
RGDS-Modified Superporous Poly(2-Hydroxyethyl Methacrylate)-Based Scaffolds as 3D In Vitro Leukemia Model

 ,
,  , ,
, ,
Abstract
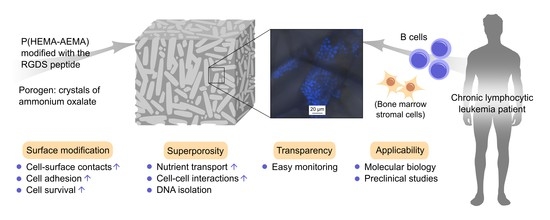
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Poly(2-Hydroxyethyl Methacrylate-co-2-Aminoethyl Methacrylate) (P(HEMA-AEMA)) Scaffolds
2.2. Immobilization of RGDS Peptide on P(HEMA-AEMA)-RGDS Scaffolds
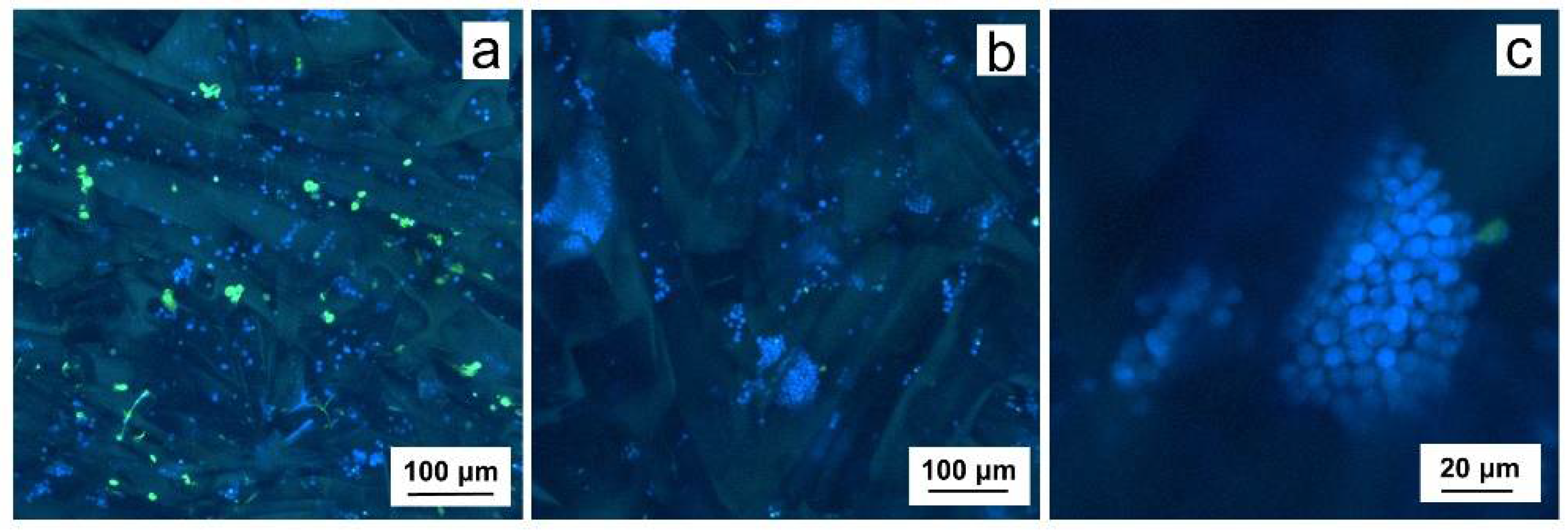
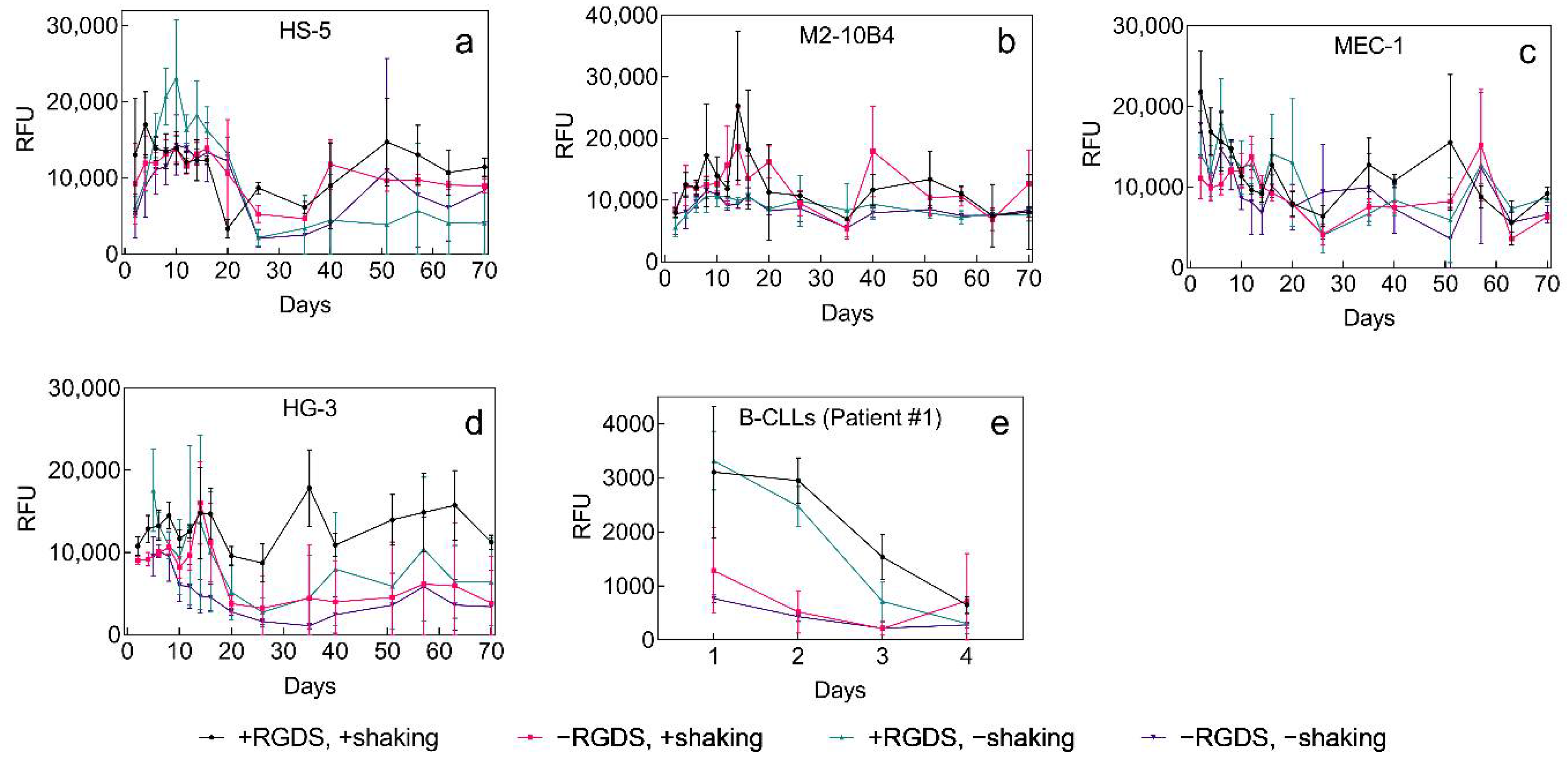
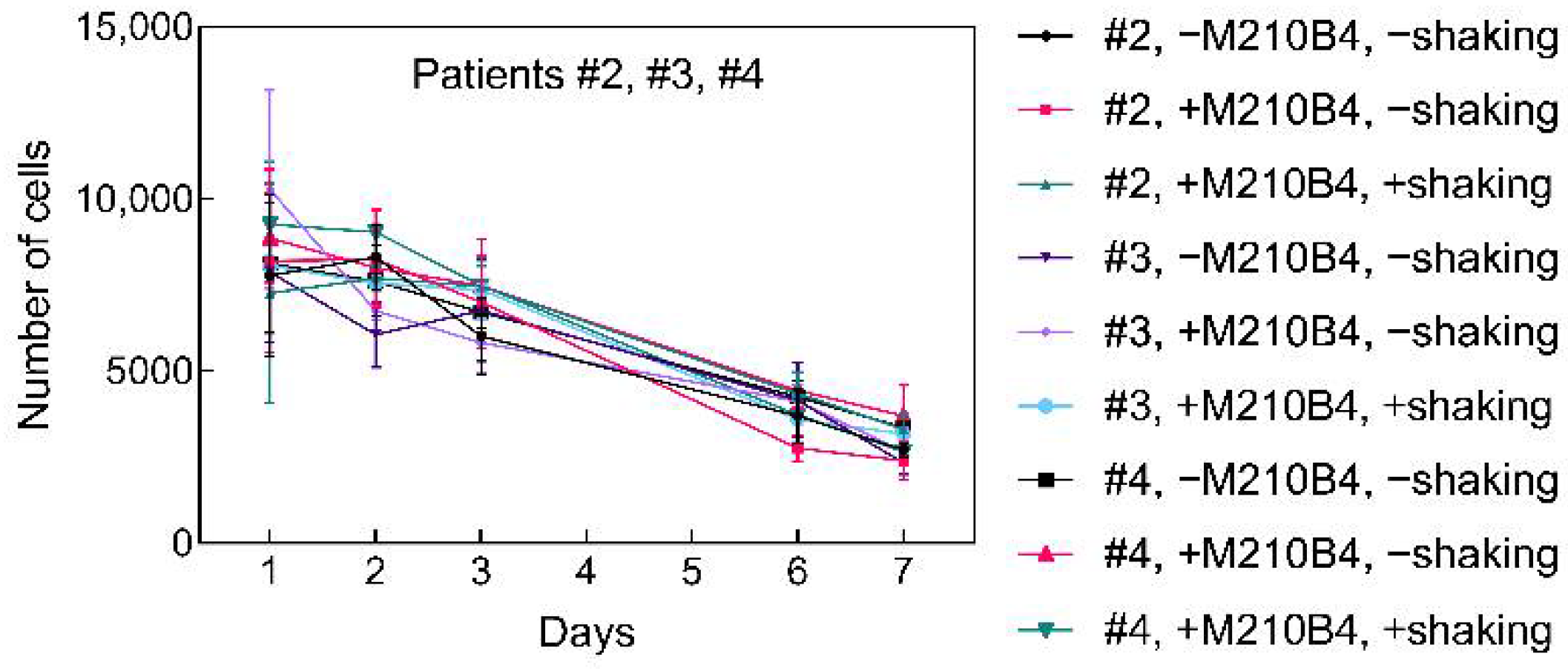
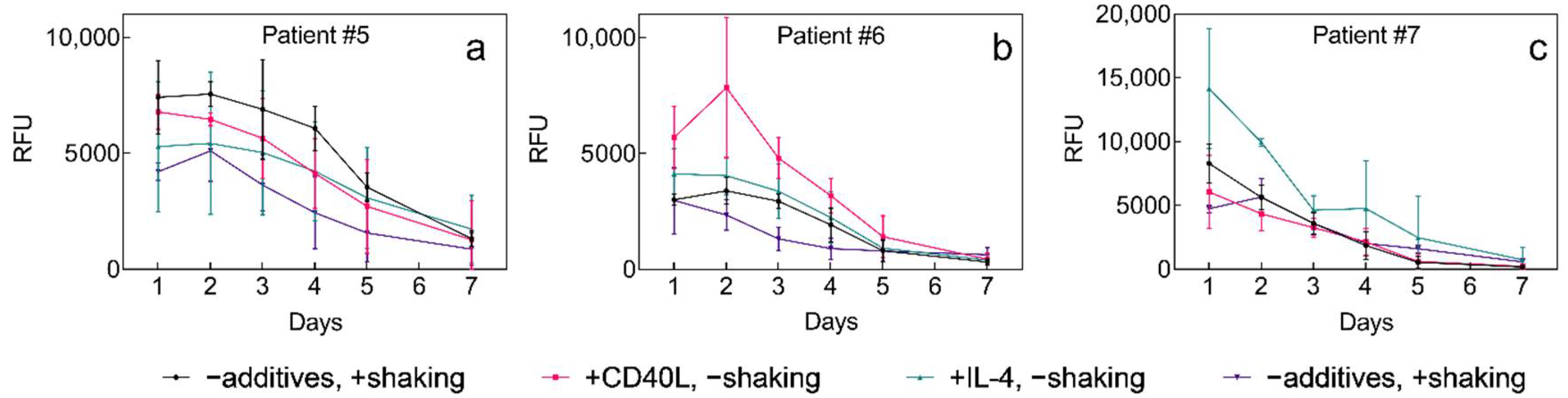
2.3. 3D Culture of Leukemia B Cells on P(HEMA-AEMA)-RGDS Scaffolds
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of P(HEMA-AEMA) Scaffolds
4.3. Synthesis of Ac-CGGGRGDSGGGY-NH2 (RGDS) Peptide
4.4. Activation of P(HEMA-AEMA) Scaffolds and Immobilization of RGDS Peptide
4.5. Primary B-CLL Cells and Cell Lines
4.6. Cell Culture Conditions
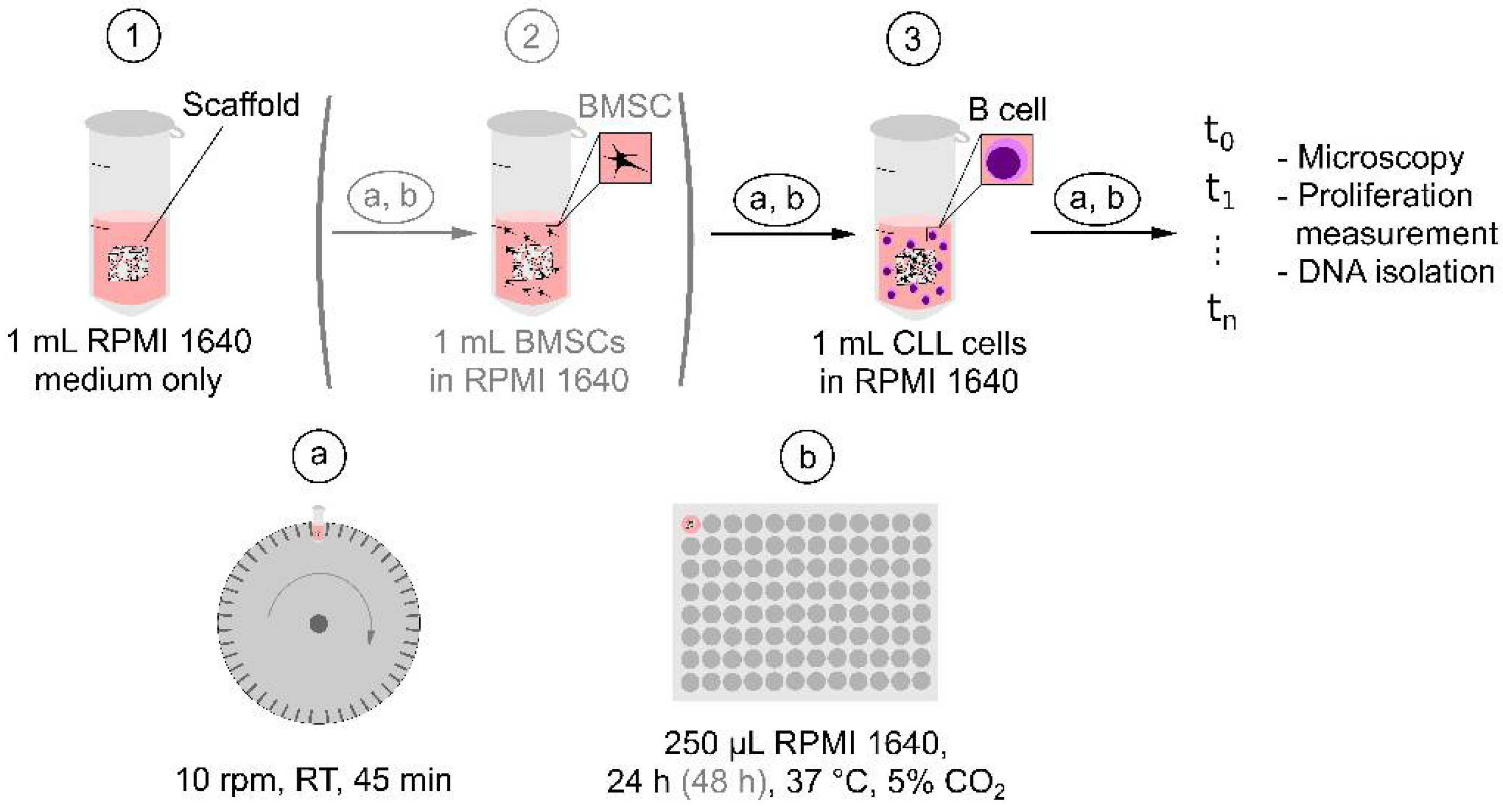
4.7. Seeding of Cells in P(HEMA-AEMA) Scaffolds
4.8. Cell Imaging and Image Analysis
4.9. Metabolic Activity
4.10. DNA Isolation and Quality Control
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Postprocessing of Microscopy Images
References
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic Lymphocytic Leukaemia. Nat. Rev. Dis. Primer 2017, 3, 16096. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M. Chronic Lymphocytic Leukemia: 2020 Update on Diagnosis, Risk Stratification and Treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [Green Version]
