
Ex uno, plures–From One Tissue to Many Cells: A Review of Single-Cell Transcriptomics in Cardiovascular Biology


Abstract
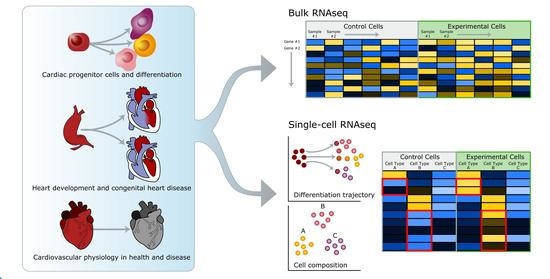
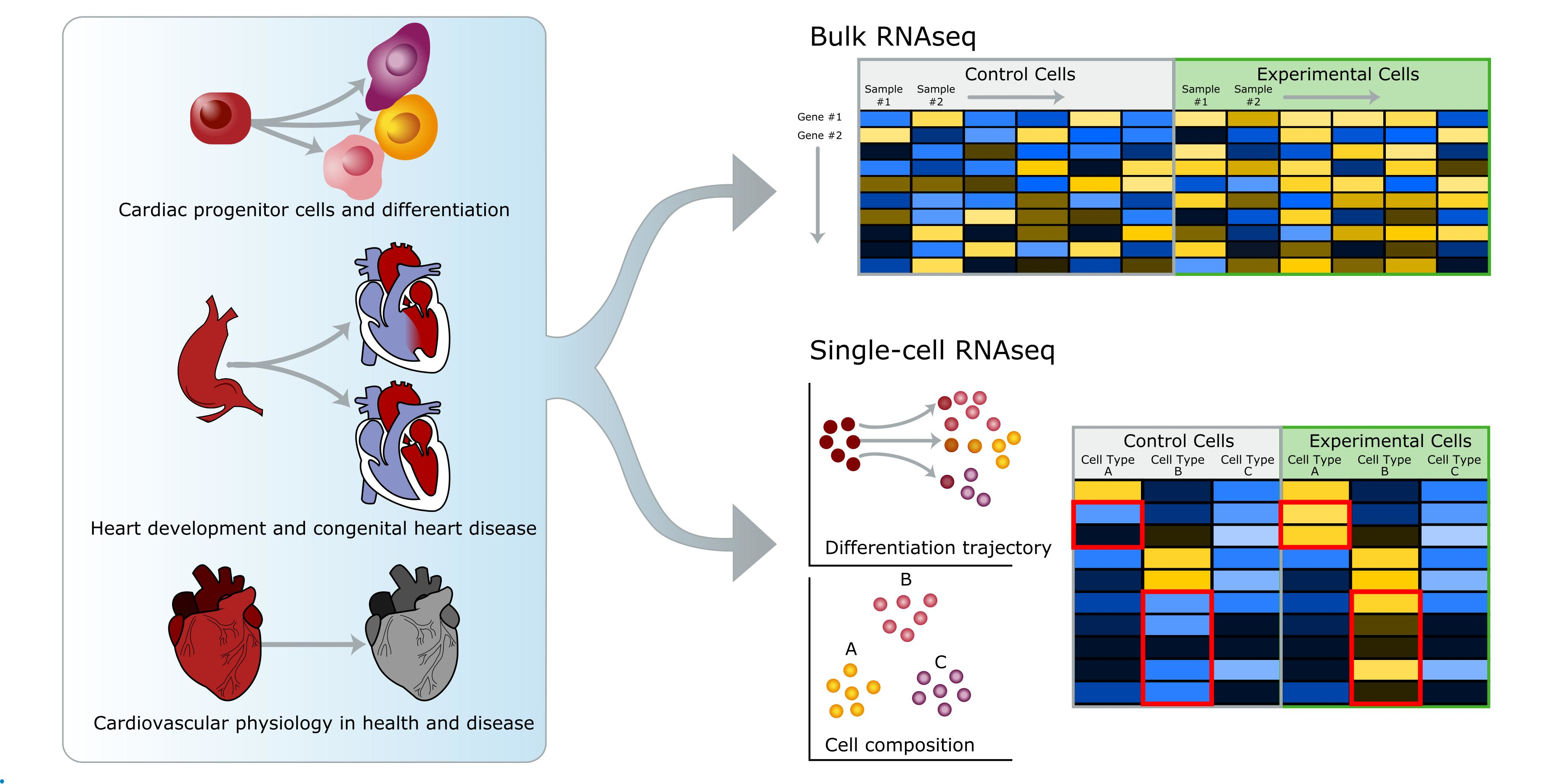
:
1. Introduction
2. Cardiac scRNAseq during Embryonic and Postnatal Development in Physiology and Disease

{kind=link}
{kind=link}
| Authors PMID | Date | # of Cells and/or Nuclei | Isolation Method | Sequencing Technology | Target Cell Types | Context |
|---|---|---|---|---|---|---|
| Li et al. [23] PMID: 25633351 | January 2015 | 448 cells | enzymatic digestion, FACS | Manual cell lysis/cDNA preparation; targeted sc-qPCR Fluidigm Dynamic Array IFCs [6] | CMs, ECs, CFs, SMCs | Comparison of embryo (E10.5)- and mESC-derived cardiac progenitor and CM differentiation |
| Kokkinopoulos et al. [24] PMID: 26469858 | October 2015 | 1088, 12 deep- sequenced | enzymatic digestion | Manual cell lysis/cDNA preparation; targeted Taqman® sc-qPCR; Illumina GA IIx | Cells from the heart forming regions in early mouse embryos | Profile the early FHF cardiac progenitors. Early EB to EHF stage (E6-8) |
| DeLaughter et al. [25] PMID: 27840107 | November 2016 | ≈1200 | enzymatic digestion | Fluidigm C1 IFCs; Illumina HiSeq2500 | embryonic and post-natal cardiac cells | Embryonic to postnatal development: E9.5, E11.5, E14.5, E18.5, P0, P3, p21 and comparison with differentiating mESC and hESC |
| Li et al. [26] PMID: 27840109 | November 2016 | 2233 | enzymatic digestion | Fluidigm C1 IFCs, Illumina HiSeq2000; targeted sc-qPCR Fluidigm 96x96 Dynamic Array | embryonic cardiac cells | Early murine embryo development: E8.5, E9.5, or E10.5 hearts dissected in multiple zones |
| Lescroart et al. [33] PMID: 29371425 | January 2018 | 672 | enzymatic digestion, FACS in lysis buffer | Smart-seq2[8]; Illumina Hi-Seq 2500 | Mouse embryonic derived cardiac progenitor cells (Mesp1+) | Early stage of cardiovascular lineage segregation: Mesp1+ progenitor from wt and Mesp1 null embryos E6.75-7.25 |
| Xiao et al. [34] PMID: 29689192 | April 2018 | 18,166 | enzymatic digestion | Drop-seq [12]; Illumina Nextseq500 | embryonic cardiac cells | Role of Hippo signaling in murine embryo development. CTR versus Lats1/2 CKO E13.5 and E14.5 embryos |
| Sereti et al. [27] PMID: 29467410 | April 2018 | 122 | mechanical and enzymatic digestion, FACS | Fluidigm C1 IFCs; Illumina NextSeq 500 | αMHC+ (αMHC-GFP) | CM heterogeneity in E9.5, E12.5, and P1 mouse hearts |
| Su et al. [31] PMID: 29973725 | July 2018 | 2384 | mechanical and enzymatic digestion, FACS | SMART-seq2, Illumina Nextera XT, NextSeq500 | ApjCreER labeled SV-cells (E12.5–14.5), and Coup-tf2OE- SV cells (E14.5) | Coronary artery specification in the SV |
| Chen et al. [38] PMID: 30128894 | August 2018 | 152 | mechanical and enzymatic digestion, mouth pipette | Smart-seq2; Illumina Hi-Seq 4000 | ventricle from E9.5 heart tube, wt and Mesp1Cre/+ x Dgcr8loxp/loxp | Effect of global microRNA KO on cardiac development |
| Hu et al. [53] PMID: 30254108 | September2018 | ≈20,000 | mechanical nuclei isolation | sNucDrop-seq [13] | early post-natal cardiac cell nuclei | Postnatal heart development in WT (p6, p10) and ERRα/γ knockout mice (p10, pediatric mitochondrial cardiomyopathy) |
| Jia et al. [28] PMID: 30451828 | November 2018 | 421 | enzymatic digestion, FACS | Fluidigm C1 IFCs, ICELL8™ Single-Cell System (Wafergen); Illumina NextSeq 500 | cardiac progenitor cells from Nkx2-5-emGFP and Isl1nGFP/+ embryos | CPs developmentE7.5, E8.5, and E9.5 embryos |
| Cui et al. [47] PMID: 30759401 | February 2019 | 4000 | enzymatic digestion, mouth-picking, FACS | Manual cell picking-lysis; STRT-seq [60]; paired-end sequencing Illumina 4000 | human cardiac fetal cells (6, 7, 13, 17 wks); ECs 22 wks, VCs 17 wks | Spatial/temporal analysis of human cardiac development Comparison with mouse data |
| Cao and Spielmann et al. [11] PMID: 30787437 | February 2019 | 7089-cardiac muscle lineage (2,058,652 total) | Nuclei from snap frozen embryos, no enzymatic digestion, dispersed in 96 well plates | In plate sci-RNA-seq3; NovaSeq (Illumina) | embryonic cells | Mouse organogenic cell atlas (MOCA) E9.5, E10.5, E11.5, E12.5, E13.5. |
| Li et al. [29] PMID: 31142541 | June 2019 | >10,500 | enzymatic digestion | Chromium Single Cells 3’ v2 (10x Genomics); Fluidigm C1 IFCs, Illumina’s HiSeq 2500 and 4000 | embryonic cardiac cells, Isl1-cre/mTmG embryos | Profiling ventricular chambers of E10.5 heart, by dissection and lineage tracing |
| Hill et al. [35] PMID: 31201182 | June 2019 | 77,122 | mechanical and enzymatic digestion | Chromium Single Cells 3’ v2 (10x Genomics); Illumina NextSeq 500 | embryonic cardiac cells | E10.5 and E13.5, control and Pitx2 mutant hearts |
| Han et al. [37] PMID: 31273086 | July 2019 | 3600 | mechanical and enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq 2500 and HiSeq X TEN | embryonic cardiac cells wt and Hand2os1-null | Effect of the lncRNA Hand2os1- on cardiac development |
| Yvanka de Soysa et al. [55] PMID: 31341279 | July 2019 | 36,654 | Micro-dissection and enzymatic digestion | Chromium Single Cells 3’ v2 (10x Genomics); Illumina NextSeq 500 and HiSeq4000 | embryonic cardiac cells | Effect of congenital mutation on cardiac development (Hand2-null versus wt, E7.75, E8.25, and E9.25 embryos) |
| Liu et al. [45] PMID: 31365875 | July 2019 | 55,611 | enzymatic digestion | Chromium Single Cells 3’ v2 (10x Genomics); Illumina NovaSeq 6000 | embryonic cells from the OFT | Murine OFT development (ps47, ps49, ps51) |
| Xiong et al. [30] PMID: 31221018 | August 2019 | 2631 | Micro-dissection and enzymatic digestion, FACS | Smart-seq2; Illumina HiSeq 4000 | embryonic cardiac cells | Differentiation trajectory and interlineage communication of cardiac progenitor cells from FHF and SHF (E8.25, 8.75, 9.25) |
| Goodyer et al. [46] PMID: 31284824 | August 2019 | >22,000 | Micro-dissection and enzymatic digestion | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq4000 | embryonic cardiac cells | Cardiac conduction system in the embryo E16.5 |
| Asp and Giacomello et al. [51] PMID: 31835037 | December 2019 | 3717 | Mechanical and enzymatic digestion, FACS in 384w plates | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq2500 | embryonic cardiac cells | Spatio-temporal transcriptomic of developing human heart at different stages |
| Weinberger et al. [32] PMID: 32084358 | February 2020 | at least 5000 cardiac B-cells | mechanical and enzymatic digestion, FACS | Smart-seq2 and TARGET-seq [61]; Illumina NextSeq500 | fluorescent epicardial reporters (e.g., tbx18:myr-eGFP) | Epicardium heterogeneity in zebrafish cardiac development |
| Holowiecki et al. [36] PMID: 32094112 | March 2020 | 5300 | mechanical and enzymatic digestion, FACS | Chromium 10x; Illumina HiSeq2500 | nkx2.5:ZsYellow+ cells at 28 hpf | pbx4 depletion and OFT development in zebrafish |
| Suryawanshi et al. [57] PMID: 31589297 | July 2020 | 17,747 | Langendorff enzymatic perfusion | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq2500 | whole human fetal hearts | Congenital heart block (CHB): comparison of healthy versus anti-SSA/Ro-associated CHB foetal hearts (mid-gestation) |
3. Cardiac scRNAseq to Elucidate In Vitro Differentiation and Reprogramming
| Authors PMID | Date | # of Cells and/or Nuclei | Isolation Method | Sequencing Technology | Target Cell Types | Context |
|---|---|---|---|---|---|---|
| Chan et al. [72] PMID: 27131741 | April 2016 | 94 | enzymatic digestion, FACS (live cells) | Fluidigm C1 IFCs; Illumina HiSeq2500 | Mesp1-induced embryoid bodies | Heterogeneity of the Mesp1+ mesoderm cells |
| Cho et al. [75] PMID: 28076798 | January 2017 | 24 | mechanical and enzymatic digestion, FACS | custom plate-based; Illumina NextSeq 500 | mESCs-derived CMs and adult CMs (αMHC-GFP) | Comparison mESC-derived CMs differentiation, in vitro or post- implantation |
| Bektik et al. [62] PMID: 28796841 | August 2017 | Does not specify | enzymatic digestion from culture, FACS | Fluidigm C1 IFCs; multiplex TaqMan® sc-qPCR | hESC-derived hCMs, hCFs and hiCMs (αMHC-mCherry+) | hESC-derived fibroblast differentiation hiCM |
| Liu et al. [64] PMID: 29072293 | October 2017 | 454 | enzymatic digestion, FACS | Fluidigm C1 IFCs; Illumina HiSeq2500 | cultured CMs and fibroblasts | fibroblasts to iCM reprogramming |
| Friedman and Nguyen et al. [68] PMID: 29072293 | October 2018 | 43,168 | enzymatic digestion from culture | Chromium Single Cells 3’ v1(10x Genomics); NextSeq 500 (Illumina). Fluidigm C1 IFCs, Illumina’s HiSeq 2000 | hiPSC-derived CMs | Multiple time stages of hiPSC differentiation to CM (day 0, 2, 5, 15, 30). Identify HOPX, signal to enhance CM differentiation |
| Churko et al. [67] PMID: 30464173 | November 2018 | 10,427 | enzymatic digestion from culture | Chromium Single Cells 3’ v2 (10x Genomics); NextSeq 500 (Illumina). | hiPSC-derived CMs | hiPSC cardiac differentiation. Multiple time points (day 0, 5, 14, and day 45) |
