
CrossTORC and WNTegration in Disease: Focus on Lymphangioleiomyomatosis


Abstract
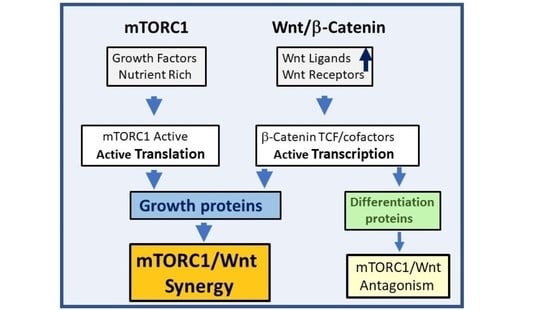
:
{kind=link}
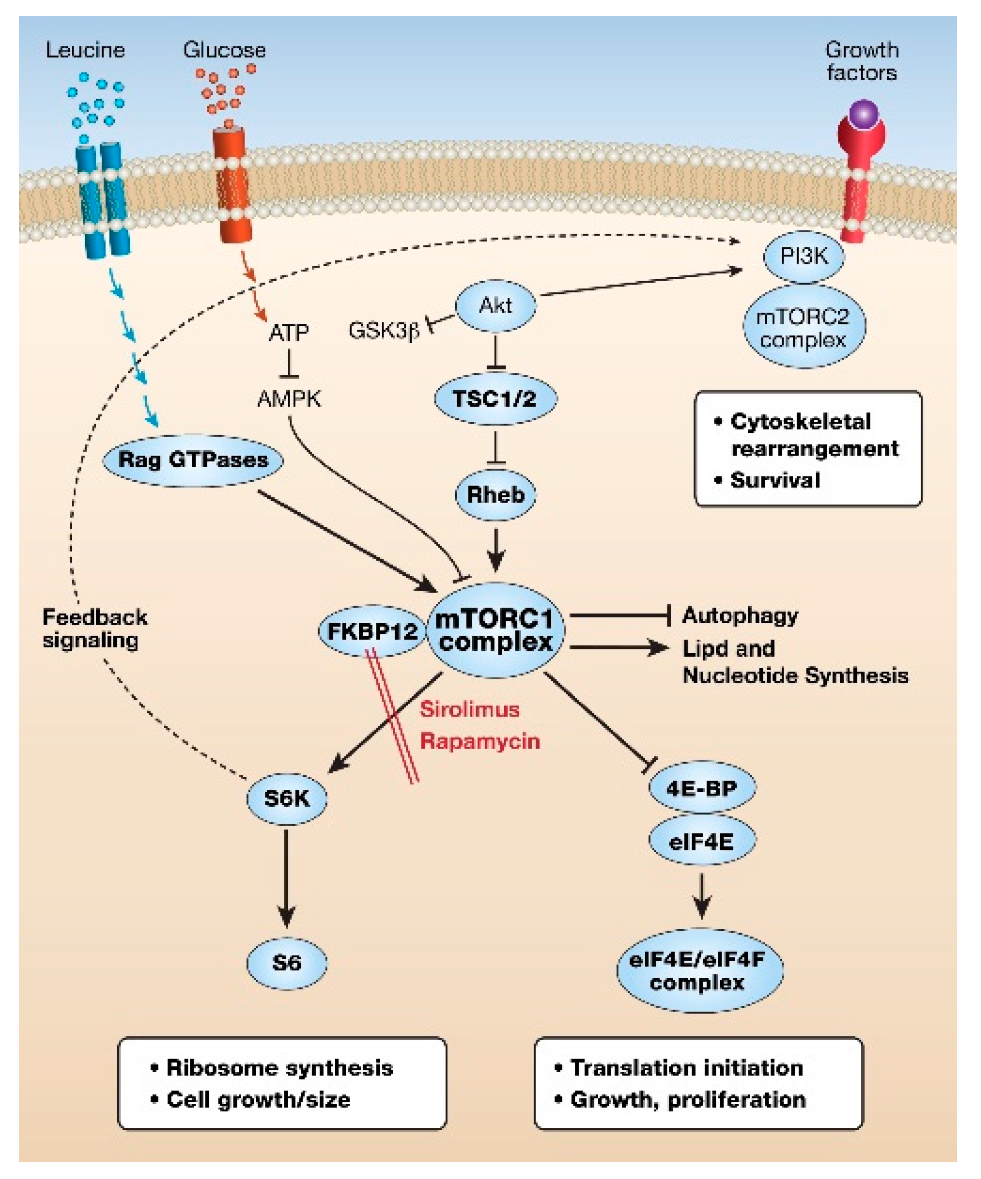{kind=link}
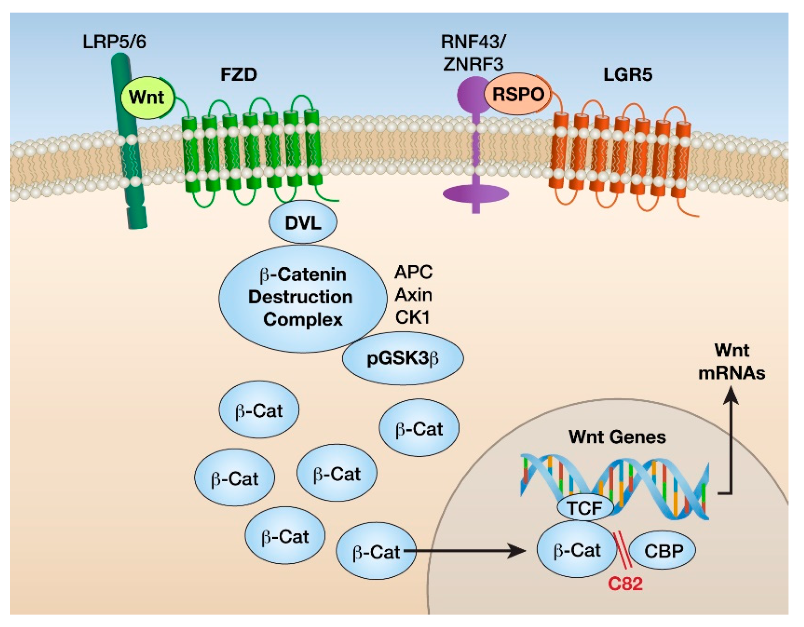{kind=link}
{kind=link}
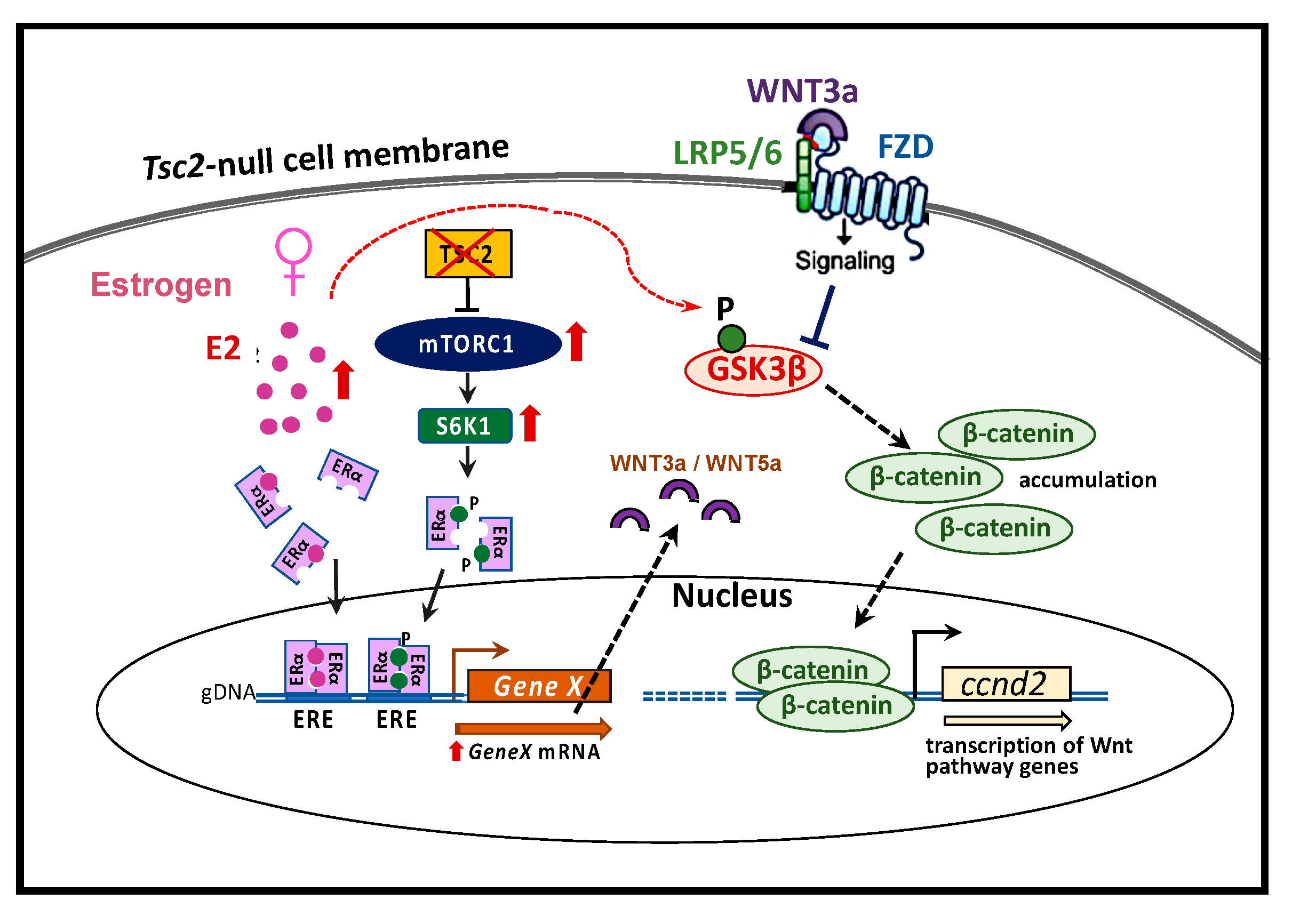{kind=link}
1. Introduction
2. mTOR Pathway Overview
3. Wnt Pathway Overview
4. mTOR and Wnt Pathway Crosstalk
5. Therapeutics for mTOR and Wnt Pathways
6. mTOR Pathway Inhibitors
7. Wnt Pathway Inhibitors
8. Targeting Multiple Growth Pathways
9. Conclusions and Potential Future Therapeutics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.; Guan, K.-L. MTOR as a Central Hub of Nutrient Signalling and Cell Growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Tee, A.R. The Target of Rapamycin and Mechanisms of Cell Growth. Int. J. Mol. Sci. 2018, 19, 880. [Google Scholar] [CrossRef] [Green Version]
- Goncharova, E.A.; Goncharov, D.A.; Li, H.; Pimtong, W.; Lu, S.; Khavin, I.; Krymskaya, V.P. MTORC2 Is Required for Proliferation and Survival of TSC2-Null Cells. Mol. Cell. Biol. 2011, 31, 2484–2498. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting MTOR for Cancer Therapy. J. Hematol. Oncol. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. MTOR Signalling and Cellular Metabolism Are Mutual Determinants in Cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Henske, E.P.; McCormack, F.X. Lymphangioleiomyomatosis—A Wolf in Sheep’s Clothing. J. Clin. Investig. 2012, 122, 3807–3816. [Google Scholar] [CrossRef] [Green Version]
- Krymskaya, V.P.; McCormack, F.X. Lymphangioleiomyomatosis: A Monogenic Model of Malignancy. Annu. Rev. Med. 2017, 68, 69–83. [Google Scholar] [CrossRef] [Green Version]
- Goncharova, E.A.; Goncharov, D.A.; Eszterhas, A.; Hunter, D.S.; Glassberg, M.K.; Yeung, R.S.; Walker, C.L.; Noonan, D.; Kwiatkowski, D.J.; Chou, M.M.; et al. Tuberin Regulates P70 S6 Kinase Activation and Ribosomal Protein S6 Phosphorylation: A Role for the TSC2 Tumor Suppressor Gene in Pulmonary Lymphangioleiomyomatosis (LAM). J. Biol. Chem. 2002, 277, 30958–30967. [Google Scholar] [CrossRef] [Green Version]
- Pond, K.W.; Doubrovinski, K.; Thorne, C.A. Wnt/β-Catenin Signaling in Tissue Self-Organization. Genes 2020, 11, 939. [Google Scholar] [CrossRef]
- Steinhart, Z.; Angers, S. Wnt Signaling in Development and Tissue Homeostasis. Dev. Camb. Engl. 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Pruitt, K. Wnt Pathway: An Integral Hub for Developmental and Oncogenic Signaling Networks. Int. J. Mol. Sci. 2020, 21, 8018. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Cohen, P. GSK3 Takes Centre Stage More than 20 Years after Its Discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef]