- Gribben, J.G. How I Treat CLL up Front. Blood 2010, 115, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Dalla-Favera, R. Chronic Lymphocytic Leukaemia: From Genetics to Treatment. Nat. Rev. Clin. Oncol. 2019, 16, 684–701. [Google Scholar] [CrossRef]
- Satpathy, A.; Datta, P.; Wu, Y.; Ayan, B.; Bayram, E.; Ozbolat, I.T. Developments with 3D Bioprinting for Novel Drug Discovery. Expert Opin. Drug Discov. 2018, 13, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.A.; Moscato, S.; George, S.; Azimi, B.; Danti, S. Liver Cancer: Current and Future Trends Using Biomaterials. Cancers 2019, 11, 2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Lin, T.L.; Lipe, B.; Hopkins, R.A.; Shinogle, H.; Aljitawi, O.S. A Novel Extracellular Matrix-Based Leukemia Model Supports Leukemia Cells with Stem Cell-like Characteristics. Leuk. Res. 2018, 72, 105–112. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Z.; Dong, D.-L.; Jang, T.-S.; Knowles, J.C.; Kim, H.-W.; Jin, G.-Z.; Xuan, Y. 3D Culture Technologies of Cancer Stem Cells: Promising Ex Vivo Tumor Models. J. Tissue Eng. 2020, 11. [Google Scholar] [CrossRef]
- Burger, J.A.; Gribben, J.G. The Microenvironment in Chronic Lymphocytic Leukemia (CLL) and Other B Cell Malignancies: Insight into Disease Biology and New Targeted Therapies. Semin. Cancer Biol. 2014, 24, 71–81. [Google Scholar] [CrossRef]
