
mTOR Signaling in Pulmonary Vascular Disease: Pathogenic Role and Therapeutic Target

Abstract
:1. Introduction
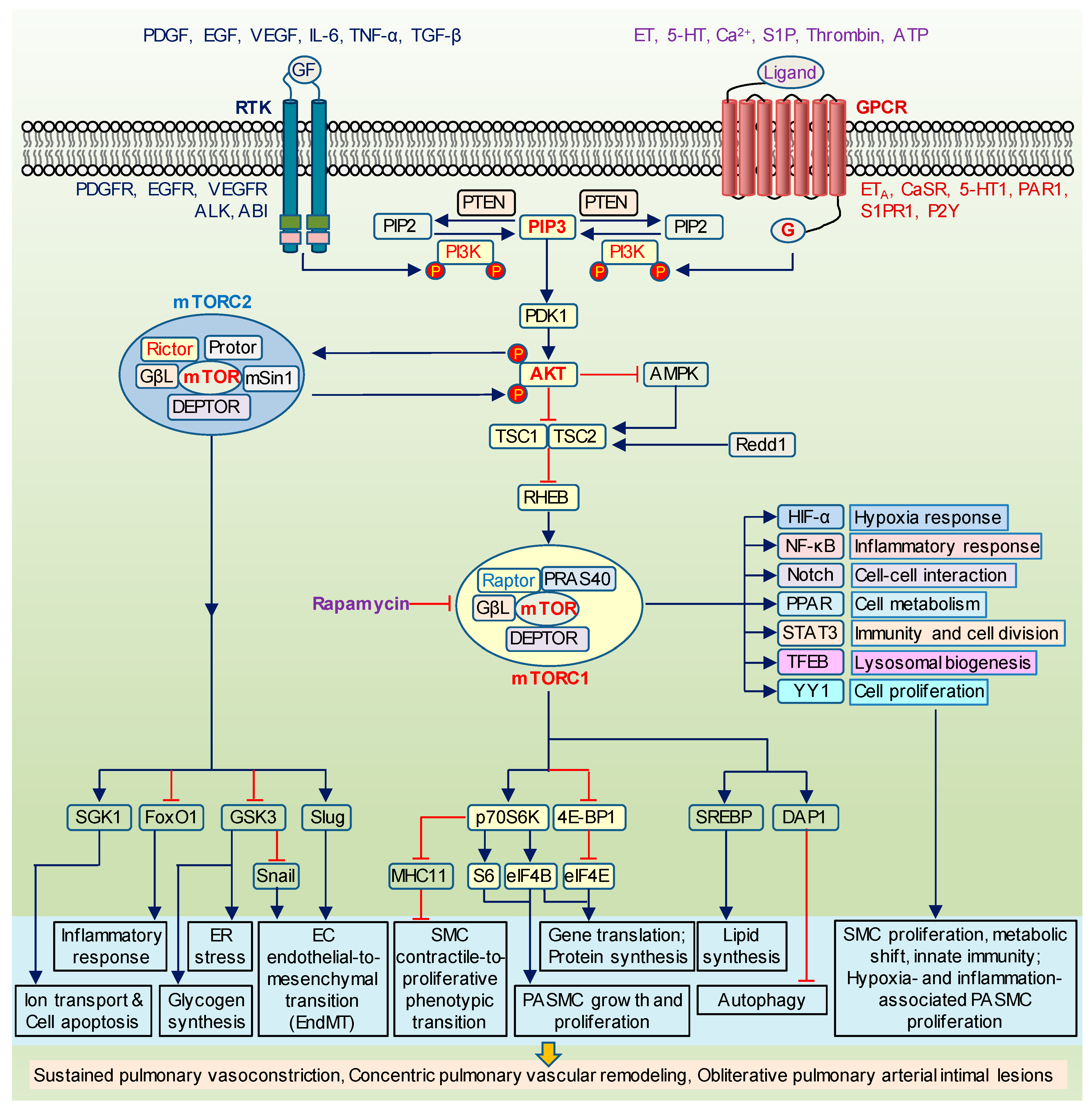
1.1. The Canonical PI3K/AKT/mTOR Signaling
1.2. Structural and Functional Differences between mTORC1 and mTORC2
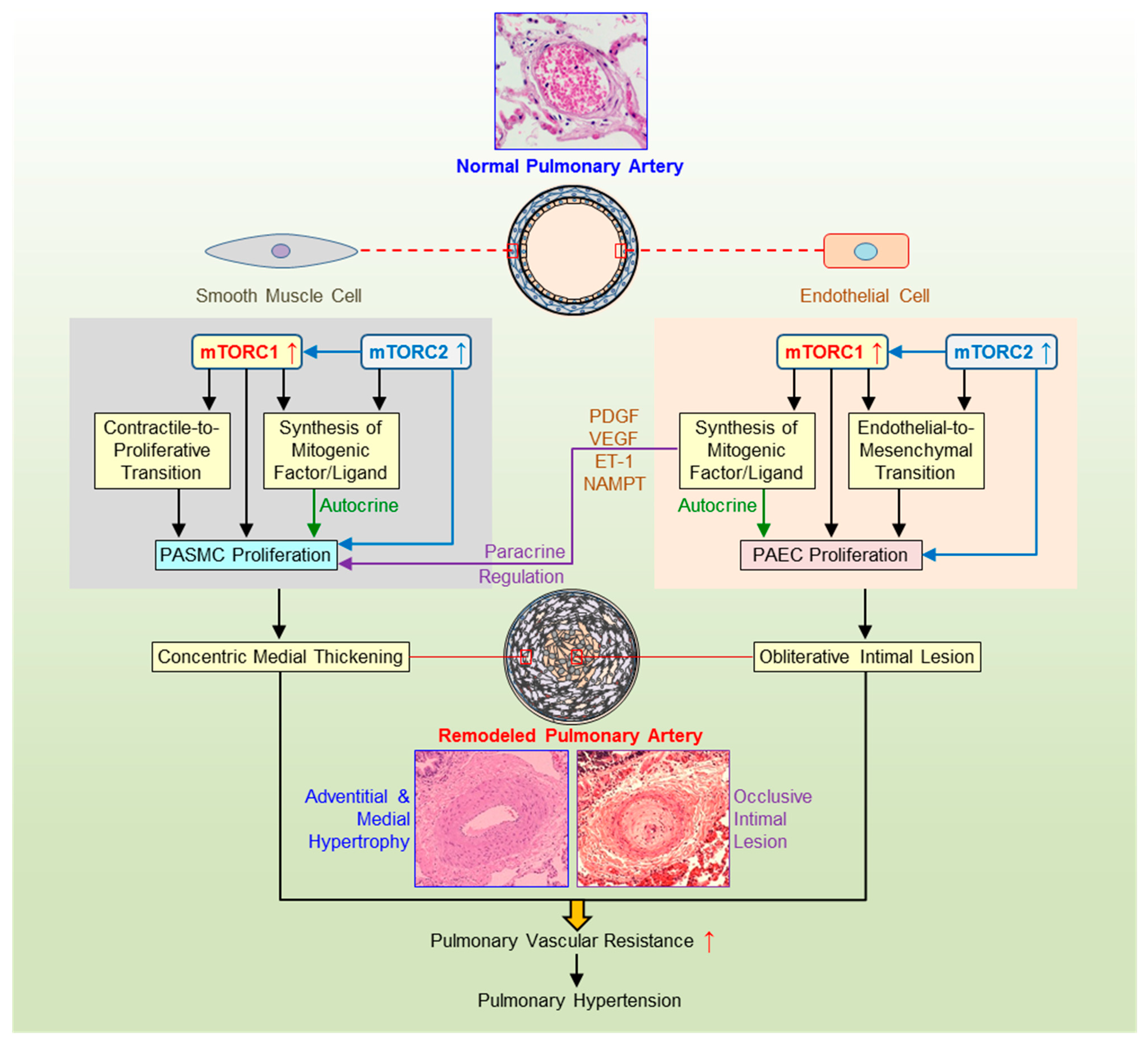
1.3. AKT/mTOR and PASMC Proliferation
1.4. AKT/mTOR and Phenotypical Transition of SMC
1.5. AKT/mTOR in SMC-EC Communication
1.6. AKT/mTOR and EC Dysfunction
1.7. AKT/mTOR Signaling as a Therapeutic Target for PH
2. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Cassady, S.J.; Ramani, G.V. Right heart failure in pulmonary hypertension. Cardiol. Clin. 2020, 38, 243–255. [Google Scholar] [CrossRef]
- Maron, B.A.; Kovacs, G.; Vaidya, A.; Bhatt, D.L.; Nishimura, R.A.; Mak, S.; Guazzi, M.; Tedford, R.J. Cardiopulmonary hemodynamics in pulmonary hypertension and heart failure: JACC review topic of the week. J. Am. Coll. Cardiol. 2020, 76, 2671–2681. [Google Scholar] [CrossRef]
- Goncharova, E.A.; Chan, S.Y.; Ventetuolo, C.E.; Weissmann, N.; Schermuly, R.T.; Mullin, C.J.; Gladwin, M.T. Update in pulmonary vascular diseases and right ventricular dysfunction 2019. Am. J. Respir. Crit. Care Med. 2020, 202, 22–28. [Google Scholar] [CrossRef]
- Maron, B.A.; Brittan, E.L.; Hess, E.; Waldo, S.W.; Barón, A.E.; Huang, S.; Goldstein, R.H.; Assad, T.; Wertheim, B.M.; Alba, G.A.; et al. Pulmonary vascular resistance and clinical outcomes in patients with pulmonary hypertension: A retrospective cohort study. Lancet Respir. Med. 2020, 8, 873–884. [Google Scholar] [CrossRef]
- Ratwatte, S.; Anderson, J.; Strange, G.; Corrigan, C.; Collins, N.; Celermajer, D.S.; Dwyer, N.; Feenstra, J.; Keating, D.; Kotlyar, E.; et al. Pulmonary arterial hypertension with below threshold pulmonary vascular resistance. Eur. Respir. J. 2020, 56, 1901654. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lui, K.O.; Zhou, B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat. Rev. Cardiol. 2018, 15, 445–456. [Google Scholar] [CrossRef]
- Lu, X.; Gong, J.; Dennery, P.A.; Yao, H. Endothelial-to-mesenchymal transition: Pathogenesis and therapeutic targets for chronic pulmonary and vascular diseases. Biochem. Pharmacol. 2019, 168, 100–107. [Google Scholar] [CrossRef]
- Leopold, J.A.; Maron, B.A. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int. J. Mol. Sci. 2016, 17, 761. [Google Scholar] [CrossRef]
- Babicheva, A.; Ayon, R.J.; Zhao, T.; Ek Vitorin, J.F.; Pohl, N.M.; Yamamura, A.; Quinton, B.A.; Ba, M.; Wu, L.; Ravellette, K.; et al. MicroRNA-mediated downregulation of K+ channels in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L10–L26. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.; Capuano, V.; Boet, A.; Tesson, L.; Bertero, T.; Nakhleh, M.K.; Remy, S.; Anegon, I.; Pechoux, C.; Hautefort, A.; et al. Characterization of Kcnk3-mutated rat, a novel model of pulmonary hypertension. Circ. Res. 2019, 125, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, J.; Fraidenburg, D.R.; Song, S.; Sysol, J.R.; Drennan, A.R.; Offermanns, S.; Ye, R.D.; Bonini, M.G.; Minshall, R.D.; et al. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L208–L220. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Wu, K.; Wang, J.; Vinjamuri, S.; Gu, Y.; Song, S.; Wang, Z.; Zhang, Q.; Balistrieri, A.; Ayon, R.J.; et al. Pathogenic role of mTORC1 and mTORC2 in pulmonary hypertension. JACC Basic Transl. Sci. 2018, 3, 744–762. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Yamamura, A.; Yamamura, H.; Song, S.; Fraidenburg, D.R.; Chen, J.; Gu, Y.; Pohl, N.M.; Zhou, T.; Jiménez-Pérez, L.; et al. Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L846–L859. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.; Capuano, V.; Olschewski, A.; Sabourin, J.; Nagaraj, C.; Girerd, B.; Weatherald, J.; Humbert, M.; Antigny, F. Ion channels in pulmonary hypertension: A therapeutic interest? Int. J. Mol. Sci. 2018, 19, 3162. [Google Scholar] [CrossRef] [Green Version]
- Rol, N.; Kurakula, K.B.; Happé, C.; Bogaard, H.J.; Goumans, M.-J. TGF-β and BMPR2 signaling in PAH: Two black sheep in one family. Int. J. Mol. Sci. 2018, 19, 2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Bórnez, M.; Galeano-Otero, I.; Del Toro, R.; Smani, T. TRPC and TRPV channels’ role in vascular remodeling and disease. Int. J. Mol. Sci. 2020, 21, 6125. [Google Scholar] [CrossRef]
- Morris, H.E.; Neves, K.B.; Montezano, A.C.; MacLean, M.R.; Touyz, R.M. Notch3 signaling and vascular remodeling in pulmonary arterial hypertension. Clin. Sci. 2019, 133, 2481–2498. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K sig-naling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumor suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Barilli, A.; Visigalli, R.; Sala, R.; Gazzola, G.C.; Parolari, A.; Tremoli, E.; Bonomini, S.; Simon, A.; Closs, E.I.; Dall’Asta, V.; et al. In human endothelial cells rapamycin causes mTORC2 inhibition and impairs cell viability and function. Cardiovasc. Res. 2008, 78, 563–571. [Google Scholar] [CrossRef]
- Zheng, M.; Zang, S.; Xie, L.; Fang, X.; Zhang, Y.; Ma, X.; Liu, J.; Lin, D.; Huang, A. Rheb phosphorylation is involved in p38-regulated/activated protein kinase-mediated tumor suppression in liver cancer. Oncol. Lett. 2015, 10, 1655–1661. [Google Scholar] [CrossRef]
- Sato, T.; Umetsu, A.; Tamanoi, F. Characterization of the Rheb-mTOR signaling pathway in mammalian cells: Constitutive active mutants of Rheb and mTOR. Methods Enzymol. 2008, 438, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of Raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Schwarzer, R.; Tondera, D.; Arnold, W.; Giese, K.; Klippel, A.; Kaufmann, J. REDD1 integrates hypoxia-mediated survival signaling downstream of phosphatidylinositol 3-kinase. Oncogene 2004, 24, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Horak, P.; Crawford, A.R.; Vadysirisack, D.D.; Nash, Z.M.; DeYoung, M.P.; Sgroi, D.; Ellisen, L.W. Negative feedback control of HIF-1 through REDD1-regulated ROS sup-presses tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 4675–4680. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G.J. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Wiza, C.; Nascimento, E.B.M.; Ouwens, D.M. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am. J. Physiol. Metab. 2012, 302, E1453–E1460. [Google Scholar] [CrossRef]
- Ruffolo, R.R.J.; Nichols, A.J. The relationship of receptor reserve and agonist efficacy to the sensitivity of α-adrenoceptor-mediated vasopressor responses to inhibition by calcium channel antagonists. Ann. N. Y. Acad. Sci. 1988, 522, 361–376. [Google Scholar] [CrossRef]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A positive feedback loop between Akt and mTORC2 via SIN1 phosphor-ylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Koren, I.; Reem, E.; Kimchi, A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr. Biol. 2010, 20, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Park, J.S.; Lee, D.H.; Han, J.; Bae, S.H. Ezetimibe ameliorates lipid accumulation during adipogenesis by regulating the AMPK-mTORC1 pathway. FASEB J. 2020, 34, 898–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [Green Version]