- Zhang, H.H.; Lipovsky, A.I.; Dibble, C.C.; Sahin, M.; Manning, B.D. S6K1 Regulates GSK3 under Conditions of MTOR-Dependent Feedback Inhibition of Akt. Mol. Cell 2006, 24, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, F.X.; Inoue, Y.; Moss, J.; Singer, L.G.; Strange, C.; Nakata, K.; Barker, A.F.; Chapman, J.T.; Brantly, M.L.; Stocks, J.M.; et al. Efficacy and Safety of Sirolimus in Lymphangioleiomyomatosis. N. Engl. J. Med. 2011, 364, 1595–1606. [Google Scholar] [CrossRef]
- Yao, J.; Taveira-DaSilva, A.M.; Jones, A.M.; Julien-Williams, P.; Stylianou, M.; Moss, J. Sustained Effects of Sirolimus on Lung Function and Cystic Lung Lesions in Lymphangioleiomyomatosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1273–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bee, J.; Fuller, S.; Miller, S.; Johnson, S.R. Lung Function Response and Side Effects to Rapamycin for Lymphangioleiomyomatosis: A Prospective National Cohort Study. Thorax 2018, 73, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Varmus, H. Three Decades of Wnts: A Personal Perspective on How a Scientific Field Developed. EMBO J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef] [Green Version]
- Willert, K.; Nusse, R. Wnt Proteins. Cold Spring Harb. Perspect. Biol. 2012, 4, a007864. [Google Scholar] [CrossRef]
- Chae, W.-J.; Bothwell, A.L.M. Canonical and Non-Canonical Wnt Signaling in Immune Cells. Trends Immunol. 2018, 39, 830–847. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Grainger, S.; Willert, K. Mechanisms of Wnt Signaling and Control. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1422. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.-X.; Jiang, X.; Cong, F. Control of Wnt Receptor Turnover by R-Spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer. Cancers 2016, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-B.; Kim, J.-W.; Baek, K.-H. Regulation of Wnt Signaling through Ubiquitination and Deubiquitination in Cancers. Int. J. Mol. Sci. 2020, 21, 3904. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, A.-B.; Cadigan, K.M. Wnt Target Genes and Where to Find Them. F1000Research 2017, 6, 746. [Google Scholar] [CrossRef]
- Peifer, M.; McCrea, P.D.; Green, K.J.; Wieschaus, E.; Gumbiner, B.M. The Vertebrate Adhesive Junction Proteins Beta-Catenin and Plakoglobin and the Drosophila Segment Polarity Gene Armadillo Form a Multigene Family with Similar Properties. J. Cell Biol. 1992, 118, 681–691. [Google Scholar] [CrossRef]
- Goentoro, L.; Kirschner, M.W. Evidence That Fold-Change, and Not Absolute Level, of Beta-Catenin Dictates Wnt Signaling. Mol. Cell 2009, 36, 872–884. [Google Scholar] [CrossRef] [Green Version]
- Gajos-Michniewicz, A.; Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 2020, 21, 4852. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Park, J.-I. Wnt Signaling in Cancer: Therapeutic Targeting of Wnt Signaling beyond β-Catenin and the Destruction Complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, W.-J.; Bothwell, A.L.M. Dickkopf1: An Immunomodulatory Ligand and Wnt Antagonist in Pathological Inflammation. Differ. Res. Biol. Divers. 2019, 108, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Maric, G.; Annis, M.G.; MacDonald, P.A.; Russo, C.; Perkins, D.; Siwak, D.R.; Mills, G.B.; Siegel, P.M. GPNMB Augments Wnt-1 Mediated Breast Tumor Initiation and Growth by Enhancing PI3K/AKT/MTOR Pathway Signaling and β-Catenin Activity. Oncogene 2019, 38, 5294–5307. [Google Scholar] [CrossRef]