- Barbaglio, F.; Belloni, D.; Scarfò, L.; Sbrana, F.V.; Ponzoni, M.; Bongiovanni, L.; Pavesi, L.; Zambroni, D.; Stamatopoulos, K.; Caiolfa, V.R.; et al. 3D Co-Culture Model of Chronic Lymphocytic Leukemia Bone Marrow Microenvironment Predicts Patient-Specific Response to Mobilizing Agents. Haematologica 2020. [Google Scholar] [CrossRef]
- Dos Santos, J.; Enfield, L.; Dos Santos, S.B.; Allenby, M.C.; Zemenides, S.; Mantalaris, A.; Panoskaltsis, N. Primary Chronic Lymphocytic Leukemia Cells Can Be Maintained Long-Term in Serum-Free, Cytokine-Free 3D Culture. Blood 2017, 130, 2989. [Google Scholar] [CrossRef]
- Verjans, E.-T.; Doijen, J.; Luyten, W.; Landuyt, B.; Schoofs, L. Three-Dimensional Cell Culture Models for Anticancer Drug Screening: Worth the Effort? J. Cell. Physiol. 2018, 233, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.; Dey, M.; Ataie, Z.; Unutmaz, D.; Ozbolat, I.T. 3D Bioprinting for Reconstituting the Cancer Microenvironment. Npj Precis. Oncol. 2020, 4, 1–13. [Google Scholar] [CrossRef]
- El-Sherbiny, I.M.; Yacoub, M.H. Hydrogel Scaffolds for Tissue Engineering: Progress and Challenges. Glob. Cardiol. Sci. Pract. 2013, 2013, 316–342. [Google Scholar] [CrossRef] [Green Version]
- Dhandayuthapani, B.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Polymeric Scaffolds in Tissue Engineering Application: A Review. Int. J. Polym. Sci. 2011. [Google Scholar] [CrossRef]