| Biendarra-Tiegs et al. [69] PMID : 30892143 | April 2019 | 85 | enzymatic digestion | Fluidigm C1 IFCs, Illumina’s HiSeq 2500 | hiPSC-derived CM (d12-d40 of differentiation) | hiPSC-derived CM maturation (Electrophysiological vs. transcriptomic profiling) |
| Zhou et al. [65] PMID: 31230860 | June 2019 | 704 | enzymatic digestion, FACS live cells | Fluidigm C1 IFCs, Illumina HiSeq 2500 | hCF-induced CMs | Time course of hCF to hiCM reprogramming (d0, d3, d5, d7, d9 post-infection) |
| Stone et al. [66] PMID: 31271750 | July 2019 | 29,718 | enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq4000 | stimulated embryonic CMs and fibroblasts | Time course of mCF to miCM reprogramming (d-1, 0, 1, 7, 14 post-infection) |
| Raghunathan et al. [79] PMID: 31678351 | October 2019 | 560 | enzymatic digestion from culture | Chromium Single Cells 3’ v2 (10x Genomics); NextSeq 500 (Illumina). | Induced cardiac pacemaker-like cells | Human CPs (derived from hASMSC) differentiation to pacemaker-like cells |
| Ruan et al. [74] PMID: 31722692 | November 2019 | 6879 | enzymatic digestion;image-based selection of live cells | Icell8 platform (Takara); Illumina NextSeq500 | embryonic cardiac cells | Human ESCs to CM differentiation (d0, 2, 5, 9, 14, and 60) |
| Gambardella et al. [73] PMID: 31767620 | December 2019 | 362 | enzymatic from culture | Smart-seq2; Illumina Nextera XT | hESC-derived epicardial cells | Characterization of epicardial cell heterogeneity |
| Selewa et al. [70] PMID: 32001747 | January 2020 | ≈50,000 | enzymatic digestion from culture, mechanical isolation; Nuclei EZ Prep isolation kit (Sigma) | Drop-seq [12], DroNc-seq [13]; Illumina NextSeq500 | hiPSC-derived CM, human cardiac nuclei | ScRNAseq versus snRNAseq on: iPSC to CM differentiation (d0, 1, 3, 7, 15), human heart tissue |
| Kamdar et al.[71] PMID: 32164890 | March 2020 | 264 | enzymatic from culture | does not specify; Illumina MiSeq | hiPSC-derived CMs | CMs derived from control and DMD patients (d30-d60) |
| He et al. [78] PMID: 32276728 | April 2020 | 11,772 | enzymatic digestion from culture | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq2000 | hESC-derived cardiac cells | Effect of nicotine on cardiac differentiation from hESCs |
| Wang et al. [76] PMID: 32444791 | May 2020 | 2497 | enzymatic digestion; image-based selection of live cells | Icell8 platform (Takara); Illumina NextSeq500 | murine heart LV; CM-fibroblasts co-cultures | Murine postnatal CM maturation: p1, 4, 7, 14, 56 hearts (LV); in vitro imCM with neonatal or adult fibroblasts |
| Miao et al. [56] PMID: 32810435 | August 2020 | 32,901 human fetal heart cells (35,284 total) | Dissection, mechanical and enzymatical digestion, MACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq4000 | Human fetal heart cell, enrichment for CD144+ endo cells; hiPSC-ECs | Hypoplastic left heart syndrome (HLHS): human fetal heart tissue, hiPSC-derived endocardium |
| Lam et al. [58] PMID: 33059525 | October 2020 | 25,079 | Enzymatic digestion | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq4000 | hiPSC-CMs and hiPSC-CMs in anisotropic sheets or cardiac strips | Pulmonary Atresia with Intact Ventricular Septum, a form of HRHS |
| Authors PMID | Date | # of Cells and/or Nuclei | Isolation Method | Sequencing Technology | Target Cell Types | Context |
|---|---|---|---|---|---|---|
| Noseda et al. [80] PMID: 25980517 | May 2015 | 128 | enzymatic digestion, FACS | Manual RNA extraction; targeted Taqman® sc-qPCR | Adult cardiac progenitor cells | Cardiac cell lineage commitment |
| Yao et al. [81] PMID: 26043119 | June 2015 | 48 | enzymatic digestion, FACS | Fluidigm Dynamic Array IFCs targeted sc-qPCR [6] | Transplanted BM- MSC | Paracrine function of injected cells. Analysis 10 days post-ligation |
| Ong et al. [82] PMID: 26304668 | August 2015 | does not specify | Langendorff enzymatic digestion, FACS | Fluidigm Dynamic Array IFCs targeted sc-qPCR [6] | Transplanted hiPSC-CM | Paracrine function of injected cells. Analysis 4 days post-ligation |
| Chen et al. [77] PMID: 27622691 | September 2016 | 6 | enzymatic digestion | Custom microfluidic chip [86]; Targeted sc- qPCR and MG430 2.0 Affimetrix single-cell transcriptome | CMs, CM-derived progenitor cells (mCPCs) | CM de-differentiation |
| Chen et al. [83] PMID: 29021323 | October 2017 | 405 | mechanical and enzymatic digestion, MACS | Fluidigm C1 IFCs; Illumina HiSeq2500 | Cardiac CD45−c-kit+ cells | Profiling the heterogeneity of c-kit+ CPs, from p1 and adult hearts |
| Kim et al. [84] PMID: 30104715 | August 2018 | 2465 (10x Chromium); 1126 (Smart-seq2) | mechanical and enzymatic digestion, MACS | 10x 3’ v2 (10x Genomics), Smart-seq2; Illumina HiSeq2500, NextSeq500. | c-Kit+/Lin− CPs freshly isolated and after 5 passages in culture | Comparison of freshly isolated and cultured CPs |
| Broughton et al. [85] PMID:31231694 | June 2019 | 1664 | mechanical and enzymatic digestion, FACS | 10x 3’ v2 (10x Genomics); Illumina HiSeq 2500 | c-kit+ interstitial non-myocytes | Ploidy in cardiac c-kit+ interstitial non-CMs |
4. Profiling Injury Models in Regenerative Heart
| Authors PMID | Date | # of Cells and/or Nuclei | Isolation Method | Sequencing Technology | Target Cell Types | Context |
|---|---|---|---|---|---|---|
| Cao et al. [90] PMID: 26657776 | December 2015 | 31 | enzymatic digestion, FACS | Fluidigm C1 platform, Illumina HiSeq 2000 | tcf21- nucEGFP+ epicardial cells | Zebrafish cardiac regeneration |
| Li and Tao et al. [97] PMID: 30143541 | September 2018 | 7849 | mechanical nuclei isolation | Chromium Single Cells 3’ v2 (10x Genomics); Illumina Nextseq 500 | adult cardiac nuclei | Analysis of Pitx2 conditional-KO with P2-MI 60 days post-sham or injury |
| Li et al. [98] PMID: 31285764 | June 2019 | 581 CD3+ heart T cells (1850 from spleen) | mechanical and enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq 2500 | CD3+ T-cells | Comparison of naïve T-cells (liver) and Treg (heart) d7 after cryoinjury in P3 mice hearts |
| Honkoop and de Bakker et al. [91] PMID: 31868166 | December 2019 | 768 | mechanical and enzymatic digestion | SORT-seq [9], plate-based | embryonic, adult zebrafish cardiac cells | Comparison of embryonic (2dpf) and regenerating CMs (7d cryoinjury) |
| Cui et al. [93] PMID: 32220304 | March 2020 | 21,737 | mechanical and enzymatic digestion, nuclei isolation, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina Nextseq 500 | CM nuclei | Neonatal and postnatal regenerative capacity: CM from P1 or P8 mice sham, d1, d3 post-MI |
| Koth et al. [92] PMID: 32341028 | April 2020 | 15,415 | mechanical and enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq 4000 | adult zebrafish cardiac cells | Runx1 KO zebrafish cardiac regeneration |
| Li et al. [99] PMID: 32724455 | June 2020 | 2431 | mechanical and enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq 2500 | cardiac and splenic T-cells | Neonatal cardiac regeneration after apical resection and cryoinfraction |
| Wang et al. [94] PMID: 33296652 | December 2020 | 17,320 | mechanical and enzymatic digestion | Chromium Single Cells 3’ v2 (10x Genomics); Illumina Nextseq 500 | Interstitial cells | Neonatal and postnatal regenerative capacity: interstitial cells from P1 or P8 mice sham, d1, d3 post-MI |
5. Profiling Cardiac Diseases and Injury Models in Non-Regenerative Hearts
| Authors PMID | Date | # of Cells and/or Nuclei | Isolation Method | Sequencing Technology | Target Cell Types | Context |
|---|---|---|---|---|---|---|
| Kanisicak et al. [116] PMID: 27447449 | July 2016 | 185 | mechanical and enzymatic digestion, FACS | Fluidigm C1 IFCs; Illumina HiSeq2500 | CD31-CD45- cardiac cells | Tcf21 lineage tracing during adult MI, TAC and/or AngII infusion |
| See et al. [100] PMID: 28790305 | August 2017 | 359 | mechanical nuclei isolation | Fluidigm C1 IFCs; Illumina HiSeq2500 | Adult human and murine CMs | CM response to heart failure: Human DCM, mouse TAC (8 weeks) |
| King et al. [122] PMID: 29106401 | November 2017 | 4215 | enzymatic digestion | InDrop [14]; Illumina HiSeq2500 | leucocytes | IFNr in leucocytes, CTR and d4 post-MI |
| Schafer et al. [113] PMID: 29160304 | November 2017 | 4548 | enzymatic digestion | Chromium Single Cells 3’ v2 (10x Genomics); NextSeq 500 (Illumina) | adult cardiac non-myocyte | wt versus PlnR9C/+ mouse (cardiac fibrosis phenotype). Il11 mediator of fibroblast activation via TGFb |
| Gladka, M.M. et al. [105] PMID: 29386203 | January 2018 | 932 | enzymatic digestion, FACS (DAPI, scatter properties) | plate-based, SORT-seq; Illumina NextSeq | adult CMs, endothelial cells, fibroblasts, and macrophages | Uninjured LV versus ischemic area 3d post-IR. Cfk4 regulator of fibroblast activation post-injury |
| Nomura et al. [101] PMID: 30375404 | October 2018 | 482 | Langendorff perfusion, manual pipette | Manual CM lysis, cDNASmart-seq2; Illumina HiSeq 2500 | adult murine CMs | CMs response to pressure-overload. Sham, 3d and 1, 2, 4, 8 wks post-TAC |
| Kretzschmar and Post et al. [106] PMID: 30530645 | December 2018 | 1939 | Mechanical and enzymatic digestion,FACS (DAPI, MitoTracker) | CEL-Seq2 and TruSeq library preparation for NextSeq500 | All murine adult ventricular cells | Ki67-RFP mouse model to assay proliferation during murine cardiac injury |
