
Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration


 and
and
Abstract
:
1. Introduction
2. Results
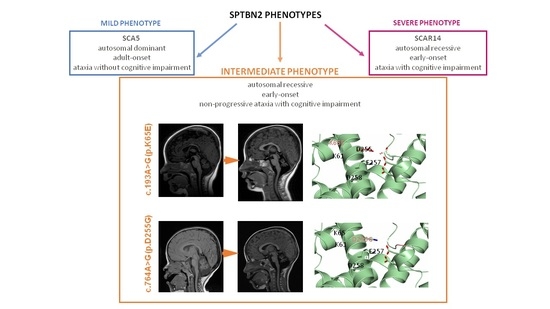
2.1. Clinical, Neuroimaging, and Genetic Findings
2.2. Modeling of the SPTBN2 p.K65E and p.D255G Mutations
2.3. Expression Analysis and Subcellular Location Studies
2.4. Phenotype to Genotype Correlations
3. Discussion
4. Materials and Methods
4.1. Patients: Clinical and Neuroimaging Assessment
4.2. Genetics and Bioinformatics
4.3. Conservation Analysis and Structural Modeling
4.4. Protein Expression
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ikeda, Y.; Dick, K.A.; Weatherspoon, M.R.; Gincel, D.; Armbrust, K.R.; Dalton, J.C.; Stevanin, G.; Durr, A.; Zuhlke, C.; Burk, K.; et al. Spectrin mutations cause spino-cerebellar ataxia type 5. Nat. Genet. 2006, 38, 184–190. [Google Scholar] [CrossRef]
- Lise, S.; Clarkson, Y.; Perkins, E.; Kwasniewska, A.; Sadighi Akha, E.; Schnekenberg, R.P.; Suminaite, D.; Hope, J.; Baker, I.; Gregory, L.; et al. Recessive mutations in SPTBN2 implicate beta-III spectrin in both cognitive and motor development. PLoS Genet. 2012, 8, e1003074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Johnson, A.K.; Nelakuditi, V.; Guidugli, L.; Fischer, D.; Arndt, K.; Ma, L.; Sandford, E.; Shakkottai, V.; Boycott, K.; et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet. Med. 2018, 21, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Nicita, F.; Nardella, M.; Bellacchio, E.; Alfieri, P.; Terrone, G.; Piccini, G.; Graziola, F.; Pignata, C.; Capuano, A.; Bertini, E.; et al. Heterozygous missense variants of SPTBN2 are a frequent cause of congenital cerebellar ataxia. Clin. Genet. 2019, 96, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, B.; Grochowalska, R.; Bogusławska, D.M.; Sikorski, A.F. The role of spectrin in cell adhesion and cell–cell contact. Exp. Biol. Med. 2019, 244, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Stankewich, M.C.; Tse, W.T.; Peters, L.L.; Ch’ng, Y.; John, K.M.; Stabach, P.R.; Devarajan, P.; Morrow, J.S.; Lux, S.E. A widely expressed betaIII spectrin associated with Golgi and cytoplasmic vesicles. Proc. Natl. Acad. Sci. USA 1998, 95, 14158–14163. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, G.; Orita, S.; Naito, A.; Maeda, M.; Igarashi, H.; Sasaki, T.; Takai, Y. A novel brain-specific isoform of beta spectrin: Isolation and its interaction with Munc13. Biochem. Biophys. Res. Commun. 1998, 248, 846–851. [Google Scholar] [CrossRef]
- Jackson, M.; Song, W.; Liu, M.-Y.; Jin, L.; Dykes-Hoberg, M.; Lin, C.-L.G.; Bowers, W.J.; Federoff, H.J.; Sternweis, P.C.; Rothstein, J.D. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nat. Cell Biol. 2001, 410, 89–93. [Google Scholar] [CrossRef]
- Stevanin, G.; Herman, A.; Brice, A.; Dürr, A. Clinical and MRI findings in spinocerebellar ataxia type 5. Neurology 1999, 53, 1355–1357. [Google Scholar] [CrossRef]
- Ranum, L.P.; Schut, L.J.; Lundgren, J.K.; Orr, H.T.; Livingston, D.M. Spinocerebellar ataxia type 5 in a family descended from the grandparents of President Lincoln maps to chromosome 11. Nat. Genet. 1994, 8, 280–284. [Google Scholar] [CrossRef]
- Rea, G.; Tirupathi, S.; Williams, J.; Clouston, P.; Morrison, P.J. Infantile Onset of Spinocerebellar Ataxia Type 5 (SCA-5) in a 6 Month-Old with Ataxic Cerebral Palsy. Cerebellum 2019, 19, 161–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, T.; Kashimada, A.; Nomura, T.; Moriyama, K.; Yokoyama, H.; Hasegawa, S.; Takagi, M.; Mizutani, S. Infantile-onset spinocerebellar ataxia type 5 associated with a novel SPTBN2 mutation: A case report. Brain Dev. 2019, 41, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, S.M.; Heller, R.; Thoenes, M.; Zaki, M.S.; Swan, D.; Elsobky, E.; Zühlke, C.; Ebermann, I.; Nürnberg, G.; Nürnberg, P.; et al. Autosomal dominant SCA5 and autosomal recessive infantile SCA are allelic conditions resulting from SPTBN2 mutations. Eur. J. Hum. Genet. 