- Lee, J.; Cuddihy, M.J.; Kotov, N.A. Three-Dimensional Cell Culture Matrices: State of the Art. Tissue Eng. Part B Rev. 2008, 14, 61–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drury, J.L.; Mooney, D.J. Hydrogels for Tissue Engineering: Scaffold Design Variables and Applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Zhu, J.; Marchant, R.E. Design Properties of Hydrogel Tissue-Engineering Scaffolds. Expert Rev. Med. Devices 2011, 8, 607–626. [Google Scholar] [CrossRef] [PubMed]
- Atzet, S.; Curtin, S.; Trinh, P.; Bryant, S.; Ratner, B. Degradable Poly(2-Hydroxyethyl Methacrylate)-co-Polycaprolactone Hydrogels for Tissue Engineering Scaffolds. Biomacromolecules 2008, 9, 3370–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kůdela, J. Hydrogels. In Encyclopedia of Polymer Science and Technology; Wiley: New York, 1987; Volume 7, pp. 783–807. [Google Scholar]
- ten Hacken, E.; Burger, J.A. Microenvironment Dependency in Chronic Lymphocytic Leukemia: The Basis for New Targeted Therapies. Pharmacol. Ther. 2014, 144, 338–348. [Google Scholar] [CrossRef]
- Lagneaux, L.; Delforge, A.; Bron, D.; De Bruyn, C.; Stryckmans, P. Chronic Lymphocytic Leukemic B Cells but Not Normal B Cells Are Rescued from Apoptosis by Contact with Normal Bone Marrow Stromal Cells. Blood 1998, 91, 2387–2396. [Google Scholar] [CrossRef] [PubMed]