- Summer, R.; Shaghaghi, H.; Schriner, D.; Roque, W.; Sales, D.; Cuevas-Mora, K.; Desai, V.; Bhushan, A.; Ramirez, M.I.; Romero, F. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L1049–L1060. [Google Scholar] [CrossRef] [PubMed]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erazo, T.; Lorente, M.; López-Plana, A.; Muñoz-Guardiola, P.; Fernández-Nogueira, P.; García-Martínez, J.A.; Bragado, P.; Fuster, G.; Salazar, M.; Espadaler, J.; et al. The new antitumor drug ABTL0812 inhibits the Akt/mTORC1 axis by upregulating Tribbles-3 pseudokinase. Clin. Cancer Res. 2015, 22, 2508–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanovska, J.; Shah, S.; Wong, M.J.; Kantores, C.; Jain, A.; Post, M.; Yeganeh, B.; Jankov, R.P. mTOR-Notch3 signaling mediates pulmonary hypertension in hypoxia-exposed neonatal rats independent of changes in autophagy. Pediatr. Pulmonol. 2017, 52, 1443–1454. [Google Scholar] [CrossRef]
- Li, Y.; Yang, L.; Dong, L.; Yang, Z.W.; Zhang, J.; Zhang, S.L.; Niu, M.-J.; Xia, J.-W.; Gong, Y.; Zhu, N.; et al. Crosstalk between the Akt/mTORC1 and NF-κB signaling pathways promotes hypoxia-induced pulmonary hypertension by increasing DPP4 expression in PASMCs. Acta Pharmacol. Sin. 2019, 40, 1322–1333. [Google Scholar] [CrossRef]
- Wang, W.; Liu, J.; Ma, A.; Miao, R.; Jin, Y.; Zhang, H.; Xu, K.; Wang, C.; Wang, J. mTORC1 is involved in hypoxia-induced pulmonary hypertension through the activation of Notch3. J. Cell Physiol. 2014, 229, 2117–2125. [Google Scholar] [CrossRef]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.; Wang, H.; Suttles, J.; Graves, D.T.; Martin, M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates Toll-like receptor 4-mediated inflammatory response via FoxO1. J. Biol. Chem. 2011, 286, 44295–44305. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, G.; Yu, K.; Jiang, Z.; Chung, A.; Yao, J.; Ha, C.; Toy, K.; Soriano, R.; Haley, B.; Blackwood, E.; et al. Phosphoproteomic analysis implicates the mTORC2-FoxO1 axis in VEGF signaling and feedback activation of receptor tyrosine kinases. Sci. Signal. 2013, 6, ra25. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Xiong, X.; Liangpunsakul, S.; Dong, X.C. Sestrin 3 protein enhances hepatic insulin sensitivity by direct activation of the mTORC2-Akt signaling. Diabetes 2014, 64, 1211–1223. [Google Scholar] [CrossRef] [Green Version]
- Schaub, T.; Gürgen, D.; Maus, D.; Lange, C.; Tarabykin, V.; Dragun, D.; Hegner, B. mTORC1 and mTORC2 differentially regulate cell fate programs to coordinate osteoblastic differentiation in mesenchymal stromal cells. Sci. Rep. 2019, 9, 20071. [Google Scholar] [CrossRef]
- Xie, J. Proud CG. Crosstalk between mTOR complexes. Nat. Cell Biol. 2013, 15, 1263–1265. [Google Scholar] [CrossRef]
- Rosner, M.; Fuchs, C.; Siegel, N.; Valli, A.; Hengstschlager, M. Functional interaction of mammalian target of rapamycin com-plexes in regulating mammalian cell size and cell cycle. Hum. Mol. Genet. 2009, 18, 3298–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebner, M.; Sinkovics, B.; Szczygieł, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [Green Version]
- Houssaïni, A.; Abid, S.; Mouraret, N.; Wan, F.; Rideau, D.; Saker, M.; Marcos, E.; Tissot, C.-M.; Dubois-Randé, J.-L.; Amsellem, V.; et al. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 48, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Liu, X.; Zhang, Y.; Qiu, H.; Ouyang, F.; He, Y. 3-Bromopyruvate ameliorates pulmonary arterial hypertension by improving mitochondrial metabolism. Life Sci. 2020, 256, 118009. [Google Scholar] [CrossRef] [PubMed]
- Krymskaya, V.P.; Snow, J.; Cesarone, G.; Khavin, I.; Goncharov, D.A.; Lim, P.N.; Veasey, S.C.; Ihida-Stansbury, K.; Jones, P.L.; Goncharova, E.A. mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J. 2011, 25, 1922–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, T.V.; Goncharov, D.A.; Pena, A.; Kelly, N.; Vanderpool, R.; Baust, J.; Kobir, A.; Shufesky, W.; Mora, A.L.; Morelli, A.E.; et al. HIPPO–integrin-linked kinase cross-talk controls self-sustaining proliferation and survival in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Gu, Y.; Luo, J.; Ye, P.; Zheng, Y.; Yu, W.; Chen, S. Inhibition of Src activation reverses pulmonary vascular remodeling in experimental pulmonary arterial hypertension via Akt/mTOR/HIF-1α signaling pathway. Exp. Cell Res. 2019, 380, 36–46. [Google Scholar] [CrossRef]
- Ogawa, A.; Firth, A.L.; Yao, W.; Madani, M.M.; Kerr, K.M.; Auger, W.R.; Jamieson, S.W.; Thistlethwaite, P.A.; Yuan, J.X.-J. Inhibition of mTOR attenuates store-operated Ca2+ entry in cells from endarterectomized tissues of patients with chronic thromboembolic pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L666–L676. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.-P.; Li, X.-H.; Yang, Y.-M.; Li, W.-Q.; Zhang, W.; Hu, C.-P.; Zhang, Z.; Li, Y.-J. A Critical role of the mTOR/eIF2α pathway in hypoxia-induced pulmonary hypertension. PLoS ONE 2015, 10, e0130806. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Yu, J.; Taylor, L.; Polgar, P. Hyperplastic growth of pulmonary artery smooth muscle cells from subjects with pulmonary arterial hypertension is activated through JNK and p38 MAPK. PLoS ONE 2015, 10, e0123662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemenoff, R.A.; Simpson, P.A.; Furgeson, S.B.; Kaplan-Albuquerque, N.; Crossno, J.; Garl, P.J.; Cooper, J.; Weiser-Evans, M.C. Targeted deletion of PTEN in smooth muscle cells results in vascular remodeling and recruitment of progenitor cells through induction of stromal cell-derived factor-1α. Circ. Res. 2008, 102, 1036–1045. [Google Scholar] [CrossRef] [Green Version]
- Horita, H.; Furgeson, S.B.; Ostriker, A.; Olszewski, K.A.; Sullivan, T.; Villegas, L.R.; Levine, M.; Parr, J.E.; Cool, C.D.; Nemenoff, R.A.; et al. Selective inactivation of PTEN in smooth muscle cells synergizes with hypoxia to induce severe pulmonary hypertension. J. Am. Heart Assoc. 2013, 2, e000188. [Google Scholar] [CrossRef] [Green Version]
- Horita, H.; Wysoczynski, C.L.; Walker, L.A.; Moulton, K.S.; Li, M.; Ostriker, A.; Tucker, R.; McKinsey, T.A.; Churchill, M.E.A.; Nemenoff, R.A.; et al. Nuclear PTEN functions as an essential regulator of SRF-dependent transcription to control smooth muscle differentiation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Di, R.; Yang, Z.; Xu, P.; Xu, Y. Silencing PDK1 limits hypoxia-induced pulmonary arterial hypertension in mice via the Akt/p70S6K signaling pathway. Exp. Ther. Med. 2019, 18, 699–704. [Google Scholar] [CrossRef]
