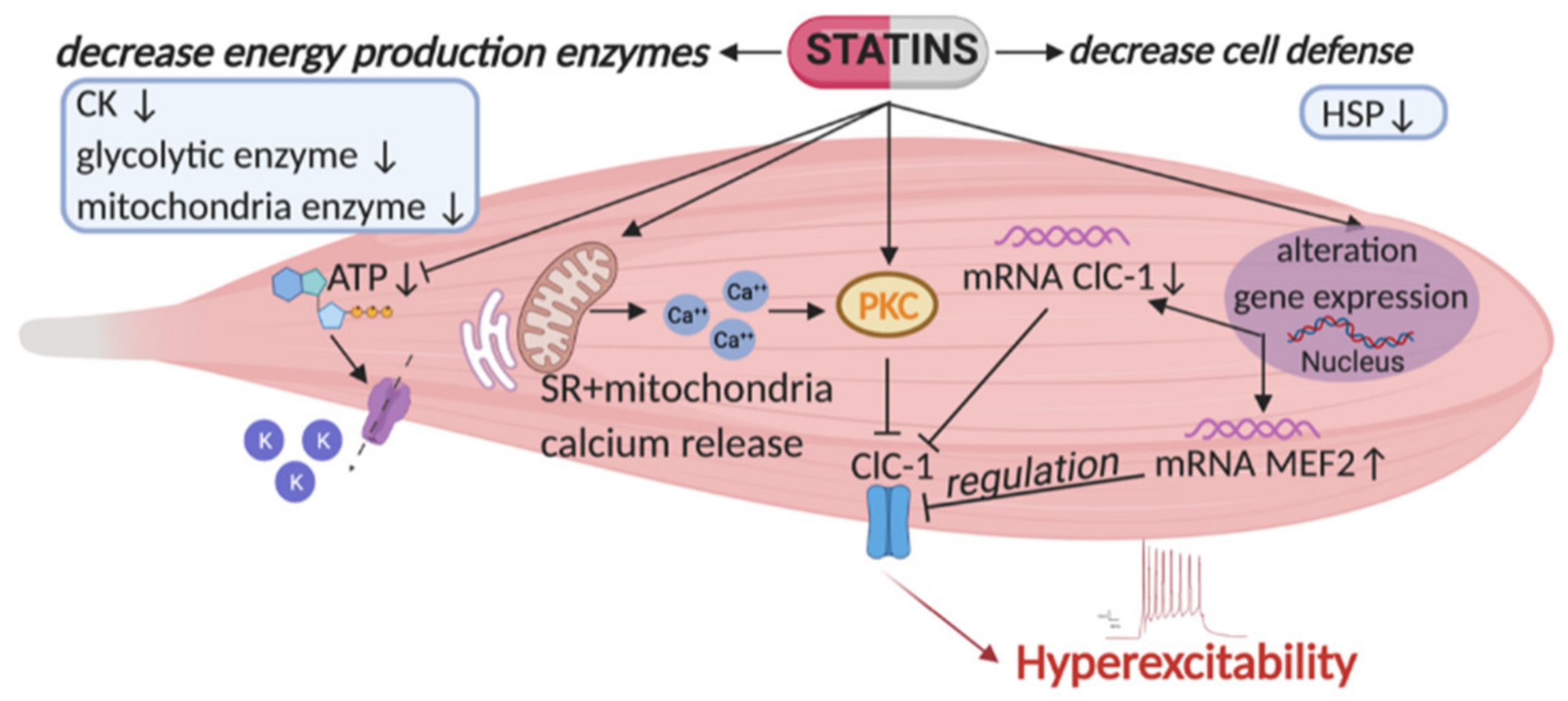{kind=link}
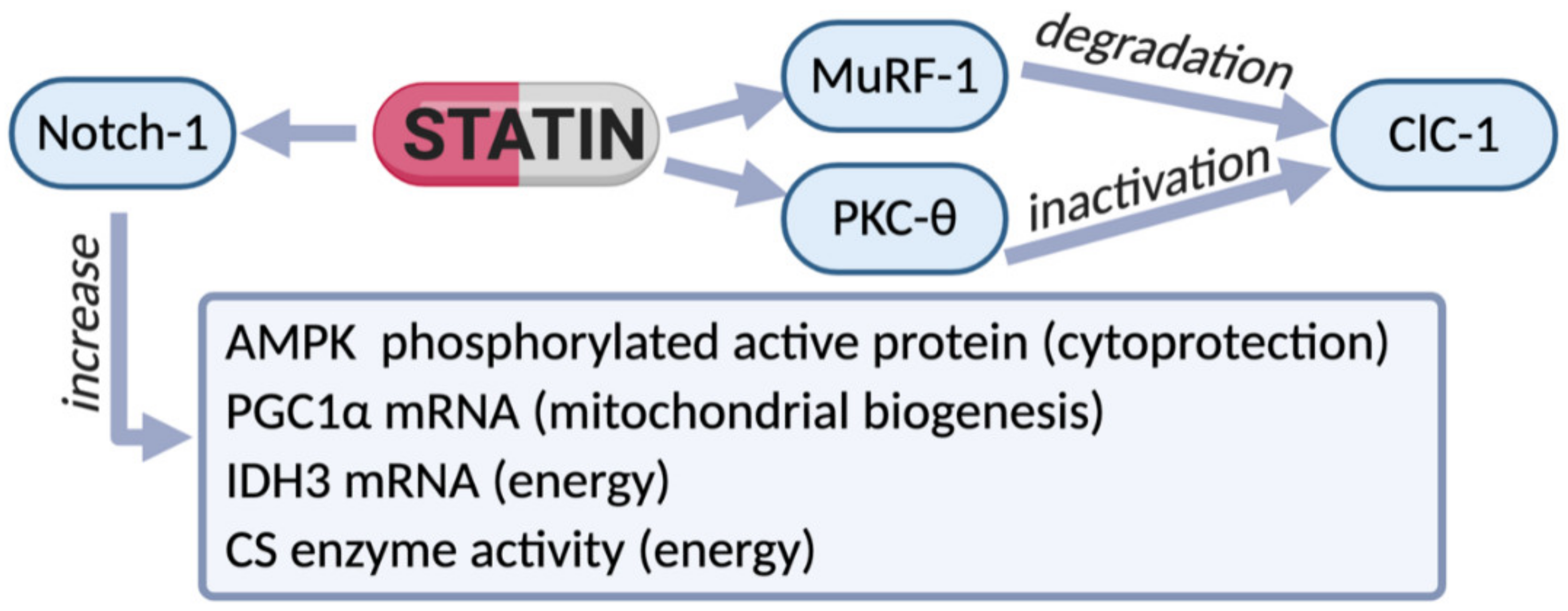{kind=link}
| Baseline Characteristics or Genetic Factors | Risk Factors | References |
|---|---|---|
| Advanced age | Decreased metabolism (reduced activity of cytochrome P450 isoenzymes); | [89] |
| Reduction of muscle chloride channel (ClC-1) activity and expression; | [50,81] | |
| Increased intracellular Ca2+; | [80,60] | |
| Energy production defect; | [96,97] | |
| Lower glomerular filtration rate | [16] | |
| Female gender | Different activity of metabolizing enzymes | [26] |
| Ethnicity and predisposing genetic variants | Polymorphism and genetic variation of metabolizing enzymes (cytochrome P450) and/or of membrane transporters (i.e., SLCO1B1); | [16] [85] [26] |
| Variant in CACNA1H encoding voltage-dependent calcium channel; | [104] | |
| Variant of ClCN-1 gene encoding for ClC1 channel protein; | [104] | |
| Genetic variants within the ryanodine and dihydropiridine receptor genes | [68] | |
| Vitamin D deficiency | Additional reduction of vitamin D synthesis due to cholesterol reduction | [10] |
| Low body mass index | Additional cholesterol decrease | [123] |
| Muscle protein degradation | [98] | |
| Pregnancy | Fetal abnormalities due to cholesterol reduction | [16] |
| Exogenous factors | ||
| Polypharmacy: drugs or food interaction | Drug–drug interaction with inhibitors of cytochrome P450 metabolic isoenzymes (fibrates, immunosuppressant drugs, cardiovascular antiplatelet or anticoagulant drugs, antimicrobials, and antiviral drugs); | [85] [26] |
| Inhibitors of OATP1B1 (gemfibrozil); | [71,123] | |