- Prossomariti, A.; Piazzi, G.; Alquati, C.; Ricciardiello, L. Are Wnt/β-Catenin and PI3K/AKT/MTORC1 Distinct Pathways in Colorectal Cancer? Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 491–506. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-MTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Adebayo Michael, A.O.; Ko, S.; Tao, J.; Moghe, A.; Yang, H.; Xu, M.; Russell, J.O.; Pradhan-Sundd, T.; Liu, S.; Singh, S.; et al. Inhibiting Glutamine-Dependent MTORC1 Activation Ameliorates Liver Cancers Driven by β-Catenin Mutations. Cell Metab. 2019, 29, 1135–1150.e6. [Google Scholar] [CrossRef]
- Kwan, H.T.; Chan, D.W.; Cai, P.C.H.; Mak, C.S.L.; Yung, M.M.H.; Leung, T.H.Y.; Wong, O.G.W.; Cheung, A.N.Y.; Ngan, H.Y.S. AMPK Activators Suppress Cervical Cancer Cell Growth through Inhibition of DVL3 Mediated Wnt/β-Catenin Signaling Activity. PLoS ONE 2013, 8, e53597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, N.; Wang, P.; Li, Y. Role of Wnt/β-Catenin Pathway in Inducing Autophagy and Apoptosis in Multiple Myeloma Cells. Oncol. Lett. 2016, 12, 4623–4629. [Google Scholar] [CrossRef] [Green Version]
- Haller, S.; Kapuria, S.; Riley, R.R.; O’Leary, M.N.; Schreiber, K.H.; Andersen, J.K.; Melov, S.; Que, J.; Rando, T.A.; Rock, J.; et al. MTORC1 Activation during Repeated Regeneration Impairs Somatic Stem Cell Maintenance. Cell Stem Cell 2017, 21, 806–818.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Lu, B.; Zamponi, R.; Yang, Z.; Wetzel, K.; Loureiro, J.; Mohammadi, S.; Beibel, M.; Bergling, S.; Reece-Hoyes, J.; et al. MTORC1 Signaling Suppresses Wnt/β-Catenin Signaling through DVL-Dependent Regulation of Wnt Receptor FZD Level. Proc. Natl. Acad. Sci. USA 2018, 115, E10362–E10369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Tyburczy, M.E.; Moss, J.; Darling, T.N.; Widlund, H.R.; Kwiatkowski, D.J. Tuberous Sclerosis Complex Inactivation Disrupts Melanogenesis via MTORC1 Activation. J. Clin. Investig. 2017, 127, 349–364. [Google Scholar] [CrossRef] [Green Version]
- Mak, B.C.; Kenerson, H.L.; Aicher, L.D.; Barnes, E.A.; Yeung, R.S. Aberrant Beta-Catenin Signaling in Tuberous Sclerosis. Am. J. Pathol. 2005, 167, 107–116. [Google Scholar] [CrossRef]
- Barnes, E.A.; Kenerson, H.L.; Mak, B.C.; Yeung, R.S. The Loss of Tuberin Promotes Cell Invasion through the SS-Catenin Pathway. Am. J. Respir. Cell Mol. Biol. 2010, 43, 617–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavin, R.J.; Cook, J.; Fiorentino, M.; Bailey, D.; Brown, M.; Loda, M.F. β-Catenin Is a Useful Adjunct Immunohistochemical Marker for the Diagnosis of Pulmonary Lymphangioleiomyomatosis. Am. J. Clin. Pathol. 2011, 135, 776–782. [Google Scholar] [CrossRef]