- Wang, H.L.; Tang, F.Q.; Jiang, Y.H.; Zhu, Y.; Jian, Z.; Xiao, Y.B. AMPKα2 deficiency exacerbates hypoxia-induced pulmonary hypertension by promoting pulmonary arterial smooth muscle cell proliferation. J. Physiol. Biochem. 2020, 76, 445–456. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, J.; Martin, M.; He, M.; Gongol, B.; Marin, T.L.; Chen, L.; Shi, X.; Yin, Y.; Shang, F.; et al. AMP-activated protein kinase phosphorylation of angiotensin-converting enzyme 2 in endothelium mitigates pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 509–520. [Google Scholar] [CrossRef]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Otsuki, T.; Kozu, K.; Numano, K.; Suzuki, K.; et al. Protective roles of endothelial AMP-activated protein kinase against hypoxia-induced pulmonary hypertension in mice. Circ. Res. 2016, 119, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Houssaini, A.; Abid, S.; Derumeaux, G.; Wan, F.; Parpaleix, A.; Rideau, D.; Marcos, E.; Kebe, K.; Czibik, G.; Sawaki, D.; et al. Selective tuberous sclerosis complex 1 gene deletion in smooth muscle activates mammalian target of rapamycin signaling and induces pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 55, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Schermuly, R.T.; Ellinghaus, P.; Milting, H.; Riedl, B.; Nikolova, S.; Pullamsetti, S.S.; Weissmann, N.; Dony, E.; Savai, R.; et al. Combined tyrosine and serine/threonine kinase inhibition by sorafenib prevents progression of experimental pulmonary hypertension and myocardial remodeling. Circulation 2008, 118, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Leong, Z.P.; Hikasa, Y. Effects of toceranib compared with sorafenib on monocrotaline-induced pulmonary arterial hypertension and cardiopulmonary remodeling in rats. Vascul. Pharmacol. 2018, 110, 31–41. [Google Scholar] [CrossRef]
- Agard, C.; Rolli-Derkinderen, M.; Dumas-De-La-Roque, E.; Rio, M.; Sagan, C.; Savineau, J.P.; Loirand, G.; Pacaud, P. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br. J. Pharmacol. 2009, 158, 1285–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, A.; Nilsen, M.; Loughlin, L.; Salt, I.P.; MacLean, M.R. Metformin reverses development of pulmonary hypertension via aromatase inhibition. Hypertension 2016, 68, 446–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Fan, R.; Lu, Y.; Yu, C.; Xu, X.; Zhang, X.; Liu, P.; Yan, S.; Chen, C.; Wang, L. Regulatory effect of AMP-activated protein kinase on pulmonary hypertension induced by chronic hypoxia in rats: In vivo and in vitro studies. Mol. Biol. Rep. 2014, 41, 4031–4041. [Google Scholar] [CrossRef] [PubMed]
- Paddenberg, R.; Stieger, P.; Von Lilien, A.-L.; Faulhammer, P.; Goldenberg, A.; Tillmanns, H.H.; Kummer, W.; Braun-Dullaeus, R.C. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir. Res. 2007, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zuo, C.; Jia, D.; Bai, P.; Kong, D.; Chen, D.; Liu, G.; Li, J.; Wang, Y.; Chen, G.; et al. Loss of DP1 aggravates vascular remodeling in pulmonary arterial hypertension via mTORC1 signaling. Am. J. Respir. Crit. Care Med. 2020, 201, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.-P.; Li, X.-H.; Gong, S.-X.; Li, W.-Q.; Hu, C.-P.; Zhang, Z.; Li, Y.-J. miR-100 suppresses mTOR signaling in hypoxia-induced pulmonary hypertension in rats. Eur. J. Pharmacol. 2015, 765, 565–573. [Google Scholar] [CrossRef]
- Liu, X.; Wang, G.; You, Z.; Qian, P.; Chen, H.; Dou, Y.; Wei, Z.; Chen, J.; Mao, C.; Zhang, J. Inhibition of hypoxia-induced proliferation of pulmonary arterial smooth muscle cells by a mTOR siRNA-loaded cyclodextrin nanovector. Biomaterials 2014, 35, 4401–4416. [Google Scholar] [CrossRef]
- Pena, A.; Kobir, A.; Goncharov, D.; Goda, A.; Kudryashova, T.V.; Ray, A.; Vanderpool, R.; Baust, J.; Chang, B.; Mora, A.L.; et al. Pharmacological inhibition of mTOR kinase reverses right ventricle remodeling and improves right ventricle structure and function in rats. Am. J. Respir. Cell Mol. Biol. 2017, 57, 615–625. [Google Scholar] [CrossRef]
- Miao, L.; Yang, L.; Huang, H.; Liang, F.; Ling, C.; Hu, Y. mTORC1 is necessary but mTORC2 and GSK3β are inhibitory for AKT3-induced axon regeneration in the central nervous system. eLife 2016, 5, e14908. [Google Scholar] [CrossRef]
- Lee, J.; Heo, J.; Kang, H. miR-92b-3p-TSC1 axis is critical for mTOR signaling-mediated vascular smooth muscle cell proliferation induced by hypoxia. Cell Death Differ. 2019, 26, 1782–1795. [Google Scholar] [CrossRef] [Green Version]
- Ke, R.; Liu, L.; Zhu, Y.; Li, S.; Xie, X.; Li, F.; Song, Y.; Yang, L.; Gao, L.; Li, M. Knockdown of AMPKα2 promotes pulmonary arterial smooth muscle cells proliferation via mTOR/Skp2/p27Kip1 signaling pathway. Int. J. Mol. Sci. 2016, 17, 844. [Google Scholar] [CrossRef] [Green Version]
- Aghamohammadzadeh, R.; Zhang, Y.; Stephens, T.E.; Arons, E.; Zaman, P.; Polach, K.J.; Matar, M.; Yung, L.; Yu, P.B.; Bowman, F.P.; et al. Up-regulation of the mammalian target of rapamycin complex 1 subunit Raptor by aldosterone induces abnormal pulmonary artery smooth muscle cell survival patterns to promote pulmonary arterial hypertension. FASEB J. 2016, 30, 2511–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Tang, H.; Lin, R.; Carr, S.G.; Wang, Z.; Babicheva, A.; Ayon, R.J.; Jain, P.P.; Xiong, M.; Rodriguez, M.; et al. Endothelial platelet-derived growth factor-mediated activation of smooth muscle platelet-derived growth factor receptors in pulmonary arterial hypertension. Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Z.; Li, J.; Ban, Y.; Mao, G.; Zhang, M.; Wang, M.; Liu, Y.; Zhao, B.; Shen, Q.; et al. Inhibition of 5-hydroxytryptamine receptor 2B reduced vascular restenosis and mitigated the β-arrestin2–mammalian target of rapamycin/p70S6K pathway. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Penumatsa, K.; Abualkhair, S.; Wei, L.; Warburton, R.; Preston, I.; Hill, N.S.; Watts, S.W.; Fanburg, B.L.; Toksoz, D. Tissue transglutaminase promotes serotonin-induced AKT signaling and mitogenesis in pulmonary vascular smooth muscle cells. Cell. Signal. 2014, 26, 2818–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hester, J.; Ventetuolo, C.; Lahm, T. Sex, gender, and sex hormones in pulmonary hypertension and right ventricular failure. Compr. Physiol. 2019, 10, 125–170. [Google Scholar] [CrossRef]
- Docherty, C.K.; Harvey, K.Y.; Mair, K.M.; Griffin, S.; Denver, N.; MacLean, M.R. The role of sex in the pathophysiology of pulmonary hypertension. Adv. Exp. Med. Biol. 2018, 1065, 511–528. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Faul, J.L.; Berry, G.J.; Veve, I.; Pearl, R.G.; Kao, P.N. 40-O-(2-Hydroxyethyl)-rapamycin attenuates pulmonary arterial hypertension and neointimal formation in rats. Am. J. Respir. Crit. Care Med. 2001, 163, 498–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, A.; Firth, A.L.; Ariyasu, S.; Yamadori, I.; Matsubara, H.; Song, S.; Fraidenburg, D.R.; Yuan, J.X.J. Thrombin-mediated activation of Akt signaling contributes to pulmonary vascular remodeling in pulmonary hypertension. Physiol. Rep. 2013, 1, e00190. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Peng, H.; Hong, C.; Chen, Z.; Deng, X.; Wang, A.; Yang, F.; Yang, L.; Chen, C.; Qin, X. PDGF promotes the Warburg effect in pulmonary arterial smooth muscle cells via activation of the PI3K/AKT/mTOR/HIF-1α signaling pathway. Cell. Physiol. Biochem. 2017, 42, 1603–1613. [Google Scholar] [CrossRef]