| Dick et al. [126] PMID: 30538339 | December 2018 | 8283 | enzymatic digestion, Ig based FACS on beads enriched CD45+ population | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq2500 | adult mononuclear phagocytes (CD45+ CD64Dim–Hi CD11b+) | Profiling macrophages post-murine MI (CTR, d2, d11, d28) |
| Satoh et al. [102] PMID: 30611794 | January 2019 | 219 | Langendorff perfusion, manual pipette | Manual cell picking, SMART-seq2, HiSeq 2500 System | adult CMs | Spatial and temporal CMs response to pressure-overload. (sham, 1, 2, 8 wks post-TAC) |
| Farbehi et al. [114] PMID: 30912746 | March 2019 | 30,118 | enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Fluidigm C1 IFCs, Illumina’s HiSeq 2500 | TIP cells and adult cardiac non-myocytes | Murine MI (Sham, d3, d7) |
| Zhang et al. [110] PMID: 31231540 | June 2019 | 31,542 | mechanical isolation and lysis from fresh frozen tissue | 10x Chromium Single Cell 5’ kit (10x Genomics); Illumina HiSeq2500 | adult cardiac nuclei | Murine MI (control and d5), tri-transgenic mouse line for CM lineage tracing |
| Li et al. [120] PMID: 31162546 | August 2019 | ≈28,000 | enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq4000 | adult cardiac endothelial cells | Murine MI (control and d7), reporter mouse for clonogenic tracing of ECs |
| Yekelchyk et al. [104] PMID: 31399804 | August 2019 | 1,301 | Langendorff enzymatic perfusion | ICell8 platform (Takara); Illumina Nextera XT | adult CMs mono- and multi-nucleated | CM profiling in CTR hearts and 8-weeks post-TAC |
| Martini et al. [123] PMID: 31661975 | October 2019 | 17,853 | mechanical and enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina NextSeq500 | Adult cardiac leukocytes | Murine pressure-overload model (1- and 4-weeks post-sham, TAC operation) |
| Wang et al. [108] PMID: 31915373 | January 2020 | 21,422 | mechanical and enzymatic digestion, image based live cell selection in 384w plates | ICell8 CellSelect (Takara), plate-based lysis and cDNA synthesis SMARTScribe; Illumina NextSeq500 | adult human CMs and non-CMs from LA/ LV, RV | Human heart failure: healthy donors, HF caused by coronary disease, partial recovery (LVAD treatment) |
| Soliman et al. [117] PMID: 31978365 | February 2020 | 32,313 | enzymatic digestion, FACS | 10x 3’ v2 (10x Genomics), Illumina NextSeq500 | Pdgfra-eGFP/ Hic1+ cells in homeostasis; Pdgfra-eGFP cells post-MI | Cardiac stromal progenitor response to injury (apical area d0, d7, d14, d28 post-MI) |
| Ren et al. [109] PMID: 32098504 | February 2020 | 11,492 | mechanical and enzymatic digestion | ICell8 CellSelect (Takara), MSND Wafergen | Murine and human heart CMs and non-CMs | Murine pressure-overload model (0, 2, 5, 8, 11 weeks); human heart failure |
| Forte et al. [115] PMID: 32130914 | March 2020 | 36,847 | mechanical and enzymatic digestion, FACS live cells [130] | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq Xten | Adult murine heart non-myocytes | Murine MI (d0, d1, d3, d5, d7, d14, d28), epicardial lineage tracing, and mouse diversity |
| Abplanalp et al. [127] PMID: 32311026 | April 2020 | 181,712 | MACS magnetic sorting | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq4000 | Human circulating monocytes (CD31+) | Effect of heart failure on circulating monocytes: Healthy versus HFrEF patients |
| Hua et al. [124] PMID: 32431172 | May 2020 | 34,665 | mechanical and enzymatic digestion, FACS live cells | Chromium Single Cells 5’ v2 (10x Genomics); Illumina HiSeq4000 | CD45+ immune cells | Experimental autoimmune myocarditis: d0, d14, d21, d60 post-induction in Balb/c mice |
| McLellan et al. [111] PMID: 32795101 | July 2020 | 29,558 | perfusion-based enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq4000 | adult cardiac non-myocyte cells and CM nuclei | Murine hypertension (Sham, AngII- 2wks post-treatment), male and female comparison |
| Vafadarnejad et al. [128] PMID: 32811295 | August 2020 | 1334 | mechanical and enzymatic digestion, FACS (CD11b+ live cells) | CITE-seq, Chromium Single Cells 3’ v2 and v3 (10x Genomics); Illumina NovaSeq6000 | adult cardiac neutrophils | Neutrophils dynamics post-murine MI (d1, d3, d5) |
| Yamaguchi et al. [103] PMID: 32868781 | August 2020 | 280 murine CMs and 514 human CMs | Langendorff enzymatic perfusion, manual pipette | Smart-seq2, Illumina HiSeq2500 | Adult murine and human CMs | Interrogation of cardiac dopamine receptor expression during arrhythmia in mice and heart failure in humans |
| Chan et al. [112] PMID: 32885678 | September 2020 | 830 mouse non-myocyte cells (additionally utilized publicly available data [100,101]) | mechanical and perfusion-based enzymatic digestion, FACS | SMART-Seq2, Illumina HiSeq2000 | LV interstitial cells 7 days post-MI, CM nuclei 8 weeks post-TAC | Identify HF biomarkers combining plasma proteomic analysis and scRNAseq |
| Ruiz-Villalba et al. [118] P MID: 32972203 | September 2020 | 36,858 | mechanical and enzymatic digestion, FACS (Col1a1-GFP, CD31, CD45) | 10x Genomics 3’ v2; Illumina NextSeq500 | Adult cardiac fibroblasts, endothelial, immune cells | Murine MI (d0, d7, d30), Cthrc1-KO MI (d7), swine MI (d7) |
| Calcagno and Ng et al. [125] PMID: 32978242 | Sept 2020 | 10,666 murine hearts (~145,000 total) | mechanical and enzymatic digestion, FACS (DAPI-, Ter119−) | inDrop [14] and 10x Genomics; Illumina HiSeq2500 and HiSeq4000 | Myeloid cells (neutrophils, monocytes, resident macrophages) | IFNr in leucocytes: human serum 28h post-NSTEMI, mouse heart d1, d2, d4 post-MI |
| Xia and Lu et al. [129] PMID: 32985264 | September 2020 | 20,755 heart T-cells (and 23,741 spleen T-cells) | mechanical and enzymatic digestion, FACS live cells | Chromium Single Cells 3’ v2 and v3 (10x Genomics); Illumina NovaSeq6000 | murine cardiac and splenic CD4+ T cell TCR sequencing | Profiling Treg in the heart, after MI, I/R, cryoinjury |
| Räsänen et al. [121] PMID: 33203221 | November 2020 | does not specify | mechanical and enzymatic digestion, FACS | Chromium Single Cells 3’ v3 (10x Genomics); Illumina NovaSeq6000 | cardiac endothelial cells (CD31+ CD45- Ter119- CD140a- DAPI-) | ECs from CTR and —V-–VEGF-B transduced adult hearts |
6. Cardiac scRNAseq Cell Atlases
| Authors PMID | Date | # of Cells and/or Nuclei | Isolation Method | Sequencing Technology | Target Cell Types | Context |
|---|---|---|---|---|---|---|
| Skelly et al. [143] PMID: 29346760 | January 2018 | 10,519 | enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina HiSeq 4000 | adult cardiac non-myocyte | homeostatic murine adult cardiac tissue. Male and female comparison |
| Han et al. [10] PMID: 29474909 | February 2018 | 5075 heart cells (Over 400,000 total) | enzymatic digestion | Microwell-Seq; Illumina HiSeq | neonatal cardiac cells | Mouse Cell Atlas |
| Tabula Muris Consortium [131] PMID:30283141 | October 2018 | 4635 heart cells (over 100,000 total) | enzymatic digestion, FACS, manual pipette | GemCode Single-Cell 3’ v2 (10x Genomics) & FACS-based full length transcriptomic; Illumina NovaSeq 6000 | adult cardiac non-myocytes, CM | Homeostatic cell profiling of 20 murine adult organs |
| Hulin et al. [140] PMID: 30796046 | March 2019 | 2840 | mechanical and enzymatic digestion | Dropseq | heart valve cells | Aortic valve and mitral valve at P7 and P30 |
| Linscheid et al. [147] PMID: 31253831 | June 2019 | 5357 | Mechanical nuclei isolation | Chromium Single Cells 3’ v3 (10x Genomics); Illumina NovaSeq 6000 | sinus node nuclei | Murine sinus node cell atlas |
| Wang et al. [148] PMID: 31455969 | August 2019 | 12,865 | mechanical and enzymatic digestion, FACS | 10x 3’ v2 (10x Genomics); Illumina HiSeq PE150 | 3D-cultured primary cells | Engineered cardiac tissues (derived from rat primary cells) |
| Vidal et al. [134] PMID: 31723062 | September 2019 | 27,808 | Mechanical nuclei isolation | Chromium Single Cells 3’ v3 (10x Genomics); Illumina Hiseq4000 | adult CM and non-myocyte nuclei | Young and aged C57BL/6JRj mice |
| Haftbaradaran Esfahani et al. [149] PMID: 31872302 | December 2019 | 435 | Enzymatic from culture and semi-automatic cell picking [157] | Smart-Seq2; Illumina HiSeq 2500 | cultured primary p2 rat CMs | Profiling of CMs with defined morphotypes through custom geometry culture chips [158] |
| Adamo et al. [141] PMID: 31945014 | January 2020 | 5571 | mechanical and enzymatic digestion, FACS | Chromium Single Cells 3’ v3 and 5’ V(D)J enriched library (10x Genomics); Illumina NovaSeq6000 | CD45+Aqua–CD19+ B-cells | B-cells from the heart and blood of 10-week-old C57BL/6J mice |
| Wolfien et al. [144] PMID:32013057 | January 2020 | 8635 | mechanical and enzymatic digestion, Nuclei PURE Prep isolation kit (Sigma) | Chromium Single Cells 3’ v3 (10x Genomics); Illumina NovaSeq6000 | adult CM and non-myocyte nuclei | snRNAseq whole murine heart (Fzt:DU outbred mice) |
| Kalucka et al. [136] PMID: 32059779 | Feb 2020 | 4612 heart endothelial cells (32,567 total cells) | mechanical and enzymatic digestion, FACS | Chromium Single Cells 3’ v2 (10x Genomics); Illumina Hiseq 4000 | Adult murine heart endothelial cells | Murine endothelial cell atlas from 11 tissues |