- Crassini, K.; Shen, Y.; Mulligan, S.; Giles Best, O. Modeling the Chronic Lymphocytic Leukemia Microenvironment in Vitro. Leuk. Lymphoma 2017, 58, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Jabs, J.; Zickgraf, F.M.; Park, J.; Wagner, S.; Jiang, X.; Jechow, K.; Kleinheinz, K.; Toprak, U.H.; Schneider, M.A.; Meister, M.; et al. Screening Drug Effects in Patient-Derived Cancer Cells Links Organoid Responses to Genome Alterations. Mol. Syst. Biol. 2017, 13, 955. [Google Scholar] [CrossRef]
- Sommerová, L.; Michalová, E.; Hrstka, R. New approaches for chemosensitivity testing in malignant diseases. Klin. Onkol. Cas. Ceske Slov. Onkol. Spolecnosti 2018, 31, 117–124. [Google Scholar] [CrossRef]
- Lee, J.; Li, M.; Milwid, J.; Dunham, J.; Vinegoni, C.; Gorbatov, R.; Iwamoto, Y.; Wang, F.; Shen, K.; Hatfield, K.; et al. Implantable Microenvironments to Attract Hematopoietic Stem/Cancer Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 19638–19643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, G.; Clarke, J.; Picard, F.; Riches, P.; Jia, L.; Han, F.; Li, B.; Shu, W. 3D Bioactive Composite Scaffolds for Bone Tissue Engineering. Bioact. Mater. 2018, 3, 278–314. [Google Scholar] [CrossRef] [Green Version]
- Kubinová, Š.; Horák, D.; Syková, E. Cholesterol-Modified Superporous Poly(2-Hydroxyethyl Methacrylate) Scaffolds for Tissue Engineering. Biomaterials 2009, 30, 4601–4609. [Google Scholar] [CrossRef]