- Song, Y.; Wu, Y.; Su, X.; Zhu, Y.; Liu, L.; Pan, Y.; Zhu, B.; Yang, L.; Gao, L.; Li, M. Activation of AMPK inhibits PDGF-induced pulmonary arterial smooth muscle cells proliferation and its potential mechanisms. Pharmacol. Res. 2016, 107, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Ogaw, A.; Firth, A.L.; Smith, K.A.; Maliakal, M.V.; Yuan, J.X. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am. J. Physiol. Cell. Physiol. 2012, 302, C405–C411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Freyhaus, H.; Berghausen, E.M.; Janssen, W.; Leuchs, M.; Zierden, M.; Murmann, K.; Klinke, A.; Vantler, M.; Caglayan, E.; Rosenkranz, S. Genetic ablation of PDGF-dependent signaling pathways abolishes vascular remodeling and experimental pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Segura-Ibarra, V.; Amione-Guerra, J.; Cruz-Solbes, A.S.; Cara, F.E.; Iruegas-Nunez, D.A.; Wu, S.; Youker, K.A.; Bhimaraj, A.; Torre-Amione, G.; Ferrari, M.; et al. Rapamycin nanoparticles localize in diseased lung vasculature and prevent pulmonary arterial hypertension. Int. J. Pharm. 2017, 524, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wei, C.; Li, H.-Z.; Bai, S.-Z.; Wang, L.-N.; Xi, Y.-H.; Yan, J.; Xu, C.-Q.; Li, H.-X. NPS2390, a selective calcium-sensing receptor antagonist controls the phenotypic modulation of hypoxic human pulmonary arterial smooth muscle cells by regulating autophagy. J. Transl. Intern. Med. 2019, 7, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Y.; Di, C.; Liu, T.; Hou, Q.; Wang, S. Protective effects of transient receptor potential canonical channels on oxy-gen-glucose deprivation-induced cell injury in neurons and PC12 cells. NeuroReport 2016, 27, 1072–1080. [Google Scholar] [CrossRef]
- Yu, Y.; Sweeney, M.; Zhang, S.; Platoshyn, O.; Landsberg, J.; Rothman, A.; Yuan, J.X.-J. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am. J. Physiol. Physiol. 2003, 284, C316–C330. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, R.A.; Wan, J.; Song, S.; Smith, K.A.; Gu, Y.; Tauseef, M.; Tang, H.; Makino, A.; Mehta, L.; Yuan, J.X.-J. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am. J. Physiol. Physiol. 2015, 308, C581–C593. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.A.; Ayon, R.J.; Tang, H.; Makino, A.; Yuan, J.X.-J. Calcium-sensing receptor regulates cytosolic [Ca2+] and plays a major role in the development of pulmonary hypertension. Front. Physiol. 2016, 7, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, S.; Heger, J.; Euler, G.; Wartenberg, M.; Piper, H.M.; Sauer, H. Platelet-derived growth factor BB stimulates vasculogenesis of embryonic stem cell-derived endothelial cells by calcium-mediated generation of reactive oxygen species. Cardiovasc. Res. 2008, 81, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenbeck, S.T.; Nelson, P.R.; Yamamura, S.; Faries, P.L.; Liu, B.; Kent, K.C. Intracellular calcium transients are necessary for platelet-derived growth factor but not extracellular matrix protein-induced vascular smooth muscle cell migration. J. Vasc. Surg. 2004, 40, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Pu, Z.; Wang, J.; Zhang, Z.; Hu, D.; Wang, J. Baicalin inhibits hypoxia-induced pulmonary artery smooth muscle cell proliferation via the AKT/HIF-1α/p27-associated pathway. Int. J. Mol. Sci. 2014, 15, 8153–8168. [Google Scholar] [CrossRef] [PubMed]
- Hibdon, E.S.; Razumilava, N.; Keeley, T.M.; Wong, G.; Solanki, S.; Shah, Y.M.; Samuelson, L.C. Notch and mTOR signaling pathways promote human gastric cancer cell proliferation. Neoplasia 2019, 21, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Okuhashi, Y.; Itoh, M.; Nara, N.; Tohda, S. NOTCH knockdown affects the proliferation and mTOR signaling of leukemia cells. Anticancer Res. 2013, 33, 4293–4298. [Google Scholar] [PubMed]
- Zhao, N.; Guo, Y.; Zhang, M.; Lin, L.; Zheng, Z. Akt-mTOR signaling is involved in Notch-1-mediated glioma cell survival and proliferation. Oncol. Rep. 2010, 23, 1443–1447. [Google Scholar] [PubMed]
- Furgeson, S.B.; Simpson, P.A.; Park, I.; VanPutten, V.; Horita, H.; Kontos, C.D.; Nemenoff, R.A.; Weiser-Evans, M.C. Inactivation of the tumor suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation. Cardiovasc. Res. 2010, 86, 274–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, A.; Firth, A.L.; Yao, W.; Rubin, L.J.; Yuan, J.X. Prednisolone inhibits PDGF-induced nuclear translocation of NF-κB in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L648–L657. [Google Scholar] [CrossRef] [Green Version]
- Langleben, D.; Orfanos, S. Vasodilator responsiveness in idiopathic pulmonary arterial hypertension: Identifying a distinct phenotype with distinct physiology and distinct prognosis. Pulm. Circ. 2017, 7, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Medarov, B.I.; Judson, M.A. The role of calcium channel blockers for the treatment of pulmonary arterial hypertension: How much do we actually know and how could they be positioned today? Respir. Med. 2015, 109, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Barst, R.J.; Maislin, G.; Fishman, A.P. Vasodilator therapy for primary pulmonary hypertension in children. Circulation 1999, 99, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, A.R.; Alnuaimat, H.; Mubarak, K. Pulmonary vasodilator testing and use of calcium channel blockers in pulmonary arterial hypertension. Respir. Med. 2010, 104, 481–496. [Google Scholar] [CrossRef] [Green Version]
- Yamaki, S.; Wagenvoort, C.A. Comparison of primary plexogenic arteriopathy in adults and children. A morphometric study in 40 patients. Br. Heart J. 1985, 54, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, E.B.; Morse, J.H.; Knowles, J.A.; Chada, K.K.; Khan, A.M.; Roberts, K.E.; McElroy, J.J.; Juskiw, N.K.; Mallory, N.C.; Rich, S.; et al. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J. Heart Lung Transplant. 2008, 27, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Zhao, M.; West, J.; Newman, J.H.; Rich, S.; Archer, S.L.; Robbins, I.M.; Blackwell, T.S.; Cogan, J.; Loyd, J.E.; et al. Critical genomic networks and vasoreactive variants in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 464–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, R.; Cai, D.; King, S.D.; Que, X.; Bath, J.M.; Chen, S.-Y. Surfactant protein A, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling—brief report. Arter. Thromb. Vasc. Biol. 2021, 41, 808–814. [Google Scholar] [CrossRef]
- Zhang, P.; Guan, Y.; Chen, J.; Li, X.; McConnell, B.K.; Zhou, W.; Boini, K.M.; Zhang, Y. Contribution of p62/SQSTM1 to PDGF-BB-induced myofibroblast-like phenotypic transition in vascular smooth muscle cells lacking Smpd1 gene. Cell Death Dis. 2018, 9, 1145. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Liu, B.; Zhu, B.; Wang, N.; Qiao, Y.; Luo, E.; Nawabi, A.Q.; Yan, G.; Tang, C. Role of integrin-linked kinase in the hypoxia-induced phenotypic transition of pulmonary artery smooth muscle cells: Implications for hypoxic pulmonary hypertension. Exp. Cell Res. 2019, 382, 111476. [Google Scholar] [CrossRef] [PubMed]