| Wolfien, Galow, and Müller et al. [145] PMID: 32243511 | April 2020 | 3464 nuclei (additionally integrated with previously published data [144]) | mechanical and enzymatic digestion, Nuclei PURE Prep isolation kit (Sigma) | Chromium Single Cells 3’v3 (10x Genomics); Illumina NovaSeq6000 | adult CM and non-myocyte nuclei | snRNAseq whole murine heart (Fzt:DU outbred vs. C57BL/6NRj mice) |
| Tucker and Chaffin et al., [150] PMID: 32403949 | May 2020 | 287, 269 | mechanical and enzymatic digestion, nuclei isolation | Chromium Single Cells 3’ v2 (10x Genomics) | all human heart cell types | Healthy human adult cardiac tissue: biopsies from four chambers |
| Rocha-Resende et al. [142] PMID: 32663200 | July 2020 | 1004 | mechanical and enzymatic digestion, FACS | Chromium 10x 3’ v3 and 5’ (10x Genomics); Illumina NovaSeq 6000 | CD45+Aqua–CD19+ B-cells | B-cells from postnatal (2 wks) and adult hearts (8 wks); comparison with other tissues (10 wks) |
| Tabula Muris Consortium [132] PMID: 32669714 | July 2020 | 18,282 heart cells over 350,000 total (9,669 cells long-reads; 8,613 short-reads) | enzymatic digestion, FACS, manual pipette | GemCode Single-Cell 3’ v2 (10x Genomics) & FACS-based full length transcriptomic; Illumina NovaSeq 6000 | adult cardiac non-myocytes, CM from all four chambers | Profiling of 23 murine adult organs over six age stages (1 to 30 months), male and female C57BL/6J. |
| Muhl et al. [138] PMID:32769974 | August 2020 | 1,279 heart cells (6158 total) | mechanical and enzymatic digestion, FACS | Smart-Seq2; Illumina HiSeq 3000 | PDGFRa+, PDGFRb+, CD31- ; | Comparison of fibroblasts and mural cells from four different organs |
| Ma et al. [135] PMID: 32913304 | September 2020 | 42,053 (109,609 additional lung nuclei) | mechanical nuclei isolation and FACS | Chromium Single Cells 3’ v3 (10x Genomics); Illumina NovaSeq6000 | young and aged primate cardiac nuclei | Lung and heart from young and aged cynomolgus monkeys |
| Litviňuková et al. [154] PMID: 32971526 | September 2020 | 487,106 | mechanical and enzymatic digestion, FACS, nuclei isolation | Chromium Single Cells 3’ v2 or v3 (10x Genomics); Illumina NextSeq 500 or Hiseq 4000 | adult human CM and non-myocyte nuclei (with selected upsampled whole cells) | Healthy human adult cardiac tissue: biopsies from four chambers plus septum and apex |
7. Single-Cell Analysis and Implications for COVID-19
8. Current Challenges and Future Prospective
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP | Action potential |
| CFs | Cardiac fibroblasts |
| CMs | Cardiomyocytes |
| CPs | Cardiac progenitors |
| DCM | Dilated cardiomyopathy |
| E | Embryonic day |
| EB | Early allontoic bud stage |
| ECM | Extracellular matrix |
| ECs | Endothelial cells |
| EHF | Early headfold stage |
| ESCs | Embryonic stem cells |
| FHF | First heart field |
| HF | Heart failure |
| HFrEF | Heart failure with reduced ejection fraction |
| HLHS | Hypoplastic left heart syndrome |
| hpf | Hours post fertilization |
| I/R | Ischemia/reperfusion |
| iCM | Induced cardiomyocytes |
| IFC | Integrated fluidic circuits |
| imCM | Immature CM (p1 hearts) |
| iPSCs | Induced pluripotent stem cells |
| KO | Knock-out |
| LRHS | Hypoplastic right heart syndrome |
| LV | Left ventricle |
| MACS | Magnetic activated cell sorting |
| MI | Myocardial Infarction. |
| MSC | Mesenchymal stem cells |
| NSTEMI | Non-ST segment elevation myocardial infarction |
| OFT | Outflow tract |
| p | Postnatal day |
| ps | Pairs of somites |
| RNAseq | Ribonucleic acid sequencing |
| ROS | Reactive oxygen species |
| RV | Right ventricle |
| SHF | Second heart field |
| SMCs | Smooth muscle cells |
| SV | Sinus venosus |
| TAC | Trans aortic constriction |
| UMI | Unique molecular identifier |
| wt | Wild type |
References
- Forte, E.; Furtado, M.B.; Rosenthal, N. The interstitium in cardiac repair: Role of the immune–stromal cell interplay. Nat. Rev. Cardiol. 2018, 15, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Eberwine, J.; Yeh, H.; Miyashiro, K.; Cao, Y.; Nair, S.; Finnell, R.; Zettel, M.; Coleman, P. Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. USA 1992, 89, 3010–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, G.; Niscove, N. [361 Construction of cDNA libraries from single cells. Methods Enzymol. 1993, 225, 611–623. [Google Scholar] [CrossRef]
- Brady, G.; Billia, F.; Knox, J.; Hoang, T.; Kirsch, I.R.; Voura, E.B.; Hawley, R.G.; Cumming, R.; Buchwald, M.; Siminovitch, K.; et al. Analysis of gene expression in a complex differentiation hierarchy by global amplification of cDNA from single cells. Curr. Biol. 1995, 5, 909–922. [Google Scholar] [CrossRef] [Green Version]
- Pieprzyk, M.; High, H. Fluidigm Dynamic Arrays provide a platform for single-cell gene expression analysis. Nat. Chem. Biol. 2009, 6. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Freire, V.; Ebert, A.D.; Kalisky, T.; Quake, S.R.; Wu, J.C. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns. Nat. Protoc. 2012, 7, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Bjorklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Muraro, M.J.; Dharmadhikari, G.; Grün, D.; Groen, N.; Dielen, T.; Jansen, E.; Van Gurp, L.; Engelse, M.A.; Carlotti, F.; De Koning, E.J.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e3. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 173, 1307. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nat. Cell Biol. 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilionis, R.; Nainys, J.; Veres, A.; Savova, V.; Zemmour, D.; Klein, R.Z.A.V.V.S.A.M.; Mazutis, R.Z.J.N.L. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 2017, 12, 44–73. [Google Scholar] [CrossRef]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, S.; Kjällquist, U.; Moliner, A.; Zajac, P.; Fan, J.-B.; Lönnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Peterson, V.M.; Zhang, K.X.; Kumar, N.; Wong, J.; Li, L.; Wilson, D.C.; Moore, R.; McClanahan, T.K.; Sadekova, S.; A Klappenbach, J. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017, 35, 936–939. [Google Scholar] [CrossRef]
- Svensson, V.; Vento-Tormo, R.; A Teichmann, S. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018, 13, 599–604. [Google Scholar] [CrossRef]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643.e4. [Google Scholar] [CrossRef] [Green Version]
- Svensson, V.; Natarajan, K.N.; Ly, L.H.; Miragaia, R.J.; Labalette, C.; Macaulay, I.C.; Cvejic, A.; Teichmann, S.A. Power analysis of single-cell RNA-sequencing experiments. Nat. Methods 2017, 14, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Karolina, P.; Plonowska, K.; Kuppusamy, R.; Sturzu, A.; Wu, S.M. Identification of cardiovascular lineage descendants at single-cell resolution. Dev. 2015, 142, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Kokkinopoulos, I.; Ishida, H.; Saba, R.; Ruchaya, P.; Cabrera, C.; Struebig, M.; Barnes, M.; Terry, A.; Kaneko, M.; Shintani, Y.; et al. Single-Cell Expression Profiling Reveals a Dynamic State of Cardiac Precursor Cells in the Early Mouse Embryo. PLoS ONE 2015, 10, e0140831. [Google Scholar] [CrossRef] [Green Version]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; Mckean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Xu, A.; Sim, S.; Priest, J.R.; Tian, X.; Khan, T.; Quertermous, T.; Zhou, B.; Tsao, P.S.; Quake, S.R.; et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev. Cell 2016, 39, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sereti, K.-I.; Nguyen, N.B.; Kamran, P.; Zhao, P.; Ranjbarvaziri, S.; Park, S.; Sabri, S.; Engel, J.L.; Sung, K.; Kulkarni, R.P.; et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat. Commun. 2018, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Preussner, J.; Chen, X.; Guenther, S.; Yuan, X.; Yekelchyk, M.; Kuenne, C.; Looso, M.; Zhou, Y.; Teichmann, S.; et al. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Tian, L.; Goodyer, W.; Kort, E.J.; Buikema, J.W.; Xu, A.; Wu, J.C.; Jovinge, S.; Wu, S.M. Single cell expression analysis reveals anatomical and cell cycle-dependent transcriptional shifts during heart development. Development 2019, 146, dev173476. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Luo, Y.; Yue, Y.; Zhang, J.; Ai, S.; Li, X.; Wang, X.; Zhang, Y.-L.; Wei, Y.; Li, H.-H.; et al. Single-Cell Transcriptomics Reveals Chemotaxis-Mediated Intraorgan Crosstalk During Cardiogenesis. Circ. Res. 2019, 125, 398–410. [Google Scholar] [CrossRef]