- Macková, H.; Plichta, Z.; Proks, V.; Kotelnikov, I.; Kučka, J.; Hlídková, H.; Horák, D.; Kubinová, Š.; Jiráková, K. RGDS- and SIKVAVS-Modified Superporous Poly(2-Hydroxyethyl Methacrylate) Scaffolds for Tissue Engineering Applications. Macromol. Biosci. 2016, 16, 1621–1631. [Google Scholar] [CrossRef]
- Singh, S.; Ghode, S.; Devi, M.R.; Limaye, L.; Kale, V. Phenotypic and Functional Characterization of a Marrow-Derived Stromal Cell Line, M210B4 and Its Comparison with Primary Marrow Stromal Cells. Biomed. Res. J. 2015, 2, 120. [Google Scholar] [CrossRef]
- Kurtova, A.V.; Balakrishnan, K.; Chen, R.; Ding, W.; Schnabl, S.; Quiroga, M.P.; Sivina, M.; Wierda, W.G.; Estrov, Z.; Keating, M.J.; et al. Diverse Marrow Stromal Cells Protect CLL Cells from Spontaneous and Drug-Induced Apoptosis: Development of a Reliable and Reproducible System to Assess Stromal Cell Adhesion-Mediated Drug Resistance. Blood 2009, 114, 4441–4450. [Google Scholar] [CrossRef]
- Stacchini, A.; Aragno, M.; Vallario, A.; Alfarano, A.; Circosta, P.; Gottardi, D.; Faldella, A.; Rege-Cambrin, G.; Thunberg, U.; Nilsson, K.; et al. MEC1 and MEC2: Two New Cell Lines Derived from B-Chronic Lymphocytic Leukaemia in Prolymphocytoid Transformation. Leuk. Res. 1999, 23, 127–136. [Google Scholar] [CrossRef]
- German Collection of Microorganisms and Cell Cultures GmbH: Details. Available online: https://www.dsmz.de/collection/catalogue/details/culture/ACC-765 (accessed on 22 October 2019).