- Aoshima, D.; Murata, T.; Hori, M.; Ozaki, H. Time-dependent phenotypic and contractile changes of pulmonary artery in chronic hypoxia–induced pulmonary hypertension. J. Pharmacol. Sci. 2009, 110, 182–190. [Google Scholar] [CrossRef]
- Sahoo, S.; Meijles, D.N.; Al Ghouleh, I.; Tandon, M.; Cifuentes-Pagano, E.; Sembrat, J.; Rojas, M.; Goncharova, E.; Pagano, P.J. MEF2C-MYOCD and leiomodin1 suppression by miRNA-214 promotes smooth muscle cell phenotype switching in pulmonary arterial hypertension. PLoS ONE 2016, 11, e0153780. [Google Scholar] [CrossRef] [Green Version]
- Hegner, B.; Lange, M.; Kusch, A.; Essin, K.; Sezer, O.; Schulze-Lohoff, E.; Luft, F.C.; Gollasch, M.; Dragun, D. mTOR regulates vascular smooth muscle cell differentiation from human bone mar-row-derived mesenchymal progenitors. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 232–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, K.J.; Tolg, C.; Panchal, T.; Leslie, B.; Yu, J.; Elkelini, M.; Sabha, N.; Tse, D.J.; Lorenzo, A.J.; Hassouna, M.; et al. Mammalian target of rapamycin (mTOR) induces proliferation and de-differentiation responses to three coordinate pathophysiologic stimuli (mechanical strain, hypoxia, and extracellular matrix remodeling) in rat bladder smooth muscle. Am. J. Pathol. 2010, 176, 304–319. [Google Scholar] [CrossRef] [Green Version]
- Garat, C.V.; Crossno, J.T.J.; Sullivan, T.M.; Reusch, J.E.; Klemm, D.J. Inhibition of phosphatidylinositol 3-kinase/Akt signaling attenuates hypoxia-induced pulmonary artery remodeling and suppresses CREB depletion in arterial smooth muscle cells. J. Cardiovasc. Pharmacol. 2013, 62, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007, 45 (Suppl. A), A25–A32. [Google Scholar] [CrossRef] [Green Version]
- Martin, K.A.; Merenick, B.L.; Ding, M.; Fetalvero, K.M.; Rzucidlo, E.M.; Kozul, C.D.; Brown, D.J.; Chiu, H.Y.; Shyu, M.; Drapeau, B.L.; et al. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J. Biol. Chem. 2007, 282, 36112–36120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, K.A.; Rzucidlo, E.M.; Merenick, B.L.; Fingar, D.C.; Brown, D.J.; Wagner, R.J.; Powell, R.J. The mTOR/p70 S6K1 pathway regulates vascular smooth muscle cell differentiation. Am. J. Physiol. Cell. Physiol. 2004, 286, C507–C517. [Google Scholar] [CrossRef]
- Zhan, J.K.; Wang, Y.J.; Wang, Y.; Wang, S.; Tan, P.; Huang, W.; Liu, Y.S. The mammalian target of rapamycin signaling pathway is involved in osteoblastic differentiation of vascular smooth muscle cells. Can. J. Cardiol. 2014, 30, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Takahashi, M.; Kimura, K.; Nishida, W.; Saga, H.; Sobue, K. Changes in the balance of phosphoinositide 3-kinase/protein kinase B (Akt) and the mitogen-activated protein kinases (ERK/p38MAPK) determine a phenotype of visceral and vascular smooth muscle cells. J. Cell Biol. 1999, 145, 727–740. [Google Scholar] [CrossRef]
- Hirschi, K.K.; Rohovsky, S.A.; D’Amore, P.A. PDGF, TGF-β, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J. Cell Biol. 1998, 141, 805–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, R.J.; Cronenwet, J.L.; Fillinger, M.F.; Wagner, R.J.; Sampson, L.N. Endothelial cell modulation of smooth muscle cell morphology and organizational growth pattern. Ann. Vasc. Surg. 1996, 10, 4–10. [Google Scholar] [CrossRef]
- Nugent, H.M.; Edelman, E.R. Endothelial implants provide long-term control of vascular repair in a porcine model of arterial injury. J. Surg. Res. 2001, 99, 228–234. [Google Scholar] [CrossRef]
- Fingerle, J.; Au, Y.P.; Clowes, A.W.; Reidy, M.A. Intimal lesion formation in rat carotid arteries after endothelial denudation in absence of medial injury. Arter. Off. J. Am. Heart Assoc. Inc. 1990, 10, 1082–1087. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.J.; Rzucidlo, E.M.; Merenick, B.L.; Wagner, R.J.; Martin, K.A.; Powell, R.J. Endothelial cell activation of the smooth muscle cell phosphoinositide 3-kinase/Akt pathway promotes differentiation. J. Vasc. Surg. 2005, 41, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Luozzo, G.; Bhargava, J.; Powell, R.J. Vascular smooth muscle cell effect on endothelial cell endothelin-1 production. J. Vasc. Surg. 2000, 31, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Holycross, B.J.; Blank, R.S.; Thompson, M.M.; Peach, M.J.; Owens, G.K. Platelet-derived growth factor-BB-induced suppression of smooth muscle cell differentiation. Circ. Res. 1992, 71, 1525–1532. [Google Scholar] [CrossRef] [Green Version]
- Mack, C.P. Signaling mechanisms that regulate smooth muscle cell differentiation. Arter. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef] [Green Version]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, K.; Zheng, Q.; Zhang, C.; Tang, H.; Babicheva, A.; Jiang, Q.; Li, M.; Chen, Y.; Carr, S.G.; et al. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L216–L228. [Google Scholar] [CrossRef]
- Li, W.; Petrimpol, M.; Molle, K.D.; Hall, M.N.; Battegay, E.J.; Humar, R. Hypoxia-induced endothelial proliferation requires both mTORC1 and mTORC2. Circ. Res. 2007, 100, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Humar, R.; Kiefer, F.N.; Berns, H.; Resink, T.J.; Battegay, E.J. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR) -dependent signaling. FASEB J. 2002, 16, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Che, D.; Yuan, Y. Effects of hypoxia on the release of PDGF-B chain from pulmonary artery endothelial cells and on growth of pulmonary artery smooth muscle cells. Zhongguo Ying Yong Sheng Li Xue Za Zhi 1997, 13, 216–219. [Google Scholar]
- Michiels, C.; De Leener, F.; Arnould, T.; Dieu, M.; Remacle, J. Hypoxia stimulates human endothelial cells to release smooth muscle cell mitogens: Role of prostaglandins and bFGF. Exp. Cell Res. 1994, 213, 43–54. [Google Scholar] [CrossRef]
- Liang, S.; Yu, H.; Chen, X.; Shen, T.; Cui, Z.; Zhang, J.; Cheng, Y.; Jia, S.; Song, S.; Zhang, X.; et al. PDGF-BB/KLF4/VEGF signaling axis in pulmonary artery endothelial cell angiogenesis. Cell. Physiol. Biochem. 2017, 41, 2333–2349. [Google Scholar] [CrossRef]
- Li, L.; Xu, M.; Li, X.; Lv, C.; Zhang, X.; Yu, H.; Zhang, M.; Fu, Y.; Meng, H.; Zhou, J. Platelet-derived growth factor-B (PDGF-B) induced by hypoxia promotes the survival of pulmonary arterial endothelial cells through the PI3K/Akt/Stat3 pathway. Cell. Physiol. Biochem. 2015, 35, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Zhang, M.; Yi, Z.; Zhang, H.; Shen, T.; Yu, X.; Zhang, C.; Zheng, X.; Yu, L.; Ma, C.; et al. The role of PDGF-B/TGF-β1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cell. Signal. 2016, 28, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, T.; Raj, J.U. Endothelial and smooth muscle cell interactions in the pathobiology of pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 54, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L256–L275. [Google Scholar]