- Su, T.; Stanley, G.; Sinha, R.; D’Amato, G.; Das, S.; Rhee, S.; Chang, A.H.; Poduri, A.; Raftrey, B.; Dinh, T.T.; et al. Single-cell analysis of early progenitor cells that build coronary arteries. Nat. Cell Biol. 2018, 559, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, M.; Simões, F.C.; Patient, R.; Sauka-Spengler, T.; Riley, P.R. Functional Heterogeneity within the Developing Zebrafish Epicardium. Dev. Cell 2020, 52, 574–590.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Hill, M.C.; Zhang, M.; Martin, T.J.; Morikawa, Y.; Wang, S.; Moise, A.R.; Wythe, J.D.; Martin, J.F. Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 2018, 45, 153–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.C.; Kadow, Z.A.; Li, L.; Tran, T.T.; Wythe, J.D.; Martin, J.F. A cellular atlas of Pitx2-dependent cardiac development. Dev. 2019, 146, dev180398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holowiecki, A.; Linstrum, K.; Ravisankar, P.; Chetal, K.; Salomonis, N.; Waxman, J.S. Pbx4 limits heart size and fosters arch artery formation by partitioning second heart field progenitors and restricting proliferation. Development 2020, 147, dev185652. [Google Scholar] [CrossRef]
- Han, X.; Zhang, J.; Liu, Y.; Fan, X.; Ai, S.; Luo, Y.; Li, X.; Jin, H.; Luo, S.; Zheng, H.; et al. The lncRNA Hand2os1/Uph locus orchestrates heart development through regulation of precise expression of Hand2. Development 2019, 146, dev176198. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, L.; Huang, R.; Qiu, H.; Wang, P.; Wu, D.; Zhu, Y.; Ming, J.; Wang, Y.; Wang, J.; et al. Dgcr8 deletion in the primitive heart uncovered novel microRNA regulating the balance of cardiac-vascular gene program. Protein Cell 2019, 10, 327–346. [Google Scholar] [CrossRef]
- Devine, W.P.; Wythe, J.D.; George, M.; Koshiba-Takeuchi, K.; Bruneau, B.G. Early patterning and specification of cardiac progenitors in gastrulating mesoderm. eLife 2014, 3, e03848. [Google Scholar] [CrossRef]
- Galdos, F.X.; Wu, S.M. Single-Cell Delineation of Who’s on First and Second Heart Fields During Development. Circ. Res. 2019, 125, 411–413. [Google Scholar] [CrossRef]
- Syeda, F.; Kirchhof, P.; Fabritz, L. PITX2 -dependent gene regulation in atrial fibrillation and rhythm control. J. Physiol. 2017, 595, 4019–4026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinor, P.T.; Lunetta, K.; Glazer, N.; Pfeufer, A.; Alonso, A.; Chung, M.; Sinner, M.; De Bakker, P.; Mueller, M.; Lubitz, S.; et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat. Genet. 2010, 42, 240–244. [Google Scholar] [CrossRef]
- Scialdone, A.; Tanaka, Y.; Bin Jawaid, W.; Moignard, V.R.; Wilson, N.K.; Macaulay, I.C.; Marioni, A.S.I.C.M.J.C.; Göttgens, Y.T.W.J.V.M.N.K.W.B. Resolving early mesoderm diversification through single-cell expression profiling. Nat. Cell Biol. 2016, 535, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Tyser, R.C.V.; Ibarra-Soria, X.; McDole, K.; Jayaram, S.A.; Godwin, J.; van den Brand, T.A.H.; Miranda, A.M.A.; Scialdone, A.; Keller, P.J.; Marioni, J.C.; et al. Characterization of a common progenitor pool of the epicardium and myocardium. Science 2021, eabb2986. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Li, W.; Li, Y.; Priest, J.R.; Zhou, B.; Wang, J.; Zhou, Z. Single-Cell RNA-Seq of the Developing Cardiac Outflow Tract Reveals Convergent Development of the Vascular Smooth Muscle Cells. Cell Rep. 2019, 28, 1346–1361.e4. [Google Scholar] [CrossRef] [Green Version]
- Goodyer, W.R.; Beyersdorf, B.M.; Paik, D.T.; Tian, L.; Li, G.; Buikema, J.W.; Chirikian, O.; Choi, S.; Venkatraman, S.; Adams, E.L.; et al. Transcriptomic Profiling of the Developing Cardiac Conduction System at Single-Cell Resolution. Circ. Res. 2019, 125, 379–397. [Google Scholar] [CrossRef]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e5. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Packer, J.S.; Ramani, V.; Cusanovich, D.A.; Huynh, C.; Daza, R.; Qiu, X.; Lee, C.; Furlan, S.N.; Steemers, F.J.; et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 357, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://atlas.gs.washington.edu/mouse-rna (accessed on 17 February 2021).
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://hdca-sweden.scilifelab.se/a-study-on-human-heart-development/ (accessed on 17 February 2021).
- Hu, P.; Liu, J.; Zhao, J.; Wilkins, B.J.; Lupino, K.; Wu, H.; Pei, L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018, 32, 1344–1357. [Google Scholar] [CrossRef]
- Luo, J.; Sladek, R.; Carrier, J.; Bader, J.-A.; Richard, D.; Giguère, V. Reduced Fat Mass in Mice Lacking Orphan Nuclear Receptor Estrogen-Related Receptor α. Mol. Cell. Biol. 2003, 23, 7947–7956. [Google Scholar] [CrossRef] [Green Version]
- De Soysa, T.Y.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Schricker, A.; Del Sol, A.; Gifford, C.A.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nat. Cell Biol. 2019, 572, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Tian, L.; Martin, M.; Paige, S.L.; Galdos, F.X.; Li, J.; Klein, A.; Zhang, H.; Ma, N.; Wei, Y.; et al. Intrinsic Endocardial Defects Contribute to Hypoplastic Left Heart Syndrome. Cell Stem Cell 2020, 27, 574–589.e8. [Google Scholar] [CrossRef]
- Suryawanshi, H.; Clancy, R.; Morozov, P.; Halushka, M.K.; Buyon, J.P.; Tuschl, T. Cell atlas of the foetal human heart and implications for autoimmune-mediated congenital heart block. Cardiovasc. Res. 2020, 116, 1446–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, Y.; Keung, W.; Chan, C.; Geng, L.; Wong, N.; Brenière-Letuffe, D.; Li, R.A.; Cheung, Y. Single-Cell Transcriptomics of Engineered Cardiac Tissues From Patient-Specific Induced Pluripotent Stem Cell–Derived Cardiomyocytes Reveals Abnormal Developmental Trajectory and Intrinsic Contractile Defects in Hypoplastic Right Heart Syndrome. J. Am. Hear. Assoc. 2020, 9, e016528. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.; Thomas, T.; Lin, Q.; Kirby, M.L.; Brown, D.D.; Olson, E.N. Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nat. Genet. 1997, 16, 154–160. [Google Scholar] [CrossRef]
- Li, L.; Dong, J.; Yan, L.; Yong, J.; Liu, X.; Hu, Y.; Fan, X.; Wu, X.; Guo, H.; Wang, X.; et al. Single-Cell RNA-Seq Analysis Maps Development of Human Germline Cells and Gonadal Niche Interactions. Cell Stem Cell 2017, 20, 891–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.-A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305.e8. [Google Scholar] [CrossRef] [Green Version]
- Bektik, E.; Dennis, A.; Prasanna, P.; Madabhushi, A.; Fu, J.-D. Single cell qPCR reveals that additional HAND2 and microRNA-1 facilitate the early reprogramming progress of seven-factor-induced human myocytes. PLoS ONE 2017, 12, e0183000. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.-D.; Stone, N.R.; Liu, L.; Spencer, C.I.; Qian, L.; Hayashi, Y.; Delgado-Olguin, P.; Ding, S.; Bruneau, B.G.; Srivastava, D. Direct Reprogramming of Human Fibroblasts toward a Cardiomyocyte-like State. Stem Cell Rep. 2013, 1, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, L.; Welch, J.D.; Ma, H.; Zhou, Y.; Vaseghi, H.R.; Yu, S.; Wall, J.B.; Alimohamadi, S.; Zheng, M.; et al. Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nat. Cell Biol. 2017, 551, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liu, Z.; Welch, J.D.; Gao, X.; Wang, L.; Garbutt, T.; Keepers, B.; Ma, H.; Prins, J.F.; Shen, W.; et al. Single-Cell Transcriptomic Analyses of Cell Fate Transitions during Human Cardiac Reprogramming. Cell Stem Cell 2019, 25, 149–164.e9. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.R.; Gifford, C.A.; Thomas, R.; Pratt, K.J.B.; Samse-Knapp, K.; Mohamed, T.M.A.; Radzinsky, E.M.; Schricker, A.; Ye, L.; Yu, P.; et al. Context-Specific Transcription Factor Functions Regulate Epigenomic and Transcriptional Dynamics during Cardiac Reprogramming. Cell Stem Cell 2019, 25, 87–102.e9. [Google Scholar] [CrossRef] [PubMed]
- Churko, J.M.; Garg, P.; Treutlein, B.; Venkatasubramanian, M.; Wu, H.; Lee, J.; Wessells, Q.N.; Chen, S.-Y.; Chen, W.-Y.; Chetal, K.; et al. Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.; Chiu, H.S.; Miklas, J.; Levy, S.; Suo, S.; Han, J.-D.J.; Osteil, P.; et al. Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell 2018, 23, 586–598.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biendarra-Tiegs, S.M.; Li, X.; Ye, D.; Brandt, E.B.; Ackerman, M.J.; Nelson, T.J. Single-Cell RNA-Sequencing and Optical Electrophysiology of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveal Discordance Between Cardiac Subtype-Associated Gene Expression Patterns and Electrophysiological Phenotypes. Stem Cells Dev. 2019, 28, 659–673. [Google Scholar] [CrossRef] [Green Version]