2013, 22, 286–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildiz Bolukbasi, E.; Afzal, M.; Mumtaz, S.; Ahmad, N.; Malik, S.; Tolun, A. Progressive SCAR14 with unclear speech, developmental delay, tremor, and behavioral problems caused by a homozygous deletion of the SPTBN2 pleckstrin ho-mology domain. Am. J. Med. Genet. A 2017, 173, 2494–2499. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, S.; Micalizzi, A.; D’Arrigo, S.; Ginevrino, M.; Biagini, T.; Mazza, T.; Valente, E.M. Between SCA5 and SCAR14: Delineation of the SPTBN2 p.R480W-associated phenotype. Eur. J. Hum. Genet. 2018, 26, 928–929. [Google Scholar] [CrossRef] [PubMed]
- Kuperberg, M.; Lev, D.; Blumkin, L.; Zerem, A.; Ginsberg, M.; Linder, I.; Carmi, N.; Kivity, S.; Lerman-Sagie, T.; Leshinsky-Silver, E. Utility of Whole Exome Sequencing for Genetic Diagnosis of Previously Undiagnosed Pediatric Neurology Patients. J. Child Neurol. 2016, 31, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Parolin Schnekenberg, R.; Perkins, E.M.; Miller, J.W.; Davies, W.I.; D’Adamo, M.C.; Pessia, M.; Fawcett, K.A.; Sims, D.; Gillard, E.; Hudspith, K.; et al. De novo point mutations in patients diagnosed with ataxic cerebral palsy. Brain 2015, 138, 1817–1832. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.-D.; Ho, E.S.; Martinez-Ojeda, M.; Darras, B.T.; Khwaja, O.S. Case of Infantile Onset Spinocerebellar Ataxia Type 5. J. Child Neurol. 2012, 28, 1292–1295. [Google Scholar] [CrossRef]
- Accogli, A.; St-Onge, J.; Addour-Boudrahem, N.; Lafond-Lapalme, J.; Laporte, A.D.; Rouleau, G.A.; Rivière, J.-B.; Srour, M. Heterozygous Missense Pathogenic Variants Within the Second Spectrin Repeat of SPTBN2 Lead to Infantile-Onset Cerebellar Ataxia. J. Child Neurol. 2019, 35, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Bolthauser, E.; Poretti, A. Clinics in Developmental Medicine. In Cerebellar Disorders in Children, 1st ed.; Mac Keith Press: London, UK, 2012; p. 415. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 1–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinlin, M.; Styger, M.; Boltshauser, E. Cognitive impairments in patients with congenital nonprogressive cerebellar ataxia. Neurology 1999, 53, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.; Aguilera-Albesa, S.; Tello, C.A.; Serrano, M.; Tomás, M.; Camino-León, R.; Fernández-Ramos, J.; Jiménez-Escrig, A.; Poó, P.; O’Callaghan, M.; et al. PLA2G6-associated neurodegeneration: New insights into brain abnormalities and disease progression. Park. Relat. Disord. 2019, 61, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Blaser, S.I.; Steinlin, M.; Al-Maawali, A.; Yoon, G. The Pediatric Cerebellum in Inherited Neurodegenerative Disorders: A Pattern-recognition Approach. Neuroimaging Clin. N. Am. 2016, 26, 373–416. [Google Scholar] [CrossRef]
- Bertini, E.; Zanni, G.; Boltshauser, E. Nonprogressive congenital ataxias. In Handbook of Clinical Neurology, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 155, pp. 91–103. [Google Scholar]
- Zambonin, J.L.; Bellomo, A.; Ben-Pazi, H.; Everman, D.B.; Frazer, L.M.; Geraghty, M.T.; Harper, A.D.; Jones, J.R.; Kamien, B.; Kernohan, K.; et al. Spinocerebellar ataxia type 29 due to mutations in ITPR1: A case series and review of this emerging congenital ataxia. Orphanet J. Rare Dis. 2017, 12, 121. [Google Scholar] [CrossRef]
- Khare, S.; Nick, J.A.; Zhang, Y.; Galeano, K.; Butler, B.; Khoshbouei, H.; Rayaprolu, S.; Hathorn, T.; Ranum, L.P.W.; Smithson, L.; et al. A KCNC3 mutation causes a neurodevelopmental, non-progressive SCA13 subtype associated with dominant negative effects and aberrant EGFR trafficking. PLoS ONE 2017, 12, e0173565. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Van Kempen, A.A.M.W.; Van Der Heide, M.; Poll-The, B.T.; Van Slooten, H.J.; Troost, D.; Rozemuller-Kwakkel, J.M. Congenital disorder of glycosylation type Ia: A clinicopathological report of a newborn infant with cerebellar pathology. Acta Neuropathol. 2005, 109, 433–442. [Google Scholar] [CrossRef]