- Hopper, R.K.; Moonen, J.R.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M.; et al. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target Slug. Circulation 2016, 133, 1783–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isobe, S.; Kataoka, M.; Endo, J.; Moriyama, H.; Okazaki, S.; Tsuchihashi, K.; Katsumata, Y.; Yamamoto, T.; Shirakawa, K.; Yoshida, N.; et al. Endothelial-mesenchymal transition drives expression of CD44 variant and xCT in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2019, 61, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, T.; Muyleart, M.; Konduri, G.G.; Mammoto, A. Twist1 in hypoxia-induced pulmonary hypertension through trans-forming growth factor-β-Smad signaling. Am. J. Respir. Cell Mol. Biol. 2018, 58, 194–207. [Google Scholar] [CrossRef]
- Rinastiti, P.; Ikeda, K.; Rahardini, E.P.; Miyagawa, K.; Tamada, N.; Kuribayashi, Y.; Hirata, K.-I.; Emoto, N. Loss of family with sequence similarity 13, member A exacerbates pulmonary hypertension through accelerating endothelial-to-mesenchymal transition. PLoS ONE 2020, 15, e0226049. [Google Scholar] [CrossRef] [Green Version]
- Dong, P.; Konno, Y.; Watari, H.; Hosaka, M.; Noguchi, M.; Sakuragi, N. The impact of microRNA-mediated PI3K/AKT signaling on epithelial-mesenchymal transition and cancer stemness in endometrial cancer. J. Transl. Med. 2014, 12, 231. [Google Scholar] [CrossRef]
- Harsha, C.; Banik, K.; Ang, H.L.; Girisa, S.; Vikkurthi, R.; Parama, D.; Rana, V.; Shabnam, B.; Khatoon, E.; Kumar, A.P.; et al. Targeting AKT/mTOR in oral cancer: Mechanisms and advances in clinical trials. Int. J. Mol. Sci. 2020, 21, 3285. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.L.; De Oca, M.K.M.; Pal, H.C.; Afaq, F. Potential therapeutic targets of epithelial–mesenchymal transition in melanoma. Cancer Lett. 2017, 391, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Ko, J.-H.; Lee, J.H.; Kim, C.; Lee, H.; Nam, D.; Lee, J.; Lee, S.-G.; Yang, W.M.; Um, J.-Y.; et al. Ginkgolic acid inhibits invasion and migration and TGF-β-induced EMT of lung cancer cells through PI3K/Akt/mTOR inactivation. J. Cell. Physiol. 2016, 232, 346–354. [Google Scholar] [CrossRef]
- Wang, Z.; Fei, S.; Suo, C.; Han, Z.; Tao, J.; Xu, Z.; Zhao, C.; Tan, R.; Gu, M. Antifibrotic effects of hepatocyte growth factor on endothelial-to-mesenchymal transition via transforming growth factor-β1 (TGF-β1)/Smad and Akt/mTOR/P70S6K signaling pathways. Ann. Transplant. 2018, 23, 1–10. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Sun, S.; Zhu, J.; Gao, S.; Pang, J.; Zhu, D.; Sun, Z. Transforming growth factor-β1 upregulation triggers pulmonary artery smooth muscle cell proliferation and apoptosis imbalance in rats with hypoxic pulmonary hypertension via the PTEN/AKT pathways. Int. J. Biochem. Cell Biol. 2016, 77, 141–154. [Google Scholar] [CrossRef]
- O’Leary, E.M.; Tian, Y.; Nigdelioglu, R.; Witt, L.J.; Cetin-Atalay, R.; Meliton, A.Y.; Woods, P.S.; Kimmig, L.M.; Sun, K.A.; Gökalp, G.A.; et al. TGF-β promotes metabolic reprogramming in lung fibroblasts via mTORC1-dependent ATF4 activation. Am. J. Respir. Cell Mol. Biol. 2020, 63, 601–612. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Yan, L.; Du, W.; Zhang, X.; Zhang, M.; Chen, H.; Zhang, Y.; Zhou, J.; Sun, H.; et al. Bone morphogenetic protein-7 inhibits endothelial-mesenchymal transition in pulmonary artery endothelial cell under hypoxia. J. Cell. Physiol. 2017, 233, 4077–4090. [Google Scholar] [CrossRef]
- Mikaelian, I.; Malek, M.; Gadet, R.; Viallet, J.; Garcia, A.; Girard-Gagnepain, A.; Hesling, C.; Gillet, G.; Gonzalo, P.; Rimokh, R.; et al. Genetic and pharmacologic inhibition of mTORC1 promotes EMT by a TGF-β–independent mechanism. Cancer Res. 2013, 73, 6621–6631. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, G.; Ren, J.-G.; Zhao, Y.-F. Bleomycin induces endothelial mesenchymal transition through activation of mTOR pathway: A possible mechanism contributing to the sclerotherapy of venous malformations. Br. J. Pharmacol. 2013, 170, 1210–1220. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Hu, F.; Li, Q.; Shen, C.; Yang, J.; Li, M. Tanshinone IIA ameliorates the bleomycin-induced endothelial-to-mesenchymal transition via the Akt/mTOR/p70S6K pathway in a murine model of systemic sclerosis. Int. Immunopharmacol. 2019, 77, 105968. [Google Scholar] [CrossRef]
- Takagi, K.; Yamakuchi, M.; Matsuyama, T.; Kondo, K.; Uchida, A.; Misono, S.; Kondo, K.; Uchida, A.; Misono, S.; Inoue, H.; et al. IL-13 enhances mesenchymal transition of pulmonary artery endothelial cells via down-regulation of miR-424/503 in vitro. Cell Signal. 2018, 42, 270–280. [Google Scholar] [CrossRef]
- Zhang, S.; Qian, G.; Zhang, Q.-Q.; Yao, Y.; Wang, D.; Chen, Z.G.; Wang, L.-J.; Chen, M.; Sun, S.-Y. mTORC2 suppresses GSK3-dependent Snail degradation to positively regulate cancer cell invasion and metastasis. Cancer Res. 2019, 79, 3725–3736. [Google Scholar] [CrossRef]
- Lau, M.-T.; Leung, P.C. The PI3K/Akt/mTOR signaling pathway mediates insulin-like growth factor 1-induced E-cadherin down-regulation and cell proliferation in ovarian cancer cells. Cancer Lett. 2012, 326, 191–198. [Google Scholar] [CrossRef]
- Kim, E.Y.; Kim, A.; Kim, S.K.; Kim, H.J.; Chang, J.; Ahn, C.M.; Chang, Y.S. Inhibition of mTORC1 induces loss of E-cadherin through AKT/GSK-3β signaling-mediated upregulation of E-cadherin repressor complexes in non-small cell lung cancer cells. Respir. Res. 2014, 15, 26. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Zhang, J.; Liu, T.; Shi, W. Rapamycin prevents endothelial cell migration by inhibiting the endothelial-to-mesenchymal transition and matrix metalloproteinase-2 and -9: An in vitro study. Mol. Vis. 2011, 17, 3406–3414. [Google Scholar]
- Mammoto, A.; Hendee, K.; Muyleart, M.; Mammoto, T. Endothelial Twist1-PDGFB signaling mediates hypoxia-induced proliferation and migration of αSMA-positive cells. Sci. Rep. 2020, 10, 7563. [Google Scholar] [CrossRef]
- Hu, J.; Xu, Q.; McTiernan, C.; Lai, Y.C.; Osei-Hwedieh, D.; Gladwin, M. Novel targets of drug treatment for pulmonary hyper-tension. Am. J. Cardiovasc. Drugs 2015, 15, 225–234. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Seeger, W.; Grimminger, F. Imatinib for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2005, 353, 1412–1413. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Morrell, N.W.; Hoeper, M.M.; Olschewski, H.; Peacock, A.J.; Barst, R.J.; Shapiro, S.; Golpon, H.; Toshner, M.; Grimminger, F.; et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am. J. Respir. Crit. Care Med. 2010, 182, 1171–1177. [Google Scholar] [CrossRef] [Green Version]
- Hoeper, M.M.; Barst, R.J.; Bourge, R.C.; Feldman, J.; Frost, A.E.; Galié, N.; Gómez-Sánchez, M.A.; Grimminger, F.; Grünig, E.; Hassoun, P.M.; et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation 2013, 127, 1128–1138. [Google Scholar] [CrossRef] [Green Version]