- Selewa, A.; Dohn, R.; Eckart, H.; Lozano, S.; Xie, B.; Gauchat, E.; Elorbany, R.; Rhodes, K.; Burnett, J.; Gilad, Y.; et al. Systematic Comparison of High-throughput Single-Cell and Single-Nucleus Transcriptomes during Cardiomyocyte Differentiation. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kamdar, F.; Das, S.; Gong, W.; Kamdar, A.K.; Meyers, T.A.; Shah, P.; Ervasti, J.M.; Townsend, D.; Kamp, T.J.; Wu, J.C.; et al. Stem Cell–Derived Cardiomyocytes and Beta-Adrenergic Receptor Blockade in Duchenne Muscular Dystrophy Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 1159–1174. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.S.-K.; Chan, H.H.W.; Kyba, M. Heterogeneity of Mesp1+ mesoderm revealed by single-cell RNA-seq. Biochem. Biophys. Res. Commun. 2016, 474, 469–475. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, L.; McManus, S.A.; Moignard, V.; Sebukhan, D.; Delaune, A.; Andrews, S.; Bernard, W.G.; Morrison, M.A.; Riley, P.R.; Göttgens, B.; et al. BNC1 regulates cell heterogeneity in human pluripotent stem cell-derived epicardium. Development 2019, 146, dev174441. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Liao, Y.; Ren, Z.; Mao, L.; Yao, F.; Yu, P.; Ye, Y.; Zhang, Z.; Li, S.; Xu, H.; et al. Single-cell reconstruction of differentiation trajectory reveals a critical role of ETS1 in human cardiac lineage commitment. BMC Biol. 2019, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cho, G.-S.; Lee, N.I.; Tampakakis, E.; Murphy, S.; Andersen, P.; Uosaki, H.; Chelko, S.; Chakir, K.; Hong, I.; Seo, K.; et al. Neonatal Transplantation Confers Maturation of PSC-Derived Cardiomyocytes Conducive to Modeling Cardiomyopathy. Cell Rep. 2017, 18, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yao, F.; Wang, L.; Li, Z.; Ren, Z.; Li, D.; Zhang, M.; Han, L.; Wang, S.-Q.; Zhou, B.; et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Chen, X.; Chakravarty, T.; Zhang, Y.; Li, X.; Zhong, J.F.; Wang, C. Single-cell transcriptome and epigenomic reprogramming of cardiomyocyte-derived cardiac progenitor cells. Sci. Data 2016, 3, 160079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Chen, J.; Tian, M.; Chen, J.; Zhou, C.; Ou, Y.; Wang, S.; Li, X.; Zhuang, J. Adverse effects of nicotine on cardiogenic differentiation from human embryonic stem cells detected by single-cell RNA sequencing. Biochem. Biophys. Res. Commun. 2020, 526, 848–855. [Google Scholar] [CrossRef]
- Raghunathan, S.; Islas, J.F.; Mistretta, B.; Iyer, D.; Shi, L.; Gunaratne, P.H.; Ko, G.; Schwartz, R.J.; McConnell, B.K. Conversion of human cardiac progenitor cells into cardiac pacemaker-like cells. J. Mol. Cell. Cardiol. 2020, 138, 12–22. [Google Scholar] [CrossRef]
- Noseda, M.; Harada, M.; McSweeney, S.; Leja, T.; Belian, E.; Stuckey, D.J.; Paiva, M.S.A.; Habib, J.; Macaulay, I.; De Smith, A.J.; et al. PDGFRα demarcates the cardiogenic clonogenic Sca1+ stem/progenitor cell in adult murine myocardium. Nat. Commun. 2015, 6, 6930. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Huang, J.; Geng, Y.; Qian, H.; Wang, F.; Liu, X.; Shang, M.; Nie, S.; Liu, N.; Du, X.; et al. Paracrine Action of Mesenchymal Stem Cells Revealed by Single Cell Gene Profiling in Infarcted Murine Hearts. PLoS ONE 2015, 10, e0129164. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.-G.; Huber, B.C.; Lee, W.H.; Kodo, K.; Ebert, A.D.; Ma, Y.; Nguyen, P.K.; Diecke, S.; Chen, W.-Y.; Wu, J.C. Microfluidic Single-Cell Analysis of Transplanted Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes After Acute Myocardial Infarction. Circulation 2015, 132, 762–771. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhu, W.; Bender, I.; Gong, W.; Kwak, I.-Y.; Yellamilli, A.; Hodges, T.J.; Nemoto, N.; Zhang, J.; Garry, D.J.; et al. Pathologic Stimulus Determines Lineage Commitment of Cardiac C-kit+ Cells. Circulation 2017, 136, 2359–2372. [Google Scholar] [CrossRef]
- Kim, T.; Echeagaray, O.H.; Wang, B.J.; Casillas, A.; Broughton, K.M.; Kim, B.-H.; Sussman, M.A. In situ transcriptome characteristics are lost following culture adaptation of adult cardiac stem cells. Sci. Rep. 2018, 8, 12060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broughton, K.M.; Khieu, T.; Nguyen, N.; Rosa, M.; Mohsin, S.; Quijada, P.; Wang, B.J.; Echeagaray, O.H.; Kubli, D.A.; Kim, T.; et al. Cardiac interstitial tetraploid cells can escape replicative senescence in rodents but not large mammals. Commun. Biol. 2019, 2, 205. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.F.; Chen, Y.; Marcus, J.S.; Scherer, A.; Quake, S.R.; Taylor, C.R.; Weiner, L.P. A microfluidic processor for gene expression profiling of single human embryonic stem cells. Lab Chip 2007, 8, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [Green Version]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart Regeneration in Zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef]
- Price, E.L.; Vieira, J.M.; Riley, P.R. Model organisms at the heart of regeneration. Dis. Model. Mech. 2019, 12, dmm040691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Navis, A.; Cox, B.D.; Dickson, A.L.; Gemberling, M.; Karra, R.; Bagnat, M.; Poss, K.D. Single epicardial cell transcriptome sequencing identifies Caveolin 1 as an essential factor in zebrafish heart regeneration. Development 2016, 143, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Honkoop, H.; De Bakker, D.E.; Aharonov, A.; Kruse, F.; Shakked, A.; Nguyen, P.D.; De Heus, C.; Garric, L.; Muraro, M.J.; Shoffner, A.; et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. eLife 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Koth, J.; Wang, X.; Killen, A.C.; Stockdale, W.T.; Potts, H.G.; Jefferson, A.; Bonkhofer, F.; Riley, P.R.; Patient, R.K.; Göttgens, B.; et al. Runx1 promotes scar deposition and inhibits myocardial proliferation and survival during zebrafish heart regeneration. Development 2020, 147, dev186569. [Google Scholar] [CrossRef]
- Cui, M.; Wang, Z.; Chen, K.; Shah, A.M.; Tan, W.; Duan, L.; Sanchez-Ortiz, E.; Li, H.; Xu, L.; Liu, N.; et al. Dynamic Transcriptional Responses to Injury of Regenerative and Non-regenerative Cardiomyocytes Revealed by Single-Nucleus RNA Sequencing. Dev. Cell 2020, 53, 102–116.e8. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, M.; Shah, A.M.; Tan, W.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Cell-Type-Specific Gene Regulatory Networks Underlying Murine Neonatal Heart Regeneration at Single-Cell Resolution. Cell Rep. 2020, 33, 108472. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Morikawa, Y.; Leach, J.; Tao, G.; Willerson, J.T.; Johnson, R.L.; Martin, J.F. Hippo signaling impedes adult heart regeneration. Development 2013, 140, 4683–4690. [Google Scholar] [CrossRef] [Green Version]
- Tao, G.; Kahr, P.C.; Morikawa, Y.; Zhang, M.; Rahmani, M.; Heallen, T.R.; Li, L.; Sun, Z.; Olson, E.N.; Amendt, Z.S.B.A.; et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nat. Cell Biol. 2016, 534, 119–123. [Google Scholar] [CrossRef]
- Li, L.; Tao, G.; Hill, M.C.; Zhang, M.; Morikawa, Y.; Martin, J.F. Pitx2 maintains mitochondrial function during regeneration to prevent myocardial fat deposition. Development 2018, 145, dev168609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yang, K.Y.; Tam, R.C.Y.; Chan, V.W.; Lan, H.Y.; Hori, S.; Zhou, B.; Lui, K.O. Regulatory T-cells regulate neonatal heart regeneration by potentiating cardiomyocyte proliferation in a paracrine manner. Theranostics 2019, 9, 4324–4341. [Google Scholar] [CrossRef]
- Li, J.; Liang, C.; Yang, K.Y.; Huang, X.; Han, M.Y.; Li, X.; Chan, V.W.; Chan, K.S.; Liu, D.; Huang, Z.-P.; et al. Specific ablation of CD4+ T-cells promotes heart regeneration in juvenile mice. Theranostics 2020, 10, 8018–8035. [Google Scholar] [CrossRef] [PubMed]
- See, K.; Tan, W.L.W.; Lim, E.H.; Tiang, Z.; Lee, L.T.; Li, P.Y.Q.; Luu, T.D.A.; Ackers-Johnson, M.; Foo, R.S. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Nomura, S.; Satoh, M.; Fujita, T.; Higo, T.; Sumida, T.; Ko, T.; Yamaguchi, T.; Tobita, T.; Naito, A.T.; Ito, M.; et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Nomura, S.; Harada, M.; Yamaguchi, T.; Ko, T.; Sumida, T.; Toko, H.; Naito, A.T.; Takeda, N.; Tobita, T.; et al. High-throughput single-molecule RNA imaging analysis reveals heterogeneous responses of cardiomyocytes to hemodynamic overload. J. Mol. Cell. Cardiol. 2019, 128, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Sumida, T.S.; Nomura, S.; Satoh, M.; Higo, T.; Ito, M.; Ko, T.; Fujita, K.; Sweet, M.E.; Sanbe, A.; et al. Cardiac dopamine D1 receptor triggers ventricular arrhythmia in chronic heart failure. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Yekelchyk, M.; Guenther, S.; Preussner, J.; Braun, T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res. Cardiol. 2019, 114, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gladka, M.M.; Molenaar, B.; De Ruiter, H.; Van Der Elst, S.; Tsui, H.; Versteeg, D.; Lacraz, G.P.A.; Huibers, M.M.H.; Van Oudenaarden, A.; Van Rooij, E. Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation. Circulation 2018, 138, 166–180. [Google Scholar] [CrossRef]
- Kretzschmar, K.; Post, Y.; Bannier-Hélaouët, M.; Mattiotti, A.; Drost, J.; Basak, O.; Li, V.S.W.; Born, M.V.D.; Gunst, Q.D.; Versteeg, D.; et al. Profiling proliferative cells and their progeny in damaged murine hearts. Proc. Natl. Acad. Sci. USA 2018, 115, E12245–E12254. [Google Scholar] [CrossRef] [Green Version]
- Ackers-Johnson, M.; Tan, W.L.W.; Foo, R.S.-Y. Following hearts, one cell at a time: Recent applications of single-cell RNA sequencing to the understanding of heart disease. Nat. Commun. 2018, 9, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Yu, P.; Li, D.; Li, Z.; Liao, Y.; Wang, Y.; Zhou, B.; Wang, L. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation 2020, 141, 1704–1719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gago-Lopez, N.; Li, N.; Zhang, Z.; Alver, N.; Liu, Y.; Martinson, A.M.; Mehri, A.; MacLellan, W.R. Single-cell imaging and transcriptomic analyses of endogenous cardiomyocyte dedifferentiation and cycling. Cell Discov. 2019, 5, 1–15. [Google Scholar] [CrossRef]