- Ghia, P.; Circosta, P.; Scielzo, C.; Vallario, A.; Camporeale, A.; Granziero, L.; Caligaris-Cappio, F. Differential effects on CLL cell survival exerted by different microenvironmental elements. In Chronic Lymphocytic Leukemia; Current Topics in Microbiology and Immunology; Springer: Berlin, Heidelberg, 2005; pp. 135–145. ISBN 978-3-540-29933-2. [Google Scholar]
- Bourgine, P.E.; Klein, T.; Paczulla, A.M.; Shimizu, T.; Kunz, L.; Kokkaliaris, K.D.; Coutu, D.L.; Lengerke, C.; Skoda, R.; Schroeder, T.; et al. In Vitro Biomimetic Engineering of a Human Hematopoietic Niche with Functional Properties. Proc. Natl. Acad. Sci. USA 2018, 115, E5688–E5695. [Google Scholar] [CrossRef] [Green Version]
- Walsby, E.; Buggins, A.; Devereux, S.; Jones, C.; Pratt, G.; Brennan, P.; Fegan, C.; Pepper, C. Development and Characterization of a Physiologically Relevant Model of Lymphocyte Migration in Chronic Lymphocytic Leukemia. Blood 2014, 123, 3607–3617. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, G.R.; Caton, M.C.; Nova, M.P.; Parandoosh, Z. Assessment of the Alamar Blue Assay for Cellular Growth and Viability in Vitro. J. Immunol. Methods 1997, 204, 205–208. [Google Scholar] [CrossRef]
- Chiaraviglio, L.; Kirby, J.E. Evaluation of Impermeant, DNA-Binding Dye Fluorescence as a Real-Time Readout of Eukaryotic Cell Toxicity in a High Throughput Screening Format. Assay Drug Dev. Technol. 2014, 12, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Rush, J.S.; Hodgkin, P.D. B Cells Activated via CD40 and IL-4 Undergo a Division Burst but Require Continued Stimulation to Maintain Division, Survival and Differentiation. Eur. J. Immunol. 2001, 31, 1150–1159. [Google Scholar] [CrossRef]
- Rombout, A.; Lust, S.; Offner, F.; Naessens, E.; Verhasselt, B.; Philippé, J. Mimicking the Tumour Microenvironment of Chronic Lymphocytic Leukaemia in Vitro Critically Depends on the Type of B-Cell Receptor Stimulation. Br. J. Cancer 2016, 114, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Natoni, A.; O’Dwyer, M.; Santocanale, C. A Cell Culture System That Mimics Chronic Lymphocytic Leukemia Cells Microenvironment for Drug Screening and Characterization. Methods Mol. Biol. Clifton NJ 2013, 986, 217–226. [Google Scholar] [CrossRef]
- Han, K.; Pierce, S.E.; Li, A.; Spees, K.; Anderson, G.R.; Seoane, J.A.; Lo, Y.-H.; Dubreuil, M.; Olivas, M.; Kamber, R.A.; et al. CRISPR Screens in Cancer Spheroids Identify 3D Growth-Specific Vulnerabilities. Nature 2020, 580, 136–141. [Google Scholar] [CrossRef]
- Rosén, A.; Bergh, A.-C.; Gogok, P.; Evaldsson, C.; Myhrinder, A.L.; Hellqvist, E.; Rasul, A.; Björkholm, M.; Jansson, M.; Mansouri, L.; et al. Lymphoblastoid Cell Line with B1 Cell Characteristics Established from a Chronic Lymphocytic Leukemia Clone by in Vitro EBV Infection. Oncoimmunology 2012, 1, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Crompot, E.; Van Damme, M.; Pieters, K.; Vermeersch, M.; Perez-Morga, D.; Mineur, P.; Maerevoet, M.; Meuleman, N.; Bron, D.; Lagneaux, L.; et al. Extracellular Vesicles of Bone Marrow Stromal Cells Rescue Chronic Lymphocytic Leukemia B Cells from Apoptosis, Enhance Their Migration and Induce Gene Expression Modifications. Haematologica 2017, 102, 1594–1604. [Google Scholar] [CrossRef] [Green Version]