- Gomberg-Maitland, M.; Barst, R.J.; Sugeng, L.; Coslet, S.; Perrino, T.J.; Bond, L.; LaCouture, M.E.; Archer, S.L.; Ratain, M.J.; Maitland, M.L. A Dosing/cross-development study of the multikinase inhibitor sorafenib in patients with pulmonary arterial hypertension. Clin. Pharmacol. Ther. 2009, 87, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Brittain, E.L.; Niswender, K.; Agrawal, V.; Chen, X.; Fan, R.; Pugh, M.E.; Rice, T.W.; Robbins, I.M.; Song, H.; Thompson, C.; et al. Mechanistic phase II clinical trial of metformin in pulmonary arterial hypertension. J. Am. Heart Assoc. 2020, 9, e018349. [Google Scholar] [CrossRef] [PubMed]
- Speich, R.; Ulrich, S.; Domenighetti, G.; Huber, L.C.; Fischler, M.; Treder, U.; Breitenstein, A. Efficacy and safety of long-term imatinib therapy for pulmonary arterial hypertension. Respiration 2015, 89, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Frost, A.E.; Barst, R.J.; Hoeper, M.M.; Chang, H.-J.; Frantz, R.P.; Fukumoto, Y.; Galié, N.; Hassoun, P.M.; Klose, H.; Matsubara, H.; et al. Long-term safety and efficacy of imatinib in pulmonary arterial hypertension. J. Heart Lung Transplant. 2015, 34, 1366–1375. [Google Scholar] [CrossRef] [Green Version]
- Ten Freyhaus, H.; Dumitrescu, D.; Berghausen, E.; Vantler, M.; Caglayan, E.; Rosenkranz, S. Imatinib mesylate for the treatment of pulmonary arterial hypertension. Expert Opin. Investig. Drugs 2012, 21, 119–134. [Google Scholar] [CrossRef]
- Maurer, B.; Reich, N.; Juengel, A.; Kriegsmann, J.; Gay, R.E.; Schett, G.; Michel, B.A.; Gay, S.; Distler, J.H.W.; Distler, O. Fra-2 transgenic mice as a novel model of pulmonary hypertension associated with systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1382–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, G.; Kataoka, M.; Inami, T.; Fukuda, K.; Yoshino, H.; Satoh, T. Sorafenib as a potential strategy for refractory pulmonary arterial hypertension. Pulm. Pharmacol. Ther. 2017, 44, 46–49. [Google Scholar] [CrossRef]
- Hong, J.H.; Lee, S.-E.; Choi, S.Y.; Kim, S.-H.; Jang, E.-J.; Bang, J.-H.; Park, J.E.; Jeon, H.-R.; Oh, Y.J.; Yi, J.-E.; et al. Reversible pulmonary arterial hypertension associated with dasatinib for chronic myeloid leukemia. Cancer Res. Treat. 2014, 47, 937–942. [Google Scholar] [CrossRef]
- Weatherald, J.; Chaumais, M.-C.; Montani, D. Pulmonary arterial hypertension induced by tyrosine kinase inhibitors. Curr. Opin. Pulm. Med. 2017, 23, 392–397. [Google Scholar] [CrossRef]
- McGee, M.; Whitehead, N.; Martin, J.; Collins, N. Drug-associated pulmonary arterial hypertension. Clin. Toxicol. 2018, 56, 801–809. [Google Scholar] [CrossRef]
- Cornet, L.; Khouri, C.; Roustit, M.; Guignabert, C.; Chaumais, M.C.; Humbert, M.; Revol, B.; Despas, F.; Montani, D.; Cracowski, J.L. Pulmonary arterial hypertension associated with protein kinase inhibitors: A pharmacovigilance-pharmacodynamic study. Eur. Respir. J. 2019, 53, 1802472. [Google Scholar] [CrossRef]
- El-Dabh, A.; Acharya, D. EXPRESS: Pulmonary hypertension with dasatinib and other tyrosine kinase inhibitors. Pulm. Circ. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Montani, D.; Chaumais, M.-C.; Guignabert, C.; Günther, S.; Girerd, B.; Jaïs, X.; Algalarrondo, V.; Price, L.C.; Savale, L.; Sitbon, O.; et al. Targeted therapies in pulmonary arterial hypertension. Pharmacol. Ther. 2014, 141, 172–191. [Google Scholar] [CrossRef]
- Minami, M.; Arita, T.; Iwasaki, H.; Muta, T.; Aoki, T.; Aoki, K.; Yamasaki, S.; Matsushima, T.; Kato, K.; Takenaka, K.; et al. Comparative analysis of pulmonary hypertension in patients treated with imatinib, nilotinib and dasatinib. Br. J. Haematol. 2017, 177, 578–587. [Google Scholar] [CrossRef]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.; Meric-Bernstam, F.; Chawla, S.; Falchook, G.; Hong, D.; Akcakanat, A.; Chen, H.; Naing, A.; Fu, S.; Kurzrock, R.; et al. Weekly nab-Rapamycin in patients with advanced nonhematologic malignancies: Final results of a phase I trial. Clin. Cancer Res. 2013, 19, 5474–5484. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Wessler, J.D.; Steingart, R.M.; Schwartz, G.K.; Harvey, B.-G.; Schaffer, W. Dramatic improvement in pulmonary hypertension with rapamycin. Chest 2010, 138, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Seyfarth, H.J.; Hammerschmidt, S.; Halank, M.; Neuhaus, P.; Wirtz, H.R. Everolimus in patients with severe pulmonary hypertension: A safety and efficacy pilot trial. Pulm. Circ. 2013, 3, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Kang, S.A.; Thoreen, C.C.; Hur, W.; Wang, J.; Chang, J.W.; Markhard, A.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Development of ATP-competitive mTOR inhibitors. Breast Cancer 2011, 821, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Schenone, S.; Brullo, C.; Musumeci, F.; Radi, M.; Botta, M. ATP-competitive inhibitors of mTOR: An update. Curr. Med. Chem. 2011, 18, 2995–3014. [Google Scholar] [CrossRef]
- Liu, Y.; Wan, W.Z.; Li, Y.; Zhou, G.L.; Liu, X.G. Recent development of ATP-competitive small molecule phosphatidylinosti-tol-3-kinase inhibitors as anticancer agents. Oncotarget 2017, 8, 7181–7200. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Target | Position | Regulation by mTOR | Role | Function |
|---|---|---|---|---|
| RTK | Upstream | --- | Activates PI3K | mTOR↑ |
| PI3K | Upstream | --- | Converts PIP2 to PIP3 | mTOR↑ |
| PTEN | Upstream | --- | Dephosphorylates PIP3 to PIP2 | mTOR↓ |
| PDK1 | Upstream | --- | Phosphorylates AKT at T308 | mTOR↑ |
| AKT | Upstream | --- | Inhibits TSC1/TSC2 | mTOR↑ |
| TSC | Upstream | --- | Inhibits RHEB | mTOR↓ |
| RHEB | Upstream | --- | Phosphorylates mTOR | mTOR↑ |
| Redd1 | Upstream | --- | Activates TSC | mTOR↓ |
| AMPK | Upstream | --- | Activates TSC | mTOR↓ |
| P70S6K | Downstream | Activation | Activates S6 | PASMC growth and proliferation↑ |
| Activates eIF4B | Gene translation↑ Protein synthesis↑ PASMC growth and proliferation↑ | |||
| Inhibits MHC11 | SMC contractile-to-proliferative transition↑ | |||
| 4E-BP1 | Downstream | Inhibition | Inhibits eIF4E | Gene translation↑ Protein synthesis↑ |
| SREBP | Downstream | Activation | --- | Lipid synthesis↑ |
| DAP1 | Downstream | Activation | --- | Autophagy↓ |
| SGK1 | Downstream | Activation | --- | Ion transport↑ Cell apoptosis↑ |
| FoxO1 | Downstream | Inhibition | --- | Inflammatory response↓ |
| GSK3 | Downstream | Inhibition | --- | ER stress↓ Glycogen synthesis↓ |
| Inhibits Snail | EndMT↑ | |||
| Slug | Downstream | Activation | --- | EndMT↑ |
| HIF-α | Downstream | Activation | --- | Hypoxia response↑ |
| NF-κB | Downstream | Activation | --- | Inflammatory response↑ |
| Notch | Downstream | Activation | --- | Cell-cell interaction↑ |
| PPAR | Downstream | Activation | --- | Glucose metabolism↓ Lipid metabolism↑ |
| STAT3 | Downstream | Activation | --- | Immune response↓ |
| TFEB | Downstream | Activation | --- | Lysosomal biogenesis↑ Autophagy↑ |