- Krencz, I.; Sebestyen, A.; Papay, J.; Jeney, A.; Hujber, Z.; Burger, C.D.; Keller, C.A.; Khoor, A. In Situ Analysis of MTORC1/2 and Cellular Metabolism-Related Proteins in Human Lymphangioleiomyomatosis. Hum. Pathol. 2018, 79, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yu, J.J.; Perl, A.K.; Wikenheiser-Brokamp, K.A.; Riccetti, M.; Zhang, E.Y.; Sudha, P.; Adam, M.; Potter, A.; Kopras, E.J.; et al. Single-Cell Transcriptomic Analysis Identifies a Unique Pulmonary Lymphangioleiomyomatosis Cell. Am. J. Respir. Crit. Care Med. 2020, 202, 1373–1387. [Google Scholar] [CrossRef]
- Obraztsova, K.; Basil, M.C.; Rue, R.; Sivakumar, A.; Lin, S.M.; Mukhitov, A.R.; Gritsiuta, A.I.; Evans, J.F.; Kopp, M.; Katzen, J.; et al. MTORC1 Activation in Lung Mesenchyme Drives Sex- and Age-Dependent Pulmonary Structure and Function Decline. Nat. Commun. 2020, 11, 5640. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Muñoz, F.J.; Metcalfe, M.J.; Hitschfeld, M.; Olivares, G.; Godoy, J.A.; Inestrosa, N.C. Trolox and 17beta-Estradiol Protect against Amyloid Beta-Peptide Neurotoxicity by a Mechanism That Involves Modulation of the Wnt Signaling Pathway. J. Biol. Chem. 2005, 280, 11615–11625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The Harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge about Genes and Proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Yamnik, R.L.; Holz, M.K. MTOR/S6K1 and MAPK/RSK Signaling Pathways Coordinately Regulate Estrogen Receptor α Serine 167 Phosphorylation. FEBS Lett. 2010, 584, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Liedert, A.; Nemitz, C.; Haffner-Luntzer, M.; Schick, F.; Jakob, F.; Ignatius, A. Effects of Estrogen Receptor and Wnt Signaling Activation on Mechanically Induced Bone Formation in a Mouse Model of Postmenopausal Bone Loss. Int. J. Mol. Sci. 2020, 21, 8301. [Google Scholar] [CrossRef] [PubMed]
- Mahalati, K.; Kahan, B.D. Clinical Pharmacokinetics of Sirolimus. Clin. Pharmacokinet. 2001, 40, 573–585. [Google Scholar] [CrossRef]
- Valianou, M.; Filippidou, N.; Johnson, D.L.; Vogel, P.; Zhang, E.Y.; Liu, X.; Lu, Y.; Yu, J.J.; Bissler, J.J.; Astrinidis, A. Rapalog Resistance Is Associated with Mesenchymal-Type Changes in Tsc2-Null Cells. Sci. Rep. 2019, 9, 3015. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, X. Research Progress of MTOR Inhibitors. Eur. J. Med. Chem. 2020, 208, 112820. [Google Scholar] [CrossRef] [PubMed]
- Lukey, P.T.; Harrison, S.A.; Yang, S.; Man, Y.; Holman, B.F.; Rashidnasab, A.; Azzopardi, G.; Grayer, M.; Simpson, J.K.; Bareille, P.; et al. A Randomised, Placebo-Controlled Study of Omipalisib (PI3K/MTOR) in Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2019, 53, 1801992. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.F.; Rue, R.W.; Mukhitov, A.R.; Obraztsova, K.; Smith, C.J.; Krymskaya, V.P. Inhibition of Growth of TSC2-Null Cells by a PI3K/MTOR Inhibitor but Not by a Selective MNK1/2 Inhibitor. Biomolecules 2019, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Choo, A.Y.; Yoon, S.