- McLellan, M.A.; Skelly, D.A.; Dona, M.S.; Squiers, G.T.; Farrugia, G.E.; Gaynor, T.L.; Cohen, C.D.; Pandey, R.; Diep, H.; Vinh, A.; et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation 2020, 142, 1448–1463. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.Y.; Efthymios, M.; Tan, S.H.; Pickering, J.W.; Troughton, R.; Pemberton, C.; Ho, H.-H.; Prabath, J.-F.; Drum, C.L.; Ling, L.H.; et al. Prioritizing Candidates of Post–Myocardial Infarction Heart Failure Using Plasma Proteomics and Single-Cell Transcriptomics. Circulation 2020, 142, 1408–1421. [Google Scholar] [CrossRef]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Moreno-Moral, A.; DeLaughter, D.M.; Kingsley, C.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nat. Cell Biol. 2017, 552, 110–115. [Google Scholar] [CrossRef]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; E Nordon, R.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Skelly, D.A.; Chen, M.; Daigle, S.; Morelli, K.A.; Hon, O.; Philip, V.M.; Costa, M.W.; Rosenthal, N.A.; Furtado, M.B. Dynamic Interstitial Cell Response during Myocardial Infarction Predicts Resilience to Rupture in Genetically Diverse Mice. Cell Rep. 2020, 30, 3149–3163.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, H.; Paylor, B.; Scott, R.W.; Lemos, D.R.; Chang, C.; Arostegui, M.; Low, M.; Lee, C.; Fiore, D.; Braghetta, P.; et al. Pathogenic Potential of Hic1-Expressing Cardiac Stromal Progenitors. Cell Stem Cell 2020, 26, 205–220.e8. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Villalba, A.; Romero, J.P.; Hernandez, S.C.; Vilas-Zornoza, A.; Fortelny, N.; Castro-Labrador, L.; Martin-Uriz, P.S.; Lorenzo-Vivas, E.; García-Olloqui, P.; Palacios, M.; et al. Single-Cell RNA-seq Analysis Reveals a Crucial Role for Collagen Triple Helix Repeat Containing 1 (CTHRC1) Cardiac Fibroblasts after Myocardial Infarction. Circulation 2020. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.W.; Arostegui, M.; Schweitzer, R.; Rossi, F.M.; Underhill, T.M. Hic1 Defines Quiescent Mesenchymal Progenitor Subpopulations with Distinct Functions and Fates in Skeletal Muscle Regeneration. Cell Stem Cell 2019, 25, 797–813.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Solomonidis, E.G.; Meloni, M.; Taylor, R.S.; Duffin, R.; Dobie, R.; Magalhaes, M.S.; Henderson, B.E.P.; A Louwe, P.; D’Amico, G.; et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 2019, 40, 2507–2520. [Google Scholar] [CrossRef] [PubMed]
- Räsänen, M.; Sultan, I.; Paech, J.; Hemanthakumar, K.A.; Yu, W.; He, L.; Tang, J.; Sun, Y.; Hlushchuk, R.; Huan, X.; et al. VEGF-B Promotes Endocardium-Derived Coronary Vessel Development and Cardiac Regeneration. Circulation 2021, 143, 65–77. [Google Scholar] [CrossRef]
- King, K.R.; Aguirre, A.D.; Ye, Y.-X.; Sun, Y.; Roh, J.D.; Ng, R.P.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef]
- Martini, E.; Kunderfranco, P.; Peano, C.; Carullo, P.; Cremonesi, M.; Schorn, T.; Carriero, R.; Termanini, A.; Colombo, F.S.; Jachetti, E.; et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload–Driven Heart Failure Reveals Extent of Immune Activation. Circulation 2019, 140, 2089–2107. [Google Scholar] [CrossRef]
- Hua, X.; Hu, G.; Hu, Q.; Chang, Y.; Hu, Y.; Gao, L.; Chen, X.; Yang, P.-C.; Zhang, Y.; Li, M.; et al. Single-Cell RNA Sequencing to Dissect the Immunological Network of Autoimmune Myocarditis. Circulation 2020, 142, 384–400. [Google Scholar] [CrossRef]
- Calcagno, D.M.; Ng, R.P.; Toomu, A.; Zhang, C.; Huang, K.; Aguirre, A.D.; Weissleder, R.; Daniels, L.B.; Fu, Z.; King, K.R. The myeloid type I interferon response to myocardial infarction begins in bone marrow and is regulated by Nrf2-activated macrophages. Sci. Immunol. 2020, 5, eaaz1974. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; AlThagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Abplanalp, W.T.; John, D.; Cremer, S.; Assmus, B.; Dorsheimer, L.; Hoffmann, J.; Becker-Pergola, G.; A Rieger, M.; Zeiher, A.M.; Vasa-Nicotera, M.; et al. Single-cell RNA-sequencing reveals profound changes in circulating immune cells in patients with heart failure. Cardiovasc. Res. 2021, 117, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Vafadarnejad, E.; Rizzo, G.; Krampert, L.; Arampatzi, P.; Arias-Loza, A.-P.; Nazzal, Y.; Rizakou, A.; Knochenhauer, T.; Bandi, S.R.; Nugroho, V.A.; et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020, 127. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Lu, Y.; Gu, M.; Li, N.; Liu, M.; Jiao, J.; Zhu, Z.; Li, J.; Li, D.; Tang, T.; et al. A Unique Population of Regulatory T Cells in Heart Potentiates Cardiac Protection From Myocardial Infarction. Circulation 2020, 142, 1956–1973. [Google Scholar] [CrossRef]
- Forte, E.; Daigle, S.; Rosenthal, N.A. Protocol for Isolation of Cardiac Interstitial Cells from Adult Murine Hearts for Unbiased Single Cell Profiling. STAR Protoc. 2020, 1, 100077. [Google Scholar] [CrossRef] [PubMed]
- The Tabula Muris Consortium; Overall Coordination; Logistical Coordination; Organ Collection and Processing; Library Preparation and Sequencing; Computational Data Analysis; Cell Type Annotation; Writing Group; Supplemental Text Writing Group; Principal Investigators. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- The Tabula Muris Consortium A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nat. Cell Biol. 2020, 583, 590–595. [CrossRef]
- Available online: https://tabula-muris-senis.ds.czbiohub.org/ (accessed on 17 February 2021).
- Vidal, R.; Wagner, J.U.G.; Braeuning, C.; Fischer, C.; Patrick, R.; Tombor, L.; Muhly-Reinholz, M.; John, D.; Kliem, M.; Conrad, T.; et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Ma, S.; Sun, S.; Li, J.; Fan, Y.; Qu, J.; Sun, L.; Wang, S.; Zhang, Y.; Yang, S.; Liu, Z.; et al. Single-cell transcriptomic atlas of primate cardiopulmonary aging. Cell Res. 2020, 1–18. [Google Scholar] [CrossRef]
- Kalucka, J.; De Rooij, L.P.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.-A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e20. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.vibcancer.be/software-tools/EC-atlas (accessed on 17 February 2021).
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Available online: https://betsholtzlab.org/Publications/FibroblastMural/database.html (accessed on 17 February 2021).
- Hulin, A.; Hortells, L.; Gomez-Stallons, M.V.; O’Donnell, A.; Chetal, K.; Adam, M.; Lancellotti, P.; Oury, C.; Potter, S.S.; Salomonis, N.; et al. Maturation of heart valve cell populations during postnatal remodeling. Development 2019, 146, dev173047. [Google Scholar] [CrossRef] [Green Version]
- Adamo, L.; Rocha-Resende, C.; Lin, C.-Y.; Evans, S.; Williams, J.; Dun, H.; Li, W.; Mpoy, C.; Andhey, P.S.; Rogers, B.E.; et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Rocha-Resende, C.; Yang, W.; Li, W.; Kreisel, D.; Adamo, L.; Mann, D.L. Developmental changes in myocardial B cells mirror changes in B cells associated with different organs. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfien, M.; Galow, A.-M.; Müller, P.; Bartsch, M.; Brunner, R.M.; Goldammer, T.; Wolkenhauer, O.; Hoeflich, A.; David, R. Single-Nucleus Sequencing of an Entire Mammalian Heart: Cell Type Composition and Velocity. Cells 2020, 9, 318. [Google Scholar] [CrossRef] [Green Version]
- Wolfien, M.; Galow, A.-M.; Müller, P.; Bartsch, M.; Brunner, R.M.; Goldammer, T.; Wolkenhauer, O.; Hoeflich, A.; David, R. Single nuclei sequencing of entire mammalian hearts: Strain-dependent cell-type composition and velocity. Cardiovasc. Res. 2020, 116, 1249–1251. [Google Scholar] [CrossRef] [PubMed]
- Galow, A.-M.; Wolfien, M.; Müller, P.; Bartsch, M.; Brunner, R.M.; Hoeflich, A.; Wolkenhauer, O.; David, R.; Goldammer, T. Integrative Cluster Analysis of Whole Hearts Reveals Proliferative Cardiomyocytes in Adult Mice. Cells 2020, 9, 1144. [Google Scholar] [CrossRef]
- Linscheid, N.; Logantha, S.J.R.J.; Poulsen, P.C.; Zhang, S.; Schrölkamp, M.; Egerod, K.L.; Thompson, J.J.; Kitmitto, A.; Galli, G.; Humphries, M.J.; et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, W.; Shen, Y.; Chen, J.; Zhu, H.; Yang, X.; Jiang, X.-X.; Wang, Y.; Zhou, J. Cardiomyocyte dedifferentiation and remodeling in 3D scaffolds to generate the cellular diversity of engineering cardiac tissues. Biomater. Sci. 2019, 7, 4636–4650. [Google Scholar] [CrossRef]
- Esfahani, P.H.; Elbeck, Z.; Sagasser, S.; Li, X.; Hossain, M.B.; Talukdar, H.A.; Sandberg, R.; Knöll, R. Cell shape determines gene expression: Cardiomyocyte morphotypic transcriptomes. Basic Res. Cardiol. 2020, 115, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.C.; Akkad, A.-D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Van Der Wijst, M.G.P.; LifeLines Cohort Study; Brugge, H.; De Vries, D.H.; Deelen, P.; Swertz, M.A.; Franke, L.; BIOS Consortium. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 2018, 50, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://singlecell.broadinstitute.org/single_cell (accessed on 17 February 2021).