| YY1 | Downstream | Activation | --- | Cell proliferation↑ |
| Target | Method | PH model | Effect | Parameters | Ref |
|---|---|---|---|---|---|
| Genetic Approach | |||||
| PTEN | Transgenic global overexpression | HPH in mice | Inhibition | Pulmonary vascular remodeling↓ RVSP↓ RV hypertrophy↓ PA wall thickness↓ | [14] |
| PTEN | Conditional KO in SMC | Spontaneous PH in mice (at the age of 20 days) | Promotion | Pulmonary vascular remodeling↑ RV hypertrophy↑ PA wall thickness↑ SMC proliferation↑ | [68] |
| PTEN | Conditional and inducible KO in SMC | --- | --- | Aorta contractility↓ SMC de-differentiation↑ | [70] |
| PTEN | Conditional and inducible KO in SMC | Spontaneous PH in mice | Partial promotion | RVSP RV hypertrophy↑ Pulmonary vascular remodeling↑ PA wall thickness↑ | [69] |
| HPH in mice | Promotion | RVSP↑ RV hypertrophy↑ Pulmonary vascular remodeling↑ PA wall thickness↑ | |||
| PDK1 | Conditional KO in EC (heterozygous) | HPH in mice | Inhibition | Pulmonary vascular remodeling↓ RVSP↓ RV hypertrophy↓ PA wall thickness↓ | [71] |
| AKT1 | Global KO | HPH in mice | Inhibition | RVSP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ PA wall thickness↓ | [14] |
| AKT2 | Global KO | HPH in mice | No effect | RVSP RV hypertrophy Pulmonary vascular remodelingPA wall thickness | [14] |
| AMPKα2 | Global KO | HPH in mice | Promotion | RVSP↑ RV hypertrophy↑ Pulmonary vascular remodeling↑ PASMC proliferation↑ | [72] |
| AMPKα2 | Conditional KO in EC | Su/Hyp-PH in mice | Promotion | RVSP↑ RV hypertrophy↑ PA wall thickness↓ Pulmonary vascular remodeling↑ | [73] |
| AMPK | Conditional KO in EC | HPH in mice | Promotion | RVSP↑ RV hypertrophy↑ PA wall thickness↓ | [74] |
| TSC1 | Conditional KO in SMC | Spontaneous PH in mice (at the age of 10–12 weeks) | Promotion | RVSP↑ RV hypertrophy↑ Pulmonary vascular remodeling↑ PA wall thickness↑ PASMC proliferation↑ SMC de-differentiation↑ | [75] |
| mTOR | Conditional and inducible KO in SMC | HPH in mice | Inhibition | RVSP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ PA wall thickness↓ Polycythemia↓ | [14,15] |
| Raptor | Conditional and inducible KO in SMC | HPH in mice | Partial inhibition | RV hypertrophy↓ | [15] |
| Rictor | Conditional and inducible KO in SMC | HPH in mice (at the age of 8–10 weeks) | Negligible inhibition | RVSP RV hypertrophy | [15] |
| Conditional and inducible KO in SMC | Spontaneous PH in mice (at the age of 6–8 months) | Promotion | RVSP↑ RV hypertrophy↑ PA wall thickness↑ | ||
| Conditional and inducible KO in EC | HPH in mice (at the age of 8–10 weeks) | No effect | --- | ||
| Pharmaceutical approach | |||||
| Sorafenib (multikinase inhibitor) | Oral gavage (10 mg/kg/day) | MCT-PH in rats | Inhibition | RVSP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ PA wall thickness↓ PASMC proliferation↓ PASMC apoptosis↑ | [76] |
| Sorafenib (multikinase inhibitor) | Oral gavage (10, 30, or 100 mg/kg/day) | MCT-PH in rats | Inhibition | RVSP↓ RV hypertrophy↓ Cardiopulmonary remodeling↓ Pulmonary vascular remodeling↓ | [77] |
| Imatinib (RTK inhibitor) | Oral gavage (50 mg/kg/day) | MCT-PH in rats | Inhibition | RVSP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ PA wall thickness↓ | [76] |
| Imatinib (RTK inhibitor) | Oral gavage (100 mg/kg/day) | MCT-PH in rats | Inhibition | PAP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ PA wall thickness↓ | [60] |
| Metformin (AMPK activator) | Intraperitoneally (100 mg/kg/day) | HPH in rats | Inhibition | Mean PAP↓ RV wall thickness↓ RV hypertrophy↓ PFAT↑ PA contraction↓ PA cell proliferation↓ Endothelial function↑ | [78] |
| MCT-PH in rats | Inhibition | Mean PAP↓ RV hypertrophy↓ Survival↑ | |||
| Metformin (AMPK activator) | Oral gavage (100 mg/kg/day) | Su/Hyp-PH in rats | Inhibition | RVSP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ | [79] |
| AICAR (AMPK activator) | Intraperitoneally (1 mg/kg/day) | HPH in rats | Inhibition | Mean PAP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ PA wall thickness↓ | [80] |
| Rapamycin (mTOR inhibitor) | Oral gavage (5 mg/kg/day) | MCT-PH in rats | Inhibition | PAP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ PASMC proliferation↓ | [60] |
| Rapamycin (mTOR inhibitor) | Oral gavage (1 mg/kg/day) | HPH in rats | Inhibition | Pulmonary vascular remodeling↓ RVSP↓mPAP↓ PA wall thickness↓ | [66] |
| Rapamycin (mTOR inhibitor) | Intraperitoneally (3 mg/kg/day) | HPH in mice | Inhibition | RV hypertrophy↓ Pulmonary vascular remodeling↓ PA wall thickness↓ | [81] |
| Drug | Dose | N of Participants | Status | Results | Effect | Adverse Effects | Identifier |
|---|---|---|---|---|---|---|---|
| Imatinib RTK inhibitor) | --- | 130 | Completed | Not available | --- | --- | NCT01092897 |
| Imatinib (RTK inhibitor) | 200–400 mg orally once daily | 59 | Completed (24-week randomized, double-blind, placebo-controlled) | Available [178] | PVR↓ CO↑ | Nausea, headache, peripheral edema | NCT00477269 |
| Imatinib (RTK inhibitor) | 200–400 mg orally once daily | 202 | Completed (24-week randomized, double-blind, placebo-controlled) | Available [179] | PVR↓ mPAP↓ CO↑ 6MWD↑ | Cardiac failure, subdural hematoma, dyspnea, worsening PAH | NCT00902174 |
| Nilotinib (RTK inhibitor) | 50–300 mg orally twice a day | 23 | Terminated (due to serious adverse events) | Not Available | --- | Cardiogenic shock, right ventricular dysfunction, gastric ulcer hemorrhage, cholecystitis, hepatitis | NCT01179737 |
| Sorafenib (multikinase inhibitor) | 200–400 mg twice daily | 12 | Completed (16-week, open-label) | Available [180] | CO↓ | Diarrhea, hand–foot syndrome, rash, alopecia | NCT00452218 |
| Metformin (AMPK activator) | 500 mg–1000 mg orally once or twice daily | 1899 | Recruiting | Preliminary [181] | RVSP 6MWD RV function↑ RV lipid content↓ | --- | NCT01884051 |
| Metformin (AMPK activator) | 1000 mg orally twice daily | 130 | Recruiting | Not available | --- | --- | NCT03617458 |
| Rapamycin (mTOR inhibitor) | --- | 25 | Recruiting | Not Available | --- | --- | NCT02587325 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babicheva, A.; Makino, A.; Yuan, J.X.-J. mTOR Signaling in Pulmonary Vascular Disease: Pathogenic Role and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 2144. https://doi.org/10.3390/ijms22042144
Babicheva A, Makino A, Yuan JX-J. mTOR Signaling in Pulmonary Vascular Disease: Pathogenic Role and Therapeutic Target. International Journal of Molecular Sciences. 2021; 22(4):2144. https://doi.org/10.3390/ijms22042144
Chicago/Turabian StyleBabicheva, Aleksandra, Ayako Makino, and Jason X.-J. Yuan. 2021. "mTOR Signaling in Pulmonary Vascular Disease: Pathogenic Role and Therapeutic Target" International Journal of Molecular Sciences 22, no. 4: 2144. https://doi.org/10.3390/ijms22042144
APA StyleBabicheva, A., Makino, A., & Yuan, J. X. -J. (2021). mTOR Signaling in Pulmonary Vascular Disease: Pathogenic Role and Therapeutic Target. International Journal of Molecular Sciences, 22(4), 2144. https://doi.org/10.3390/ijms22042144