| High consumption of grapefruit juice (containing furanocoumarins) that inhibits CYP3A4 and increases the plasma concentration of statins | ||
| Strenuous exercise | Amplification of the Creatine Kinase (CK) increase that commonly occurs after strenuous exercise | [123,124] |
| Alcohol abuse | Liver disease (additive transaminases increase) | [120,123] |
| Energy production defect | ||
| Mitochondrial gene defect | ATP synthesis reduction | [96,97] |
| Comorbidity/Pre-existing diseases | ||
| Hypertrigliceridemia | Fibrates administration | [101] |
| Intracerebral hemorrhage | Statin may potentiate the anticoagulant effect of coadministered drugs (i.e., warfarin) | [16] |
| Amyotrophic lateral sclerosis | Skeletal muscle hypermetabolism; | |
| Muscle mitochondria energy defect; | ||
| Decrease of nutrients supply to muscle; | [106] | |
| Cholesterol synthesis inhibition and muscle membrane instability; | ||
| Reduction of muscle chloride channel (ClC-1) activity and expression with hyperexcitability and alteration of contraction | [16] | |
| [105] | ||
| Myotonia congenita | ClC-1 malfunction and skeletal muscle involvement | [53] |
| Huntington disease | ClC-1 malfunction and skeletal muscle involvement | [107] |
| Guillain–Barrè syndrome | Additional elevated CK | [114] |
| Diabetes | GLUT4 mRNA reduction; | |
| Reduced secretion of insulin | [50] | |
| Inhibition of insulin release due to the opening of KATP channels in pancreatic beta cells | [120] [59] | |
| Hypothyroidism | Hypothyroidism can cause hypercholesterolemia and raised serum CK | [17,101] |
| Chronic kidney disease | Renal function impairment with increased drug systemic exposure | [16] |
| Liver disease | Elevated transaminases | [10] |