-O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin Differentially Inhibits S6Ks and 4E-BP1 to Mediate Cell-Type-Specific Repression of MRNA Translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.K.; Arya, A.; Malecek, M.-K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing Metformin for Cancer Treatment: Current Clinical Studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef] [Green Version]
- El-Chemaly, S.; Taveira-Dasilva, A.; Goldberg, H.J.; Peters, E.; Haughey, M.; Bienfang, D.; Jones, A.M.; Julien-Williams, P.; Cui, Y.; Villalba, J.A.; et al. Sirolimus and Autophagy Inhibition in Lymphangioleiomyomatosis: Results of a Phase I Clinical Trial. Chest 2017, 151, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- El-Chemaly, S.; Taveira-DaSilva, A.; Bagwe, S.; Klonowska, K.; Machado, T.; Lamattina, A.M.; Goldberg, H.J.; Jones, A.M.; Julien-Williams, P.; Maurer, R.; et al. Celecoxib in lymphangioleiomyomatosis: Results of a phase I clinical trial. Eur. Respir. J. 2020, 55, 1902370. [Google Scholar] [CrossRef]
- Valvezan, A.J.; Turner, M.; Belaid, A.; Lam, H.C.; Miller, S.K.; McNamara, M.C.; Baglini, C.; Housden, B.E.; Perrimon, N.; Kwiatkowski, D.J.; et al. MTORC1 Couples Nucleotide Synthesis to Nucleotide Demand Resulting in a Targetable Metabolic Vulnerability. Cancer Cell 2017, 32, 624–638.e5. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; Clevers, H. WNT Signalling Events near the Cell Membrane and Their Pharmacological Targeting for the Treatment of Cancer. Br. J. Pharmacol. 2017, 174, 4547–4563. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-Catenin Signaling Pathway in Cancer. J. Hematol. Oncol.J Hematol Oncol 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Molecular Genetics and Targeted Therapy of WNT-Related Human Diseases (Review). Int. J. Mol. Med. 2017, 40, 587–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, H.-J.; Kahn, M. Safely Targeting Cancer Stem Cells via Selective Catenin Coactivator Antagonism. Cancer Sci. 2014, 105, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Lai, K.K.Y.; Wu, K.; Nguyen, C.; Lin, D.P.; Murali, R.; Kahn, M. Nuclear Receptor/Wnt Beta-Catenin Interactions Are Regulated via Differential CBP/P300 Coactivator Usage. PLoS ONE 2018, 13, e0200714. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Ning, Y.; Yang, D.; Cole, S.; Kahn, M.; Zoghbi, M.; Berg, J.; Fujimori, M.; Inada, T.; Kouji, H.; et al. Phase I First-in-Human Study of PRI-724 in Patients (Pts) with Advanced Solid Tumors. J. Clin. Oncol. 2013, 31 (Suppl. 15), 2501. [Google Scholar] [CrossRef]