- Clarkson, Y.L.; Gillespie, T.; Perkins, E.M.; Lyndon, A.R.; Jackson, M. Beta-III spectrin mutation L253P associated with spinocerebellar ataxia type 5 interferes with binding to Arp1 and protein trafficking from the Golgi. Hum. Mol. Genet. 2010, 19, 3634–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, E.M.; Clarkson, Y.L.; Sabatier, N.; Longhurst, D.M.; Millward, C.P.; Jack, J.; Toraiwa, J.; Watanabe, M.; Rothstein, J.D.; Lyndon, A.R.; et al. Loss of beta-III spectrin leads to Purkinje cell dysfunction re-capitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J. Neurosci. 2010, 30, 4857–4867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammarlund, M.; Jorgensen, E.M.; Bastiani, M.J. Axons break in animals lacking beta-spectrin. J. Cell Biol. 2007, 176, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Avery, A.W.; Crain, J.; Thomas, D.D.; Hays, T.S. A human beta-III-spectrin spinocerebellar ataxia type 5 mutation causes high-affinity F-actin binding. Sci. Rep. 2016, 6, 21375. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Bross, P.; Jørgensen, M.M.; Corydon, T.J.; Andresen, B.S. Defective folding and rapid degradation of mutant proteins is a common disease mechanism in genetic disorders. J. Inherit. Metab. Dis. 2000, 23, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Le Guerroué, F.; Youle, R.J. Ubiquitin signaling in neurodegenerative diseases: An autophagy and proteasome perspective. Cell Death Differ. 2021, 28, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Ułamek-Kozioł, M.; Furmaga-Jabłońska, W.; Januszewski, S.; Brzozowska, J.; Ściślewska, M.; Jabłoński, M.; Pluta, R. Neuronal Autophagy: Self-eating or Self-cannibalism in Alzheimer’s Disease. Neurochem. Res. 2013, 38, 1769–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, Spectrin, and Associated Proteins Form a Periodic Cytoskeletal Structure in Axons. Science 2013, 339, 452–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Zhou, R.; Xia, C.; Zhuang, X. Structural organization of the actin-spectrin–based membrane skeleton in dendrites and soma of neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E6678–E6685. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Hubsch, T.; Du Montcel, S.T.; Baliko, L.; Berciano, J.; Boesch, S.; Depondt, C.; Giunti, P.; Globas, C.; Infante, J.; Kang, J.-S.; et al. Scale for the assessment and rating of ataxia: Development of a new clinical scale. Neurology 2006, 66, 1717–1720. [Google Scholar] [CrossRef]
- Lawerman, T.F.; Brandsma, R.; Verbeek, R.J.; van der Hoeven, J.H.; Lunsing, R.J.; Kremer, H.P.H.; Sival, D.A. Construct Validity and Reliability of the SARA Gait and Posture Sub-scale in Early Onset Ataxia. Front. Hum. Neurosci. 2017, 11, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandsma, R.; Lawerman, T.F.; Kuiper, M.J.; Lunsing, R.J.; Sival, D.A.; Burger, H. Reliability and discriminant validity of ataxia rating scales in early onset ataxia. Dev. Med. Child Neurol. 2016, 59, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Trouillas, P.; Takayanagi, T.; Hallett, M.; Currier, R.; Subramony, S.; Wessel, K.; Bryer, A.; Diener, H.; Massaquoi, S.; Gomez, C.; et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. J. Neurol. Sci. 1997, 145, 205–211. [Google Scholar] [CrossRef]
- Storey, E.; Tuck, K.; Hester, R.; Hughes, A.; Churchyard, A. Inter-rater reliability of the International Cooperative Ataxia Rating Scale (ICARS). Mov. Disord. 2004, 19, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Monseny, A.F.; Bolasell, M.; Callejón-Póo, L.; Cuadras, D.; Freniche, V.; Itzep, D.C.; Gassiot, S.; Arango, P.; Casas-Alba, D.; De La Morena, E.; et al. AZATAX: Acetazolamide safety and efficacy in cerebellar syndrome in PMM2 congenital disorder of glycosylation (PMM2-CDG). Ann. Neurol. 2019, 85, 740–751. [Google Scholar] [CrossRef]