| Mitochondrial myopathies | Additive mitochondrial function deficit | [123] |
| Glycogen storage disease GSD V (McArdle disease) GSD VII (Tarui’s disease) GSD XIII | Impairment of glucose and glycogen metabolism Potentiation of myalgia, cramps, fatigue Rhabdomyolysis with myoglobinuria (dark urine) Muscle cramps, exercise intolerance and rhabdomyolysis, symptoms that can worsen during statin therapy | [109] [110] [111] [108,112] |
| Lipid Storage myopathies CPT2 deficiency | Impairment of lipid metabolism Increasing of rhadomyolysis episodes | [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camerino, G.M.; Tarantino, N.; Canfora, I.; De Bellis, M.; Musumeci, O.; Pierno, S. Statin-Induced Myopathy: Translational Studies from Preclinical to Clinical Evidence. Int. J. Mol. Sci. 2021, 22, 2070. https://doi.org/10.3390/ijms22042070
Camerino GM, Tarantino N, Canfora I, De Bellis M, Musumeci O, Pierno S. Statin-Induced Myopathy: Translational Studies from Preclinical to Clinical Evidence. International Journal of Molecular Sciences. 2021; 22(4):2070. https://doi.org/10.3390/ijms22042070
Chicago/Turabian StyleCamerino, Giulia Maria, Nancy Tarantino, Ileana Canfora, Michela De Bellis, Olimpia Musumeci, and Sabata Pierno. 2021. "Statin-Induced Myopathy: Translational Studies from Preclinical to Clinical Evidence" International Journal of Molecular Sciences 22, no. 4: 2070. https://doi.org/10.3390/ijms22042070
APA StyleCamerino, G. M., Tarantino, N., Canfora, I., De Bellis, M., Musumeci, O., & Pierno, S. (2021). Statin-Induced Myopathy: Translational Studies from Preclinical to Clinical Evidence. International Journal of Molecular Sciences, 22(4), 2070. https://doi.org/10.3390/ijms22042070