- Kimura, K.; Ikoma, A.; Shibakawa, M.; Shimoda, S.; Harada, K.; Saio, M.; Imamura, J.; Osawa, Y.; Kimura, M.; Nishikawa, K.; et al. Safety, Tolerability, and Preliminary Efficacy of the Anti-Fibrotic Small Molecule PRI-724, a CBP/β-Catenin Inhibitor, in Patients with Hepatitis C Virus-Related Cirrhosis: A Single-Center, Open-Label, Dose Escalation Phase 1 Trial. EBioMedicine 2017, 23, 79–87. [Google Scholar] [CrossRef]
- Lafyatis, R.; Mantero, J.C.; Gordon, J.; Kishore, N.; Carns, M.; Dittrich, H.; Spiera, R.; Simms, R.W.; Varga, J. Inhibition of β-Catenin Signaling in the Skin Rescues Cutaneous Adipogenesis in Systemic Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Trial of C-82. J. Investig. Dermatol. 2017, 137, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhou, Y.; Rychahou, P.; Harris, J.W.; Zaytseva, Y.Y.; Liu, J.; Wang, C.; Weiss, H.L.; Liu, C.; Lee, E.Y.; et al. Deptor Is a Novel Target of Wnt/β-Catenin/c-Myc and Contributes to Colorectal Cancer Cell Growth. Cancer Res. 2018, 78, 3163–3175. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Sepramaniam, S.; Chew, X.H.; Wood, K.; Lee, M.A.; Madan, B.; Virshup, D.M. PORCN Inhibition Synergizes with PI3K/MTOR Inhibition in Wnt-Addicted Cancers. Oncogene 2019, 38, 6662–6677. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Toh, S.H.-M.; Chan, Z.-L.; Quah, J.Y.; Chooi, J.-Y.; Tan, T.Z.; Chong, P.S.Y.; Zeng, Q.; Chng, W.-J. A Loss-of-Function Genetic Screening Reveals Synergistic Targeting of AKT/MTOR and WTN/β-Catenin Pathways for Treatment of AML with High PRL-3 Phosphatase. J. Hematol. Oncol. J Hematol Oncol. 2018, 11, 36. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Lazar, V.; Rubin, E.; Depil, S.; Pawitan, Y.; Martini, J.-F.; Gomez-Navarro, J.; Yver, A.; Kan, Z.; Dry, J.R.; Kehren, J.; et al. A Simplified Interventional Mapping System (SIMS) for the Selection of Combinations of Targeted Treatments in Non-Small Cell Lung Cancer. Oncotarget 2015, 6, 14139–14152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.; Lee, H.-S.; Inoue, Y.; Moss, J.; Singer, L.G.; Strange, C.; Nakata, K.; Barker, A.F.; Chapman, J.T.; Brantly, M.L.; et al. Serum VEGF-D a Concentration as a Biomarker of Lymphangioleiomyomatosis Severity and Treatment Response: A Prospective Analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) Trial. Lancet Respir. Med. 2013, 1, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Hirose, M.; Matsumuro, A.; Arai, T.; Sugimoto, C.; Akira, M.; Kitaichi, M.; Young, L.R.; McCormack, F.X.; Inoue, Y. Serum Vascular Endothelial Growth Factor-D as a Diagnostic and Therapeutic Biomarker for Lymphangioleiomyomatosis. PLoS ONE 2019, 14, e0212776. [Google Scholar] [CrossRef] [PubMed]
- Lamattina, A.M.; Taveira-Dasilva, A.; Goldberg, H.J.; Bagwe, S.; Cui, Y.; Rosas, I.O.; Moss, J.; Henske, E.P.; El-Chemaly, S. Circulating Biomarkers From the Phase 1 Trial of Sirolimus and Autophagy Inhibition for Patients With Lymphangioleiomyomatosis. Chest 2018, 154, 1070–1082. [Google Scholar] [CrossRef]