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nat. Cell Biol. 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Mirkov, M.U.; De Leeuw, C.A.; Heuvel, M.P.V.D.; Posthuma, D. Genetic mapping of cell type specificity for complex traits. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.heartcellatlas.org/ (accessed on 17 February 2021).
- Környei, Z.; Beke, S.; Mihálffy, T.; Jelitai, M.; Kovács, K.; Szabó, Z.; Szabó, B. Cell sorting in a Petri dish controlled by computer vision. Sci. Rep. 2013, 3, srep01088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esfahani, P.H.; Knöll, R. An Approach to Study Shape-Dependent Transcriptomics at a Single Cell Level. J. Vis. Exp. 2020, 2020, e61577. [Google Scholar] [CrossRef]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Zheng, Y.-Y.; Ma, Y.-T.; Zhang, J.-Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Madjid, M.; Safavi-Naeini, P.; Solomon, S.D.; Vardeny, O. Potential Effects of Coronaviruses on the Cardiovascular System. JAMA Cardiol. 2020, 5, 831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmerov, A.; Marbán, E. COVID-19 and the Heart. Circ. Res. 2020, 126, 1443–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuomo, V.; Esposito, R.; Santoro, C. Fulminant myocarditis in the time of coronavirus. Eur. Heart J. 2020, 41, 2121. [Google Scholar] [CrossRef]
- Hu, H.; Ma, F.; Wei, X.; Fang, Y. Coronavirus fulminant myocarditis treated with glucocorticoid and human immunoglobulin. Eur. Heart J. 2021, 42, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Fang, Y.; Hu, H. Immune-mediated mechanism in coronavirus fulminant myocarditis. Eur. Heart J. 2020, 41, 1855. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Lv, J.; Lin, L. Coagulopathy in COVID-19: Focus on vascular thrombotic events. J. Mol. Cell. Cardiol. 2020, 146, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini, P.E.; Boutjdir, M.; Capecchi, P.L. COVID-19, Arrhythmic Risk, and Inflammation. Circulation 2020, 142, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, H.; Hang, W.; Wang, D.W. Cardiac injuries in coronavirus disease 2019 (COVID-19). J. Mol. Cell. Cardiol. 2020, 145, 25–29. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Wang, K.; Gheblawi, M.; Oudit, G.Y. Angiotensin Converting Enzyme 2: A Double-Edged Sword. Circulation 2020, 142. [Google Scholar] [CrossRef]
- Anguiano, L.; Riera, M.; Pascual, J.; Valdivielso, J.M.; Barrios, C.; Betriu, A.; Mojal, S.; Fernández, E.; Soler, M.J. Circulating angiotensin-converting enzyme 2 activity in patients with chronic kidney disease without previous history of cardiovascular disease. Nephrol. Dial. Transplant. 2015, 30, 1176–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.; Bridget Tallant, E.; Ann Diz Debra, I.; Gallagher Patricia, E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Ruiz, I. RAAS inhibitors do not increase the risk of COVID-19. Nat. Rev. Cardiol. 2020, 17, 383. [Google Scholar] [CrossRef] [PubMed]
- Ravindra, N.G.; Alfajaro, M.M.; Gasque, V.; Wei, J.; Filler, R.B.; Huston, N.C.; Wan, H.; Szigeti-Buck, K.; Wang, B.; Montgomery, R.R.; et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human bronchial epithelial cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Sharma, A.; Garcia, G.; Wang, Y.; Plummer, J.T.; Morizono, K.; Arumugaswami, V.; Svendsen, C.N. Human iPSC-Derived Cardiomyocytes Are Susceptible to SARS-CoV-2 Infection. Cell Rep. Med. 2020, 1, 100052. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wei, X.; Li, Q.; Li, L.; Yang, Z.; Shi, Y.; Qin, Y.; Zhang, X.; Wang, X.; Zhi, X.; et al. Single-cell RNA analysis on ACE2 expression provides insights into SARS-CoV-2 potential entry into the bloodstream and heart injury. J. Cell. Physiol. 2020, 235, 9884–9894. [Google Scholar] [CrossRef]
- Liu, H.; Gai, S.; Wang, X.; Zeng, J.; Sun, C.; Zhao, Y.; Zheng, Z. Single-cell analysis of SARS-CoV-2 receptor ACE2 and spike protein priming expression of proteases in the human heart. Cardiovasc. Res. 2020, 116, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, Y.; Song, X.; Guo, X.; Pang, J.; Wang, J.; Zhang, S.; Wang, C. Single-cell transcriptional profile of ACE2 in healthy and failing human hearts. Sci. China Life Sci. 2020, 1–4. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, J.D.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, N.R.; Chaffin, M.; Bedi, K.C., Jr.; Papangeli, I.; Akkad, A.-D.; Arduini, A.; Hayat, S.; Eraslan, G.; Muus, C.; Bhattacharyya, R.P.; et al. Myocyte Specific Upregulation of ACE2 in Cardiovascular Disease: Implications for SARS-CoV-2 Mediated Myocarditis. Circulation 2020, 142, 708–710. [Google Scholar] [CrossRef]
- Stoeckius, M.; Zheng, S.; Houck-Loomis, B.; Hao, S.; Yeung, B.Z.; Mauck, W.M.; Smibert, P.; Satija, R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Chen, Y.; Skelly, D.A.; Churchill, G.A. Bayesian model selection reveals biological origins of zero inflation in single-cell transcriptomics. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Gupta, I.; Collier, P.G.; Haase, B.; Mahfouz, A.; Joglekar, A.; Floyd, T.; Koopmans, F.; Barres, B.; Smit, A.B.; A Sloan, S.; et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 2018, 36, 1197–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramsköld, D.; Hendriks, G.-J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef]
- Joglekar, A.; Prjibelski, A.; Mahfouz, A.; Collier, P.; Lin, S.; Anna, K.S.; Jordan, M.; Stephen, R.W.; Bettina, H.; Ashley, H.; et al. Cell-type, single-cell, and spatial signatures of brain-region specific splicing in postnatal development. BioRxiv 2020. [Google Scholar] [CrossRef]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nat. Cell Biol. 2018, 560, 494–498. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.D.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367, eaaw3381. [Google Scholar] [CrossRef] [PubMed]
- Spanjaard, B.; Hu, B.; Mitic, N.; Olivares-Chauvet, P.; Janjuha, S.; Ninov, N.; Junker, J.P. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat. Biotechnol. 2018, 36, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.M.; Smith, Z.D.; Grosswendt, S.; Kretzmer, H.; Norman, T.M.; Adamson, B.; Jost, M.; Quinn, J.J.; Yang, D.; Jones, M.G.; et al. Molecular recording of mammalian embryogenesis. Nat. Cell Biol. 2019, 570, 77–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially Resolved Transcriptomes—Next Generation Tools for Tissue Exploration. BioEssays 2020, 42, e1900221. [Google Scholar] [CrossRef] [PubMed]
- Kuppe, C.; Ramirez, F.R.O.; Li, Z.; Hannani, M.T.; Tanevski, J.; Halder, M.; Cheng, M.; Ziegler, S.; Zhang, X.; Preisker, F.; et al. Spatial mul-ti-omic map of human myocardial infarction. BioRxiv 2020. [Google Scholar] [CrossRef]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]
- Füllgrabe, A.; George, N.; Green, M.; Nejad, P.; Aronow, B.; Fexova, S.K.; Fischer, C.; Freeberg, M.A.; Huerta, L.; Morrison, N.; et al. Guidelines for reporting single-cell RNA-seq experiments. Nat. Biotechnol. 2020, 38, 1–3. [Google Scholar] [CrossRef]

| Authors PMID | Date | # of Cells and/or Nuclei | Isolation Method | Sequencing Technology | Target Cell Types | Context |
|---|---|---|---|---|---|---|
| Chen et al. [180] PMID: 32227090 | March 2020 | does not specify | enzymatic digestion, nuclei isolation | Chromium Single Cells 3’ v3 (10x Genomics); Illumina Hi-seq Xten | all human heart cell types | hACE2 expression in healthy and failing human hearts |
| Nicin et al. [181] PMID: 32293672 | April 2020 | 57,601 | enzymatic digestion, nuclei isolation | does not specify | all human heart cell types | hACE2 expression in healthy and failing human hearts (1 healthy, 5 aortic stenosis, 2 HFrEF patients) |
| Tucker and Chaffin et al. [182] PMID: 32795091 | June 2020 | 677,785 | mechanical and enzymatic digestion, nuclei isolation | Chromium Single Cells 3’ v3 (10x Genomics) | All human LV cell types | Healthy (n = 11) and failing adult human heart (11 dilated-, 15 hypertrophic- cardiomyopathy) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forte, E.; McLellan, M.A.; Skelly, D.A.; Rosenthal, N.A. Ex uno, plures–From One Tissue to Many Cells: A Review of Single-Cell Transcriptomics in Cardiovascular Biology. Int. J. Mol. Sci. 2021, 22, 2071. https://doi.org/10.3390/ijms22042071
Forte E, McLellan MA, Skelly DA, Rosenthal NA. Ex uno, plures–From One Tissue to Many Cells: A Review of Single-Cell Transcriptomics in Cardiovascular Biology. International Journal of Molecular Sciences. 2021; 22(4):2071. https://doi.org/10.3390/ijms22042071
Chicago/Turabian StyleForte, Elvira, Micheal A. McLellan, Daniel A. Skelly, and Nadia A. Rosenthal. 2021. "Ex uno, plures–From One Tissue to Many Cells: A Review of Single-Cell Transcriptomics in Cardiovascular Biology" International Journal of Molecular Sciences 22, no. 4: 2071. https://doi.org/10.3390/ijms22042071
APA StyleForte, E., McLellan, M. A., Skelly, D. A., & Rosenthal, N. A. (2021). Ex uno, plures–From One Tissue to Many Cells: A Review of Single-Cell Transcriptomics in Cardiovascular Biology. International Journal of Molecular Sciences, 22(4), 2071. https://doi.org/10.3390/ijms22042071