- Wolf, N.I.; Boltshauser, E.; Poretti, A. Differential Diagnosis of Cerebellar Atrophy in Childhood: An Update. Neuropediatrics 2015, 46, 359–370. [Google Scholar] [CrossRef] [PubMed]
- De Diego, V.; Martinz-Monseny, A.F.; Muchart, J.; Cuadras, D.; Montero, R.; Artuch, R.; Perez-Cerda, C.; Perez, B.; Perez-Duenas, B.; Poretti, A.; et al. Longitudinal volumetric and 2D assessment of cerebellar atrophy in a large cohort of children with phosphomannomutase deficiency (PMM2-CDG). J. Inherit. Metab. Dis. 2017, 40, 709–713. [Google Scholar] [CrossRef]
- Correa-Vela, M.; Lupo, V.; Montpeyó, M.; Sancho, P.; Marcé-Grau, A.; Hernández-Vara, J.; Darling, A.; Jenkins, A.; Fernández-Rodríguez, S.; Tello, C.; et al. Impaired proteasome activity and neurodegeneration with brain iron accumulation in FBXO7 defect. Ann. Clin. Transl. Neurol. 2020, 7, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Monteagudo, A.; Álvarez-Sauco, M.; Sastre, I.; Martínez-Torres, I.; Lupo, V.; Berenguer, M.; Espinós, C. Genetics of Wilson disease and Wilson-like phenotype in a clinical series from eastern Spain. Clin. Genet 2020, 97, 758–763. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | 1 (MD-219) | 2 (MD-207) |
|---|---|---|
| Gender, age (years) | M, 11 | M, 8 |
| SPTBN2 variant | c.193A > G | c.764A > G |
| Position | chr11:66483417 | chr11:66481110 |
| Mutation type | Missense | Missense |
| Protein change | p.K65E | p.D255G |
| Protein domain | CH1 | CH2 |
| Age at onset | 4 months | 12 months |
| First clinical features | Hypotonia and transient upgaze deviation | Motor delay |
| Head control achieved | 9 months | 3 months |
| Sitting unsupported | 24 months | 9 months |
| Walking features | Ataxic gait, supported | Ataxic gait, unsupported |
| Speech | Impaired, understandable | Impaired, difficult to understand |
| Ocular anomalies | Strabismus, horizontal nystagmus, dysmetria | Strabismus |
| Hypotonia | + | + |
| Corticospinal signs | - | - |
| Tremor | Action (mild) | Action (moderate) |
| Dystonia | - | - |
| Bradykinesia | + (mild) | - |
| Bulbar dysfunction | - | - |
| SARA total score 1 | 19/40 | 17/40 |
| ICARS total score 1 | 38/100 | 26/100 |
| Global IQ (z-score) | 51 (z = −3) | 76 (z = −1.6) |
| Behavioral problems | No | ADHD |
| Clinical course | Non-progressive | Non-progressive |
| Cerebellar atrophy | Progressive | Progressive |
| Cortical hyperintensity on FLAIR images | + | + |
| Additional MRI findings | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sancho, P.; Andrés-Bordería, A.; Gorría-Redondo, N.; Llano, K.; Martínez-Rubio, D.; Yoldi-Petri, M.E.; Blumkin, L.; Rodríguez de la Fuente, P.; Gil-Ortiz, F.; Fernández-Murga, L.; et al. Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 2505. https://doi.org/10.3390/ijms22052505
Sancho P, Andrés-Bordería A, Gorría-Redondo N, Llano K, Martínez-Rubio D, Yoldi-Petri ME, Blumkin L, Rodríguez de la Fuente P, Gil-Ortiz F, Fernández-Murga L, et al. Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration. International Journal of Molecular Sciences. 2021; 22(5):2505. https://doi.org/10.3390/ijms22052505
Chicago/Turabian StyleSancho, Paula, Amparo Andrés-Bordería, Nerea Gorría-Redondo, Katia Llano, Dolores Martínez-Rubio, María Eugenia Yoldi-Petri, Luba Blumkin, Pablo Rodríguez de la Fuente, Fernando Gil-Ortiz, Leonor Fernández-Murga, and et al. 2021. "Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration" International Journal of Molecular Sciences 22, no. 5: 2505. https://doi.org/10.3390/ijms22052505
APA StyleSancho, P., Andrés-Bordería, A., Gorría-Redondo, N., Llano, K., Martínez-Rubio, D., Yoldi-Petri, M. E., Blumkin, L., Rodríguez de la Fuente, P., Gil-Ortiz, F., Fernández-Murga, L., Sánchez-Monteagudo, A., Lupo, V., Pérez-Dueñas, B., Espinós, C., & Aguilera-Albesa, S. (2021). Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration. International Journal of Molecular Sciences, 22(5), 2505. https://doi.org/10.3390/ijms22052505