- Klover, P.J.; Thangapazham, R.L.; Kato, J.; Wang, J.-A.; Anderson, S.A.; Hoffmann, V.; Steagall, W.K.; Li, S.; McCart, E.; Nathan, N.; et al. Tsc2 Disruption in Mesenchymal Progenitors Results in Tumors with Vascular Anomalies Overexpressing Lgals3. eLife 2017, 6, e23202. [Google Scholar] [CrossRef]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of Transforming Growth Factor-Β1-Driven Lung Fibrosis by Galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Alkhatib, A.; Kolls, J.K.; Kondoh, Y.; Lasky, J.A. Pharmacotherapy and Adjunctive Treatment for Idiopathic Pulmonary Fibrosis (IPF). J. Thorac. Dis. 2019, 11 (Suppl. 14), S1740–S1754. [Google Scholar] [CrossRef]
- El-Chemaly, S.; Henske, E.P. The next Breakthrough in LAM Clinical Trials May Be Their Design: Challenges in Design and Execution of Future LAM Clinical Trials. Expert Rev. Respir. Med. 2015, 9, 195–204. [Google Scholar] [CrossRef]
- Burnett, T.; Mozgunov, P.; Pallmann, P.; Villar, S.S.; Wheeler, G.M.; Jaki, T. Adding Flexibility to Clinical Trial Designs: An Example-Based Guide to the Practical Use of Adaptive Designs. BMC Med. 2020, 18, 352. [Google Scholar] [CrossRef]
- Evans, J.F.; Hutchinson, J.H. Seeing the Future of Bioactive Lipid Drug Targets. Nat. Chem. Biol. 2010, 6, 476–479. [Google Scholar] [CrossRef]
- Feinberg, E.N.; Joshi, E.; Pande, V.S.; Cheng, A.C. Improvement in ADMET Prediction with Multitask Deep Featurization. J. Med. Chem. 2020, 63, 8835–8848. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, A.; Pandiella, A. Proteolysis Targeting Chimeras (PROTACs) in Cancer Therapy. J. Exp. Clin. Cancer Res. CR 2020, 39, 189. [Google Scholar] [CrossRef] [PubMed]
- Pecetta, S.; Finco, O.; Seubert, A. Quantum Leap of Monoclonal Antibody (MAb) Discovery and Development in the COVID-19 Era. Semin. Immunol. 2020, 50, 101427. [Google Scholar] [CrossRef]
- Gary, E.N.; Weiner, D.B. DNA Vaccines: Prime Time Is Now. Curr. Opin. Immunol. 2020, 65, 21–27. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Roose, K. How MRNA Therapeutics Are Entering the Monoclonal Antibody Field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisel, K.; Merrilees, M.J.; Atochina-Vasserman, E.N.; Lian, L.; Obraztsova, K.; Rue, R.; Vasserman, A.N.; Zuo, N.; Angel, L.F.; Gow, A.J.; et al. Immune Checkpoint Ligand PD-L1 Is Upregulated in Pulmonary Lymphangioleiomyomatosis (LAM). Am. J. Respir. Cell Mol. Biol. 2018, 59, 723–732. [Google Scholar] [CrossRef]
- Liu, H.-J.; Lizotte, P.H.; Du, H.; Speranza, M.C.; Lam, H.C.; Vaughan, S.; Alesi, N.; Wong, K.-K.; Freeman, G.J.; Sharpe, A.H.; et al. TSC2-Deficient Tumors Have Evidence of T Cell Exhaustion and Respond to Anti-PD-1/Anti-CTLA-4 Immunotherapy. JCI Insight 2018, 3, e98674. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Massi, D.; Teng, M.W.L.; Mandala, M. PI3K-AKT-MTOR Inhibition in Cancer Immunotherapy, Redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni, V.; Sharma, M.R.; Patnaik, A. The Resurgence of Antibody Drug Conjugates in Cancer Therapeutics: Novel Targets and Payloads. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e58–e74. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Azhdarinia, A.; Ghosh, S.C.; Xiong, W.; An, Z.; Liu, Q.; Carmon, K.S. LGR5-Targeted Antibody-Drug Conjugate Eradicates Gastrointestinal Tumors and Prevents Recurrence. Mol. Cancer Ther. 2016, 15, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Khalil, A.M. The Genome Editing Revolution: Review. J. Genet. Eng. Biotechnol. 2020, 18, 68. [Google Scholar] [CrossRef]
- Liu, H.-J.; Lam, H.C.; Baglini, C.V.; Nijmeh, J.; Cottrill, A.A.; Chan, S.Y.; Henske, E.P. Rapamycin-Upregulated MiR-29b Promotes MTORC1-Hyperactive Cell Growth in TSC2-Deficient Cells by Downregulating Tumor Suppressor Retinoic Acid Receptor β (RARβ). Oncogene 2019, 38, 7367–7383. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Luo, S.; Wu, M.; Huang, C.; Shi, H.; Song, X. LncRNA GHET1 Promotes Cervical Cancer Progression through Regulating AKT/MTOR and Wnt/β-Catenin Signaling Pathways. Biosci. Rep. 2020, 40, BSR20191265. [Google Scholar] [CrossRef]
- Miller, A.J.; Dye, B.R.; Ferrer-Torres, D.; Hill, D.R.; Overeem, A.W.; Shea, L.D.; Spence, J.R. Generation of Lung Organoids from Human Pluripotent Stem Cells in Vitro. Nat. Protoc. 2019, 14, 518–540. [Google Scholar] [CrossRef] [PubMed]
- Nawroth, J.C.; Barrile, R.; Conegliano, D.; van Riet, S.; Hiemstra, P.S.; Villenave, R. Stem Cell-Based Lung-on-Chips: The Best of Both Worlds? Adv. Drug Deliv. Rev. 2019, 140, 12–32. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Katzen, J.; Engler, A.E.; Guo, M.; Herriges, M.J.; Kathiriya, J.J.; Windmueller, R.; Ysasi, A.B.; Zacharias, W.J.; Chapman, H.A.; et al. The Cellular and Physiological Basis for Lung Repair and Regeneration: Past, Present, and Future. Cell Stem Cell 2020, 26, 482–502. [Google Scholar] [CrossRef]
- Lund-Ricard, Y.; Cormier, P.; Morales, J.; Boutet, A. MTOR Signaling at the Crossroad between Metazoan Regeneration and Human Diseases. Int. J. Mol. Sci. 2020, 21, 2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, J.F.; Obraztsova, K.; Lin, S.M.; Krymskaya, V.P. CrossTORC and WNTegration in Disease: Focus on Lymphangioleiomyomatosis. Int. J. Mol. Sci. 2021, 22, 2233. https://doi.org/10.3390/ijms22052233
Evans JF, Obraztsova K, Lin SM, Krymskaya VP. CrossTORC and WNTegration in Disease: Focus on Lymphangioleiomyomatosis. International Journal of Molecular Sciences. 2021; 22(5):2233. https://doi.org/10.3390/ijms22052233
Chicago/Turabian StyleEvans, Jilly Frances, Kseniya Obraztsova, Susan M. Lin, and Vera P. Krymskaya. 2021. "CrossTORC and WNTegration in Disease: Focus on Lymphangioleiomyomatosis" International Journal of Molecular Sciences 22, no. 5: 2233. https://doi.org/10.3390/ijms22052233
APA StyleEvans, J. F., Obraztsova, K., Lin, S. M., & Krymskaya, V. P. (2021). CrossTORC and WNTegration in Disease: Focus on Lymphangioleiomyomatosis. International Journal of Molecular Sciences, 22(5), 2233. https://doi.org/10.3390/ijms22052233