- Roecklein, B.A.; Torok-Storb, B. Functionally Distinct Human Marrow Stromal Cell Lines Immortalized by Transduction with the Human Papilloma Virus E6/E7 Genes. Blood 1995, 85, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, F.M.; Humphries, R.K.; Abraham, S.D.; Krystal, G.; Eaves, C.J. Partial Characterization of a Novel Stromal Cell-Derived Pre-B-Cell Growth Factor Active on Normal and Immortalized Pre-B Cells. Exp. Hematol. 1988, 16, 718–726. [Google Scholar]
- Thevenot, P.; Nair, A.; Dey, J.; Yang, J.; Tang, L. Method to Analyze Three-Dimensional Cell Distribution and Infiltration in Degradable Scaffolds. Tissue Eng. Part C Methods 2008, 14, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, B.; Liminga, G.; Csoka, K.; Fridborg, H.; Dhar, S.; Nygren, P.; Larsson, R. Cytotoxic Activity of Calcein Acetoxymethyl Ester (Calcein/AM) on Primary Cultures of Human Haematological and Solid Tumours. Eur. J. Cancer Oxf. Engl. 1990 1996, 32A, 883–887. [Google Scholar] [CrossRef]
- Durand, R.E.; Olive, P.L. Cytotoxicity, Mutagenicity and DNA Damage by Hoechst 33342. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1982, 30, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munshi, S.; Twining, R.C.; Dahl, R. Alamar Blue Reagent Interacts with Cell-Culture Media Giving Different Fluorescence over Time: Potential for False Positives. J. Pharmacol. Toxicol. Methods 2014, 70, 195–198. [Google Scholar] [CrossRef] [PubMed]
- FastDNATM SPIN Kit for Soil, MP Biomedicals—Instruction Manual. Available online: https://media.mpbio.com/productattachment/LS082019-EN-FastDNA-SPIN-Kit-for-Soil-116560200-Manual.pdf (accessed on 22 January 2021).
- Agilent Genomic DNA Screentape—Quick Guide for TapeStation Systems. Available online: https://www.agilent.com/cs/library/usermanuals/public/gDNA_QuickGuide.pdf (accessed on 3 December 2019).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svozilová, H.; Plichta, Z.; Proks, V.; Studená, R.; Baloun, J.; Doubek, M.; Pospíšilová, Š.; Horák, D. RGDS-Modified Superporous Poly(2-Hydroxyethyl Methacrylate)-Based Scaffolds as 3D In Vitro Leukemia Model. Int. J. Mol. Sci. 2021, 22, 2376. https://doi.org/10.3390/ijms22052376
Svozilová H, Plichta Z, Proks V, Studená R, Baloun J, Doubek M, Pospíšilová Š, Horák D. RGDS-Modified Superporous Poly(2-Hydroxyethyl Methacrylate)-Based Scaffolds as 3D In Vitro Leukemia Model. International Journal of Molecular Sciences. 2021; 22(5):2376. https://doi.org/10.3390/ijms22052376
Chicago/Turabian StyleSvozilová, Hana, Zdeněk Plichta, Vladimír Proks, Radana Studená, Jiří Baloun, Michael Doubek, Šárka Pospíšilová, and Daniel Horák. 2021. "RGDS-Modified Superporous Poly(2-Hydroxyethyl Methacrylate)-Based Scaffolds as 3D In Vitro Leukemia Model" International Journal of Molecular Sciences 22, no. 5: 2376. https://doi.org/10.3390/ijms22052376
APA StyleSvozilová, H., Plichta, Z., Proks, V., Studená, R., Baloun, J., Doubek, M., Pospíšilová, Š., & Horák, D. (2021). RGDS-Modified Superporous Poly(2-Hydroxyethyl Methacrylate)-Based Scaffolds as 3D In Vitro Leukemia Model. International Journal of Molecular Sciences, 22(5), 2376. https://doi.org/10.3390/ijms22052376