
Characterization and Comparative Analyses of Mitochondrial Genomes in Single-Celled Eukaryotes to Shed Light on the Diversity and Evolution of Linear Molecular Architecture

 ,
,
Abstract
:1. Introduction
2. Results
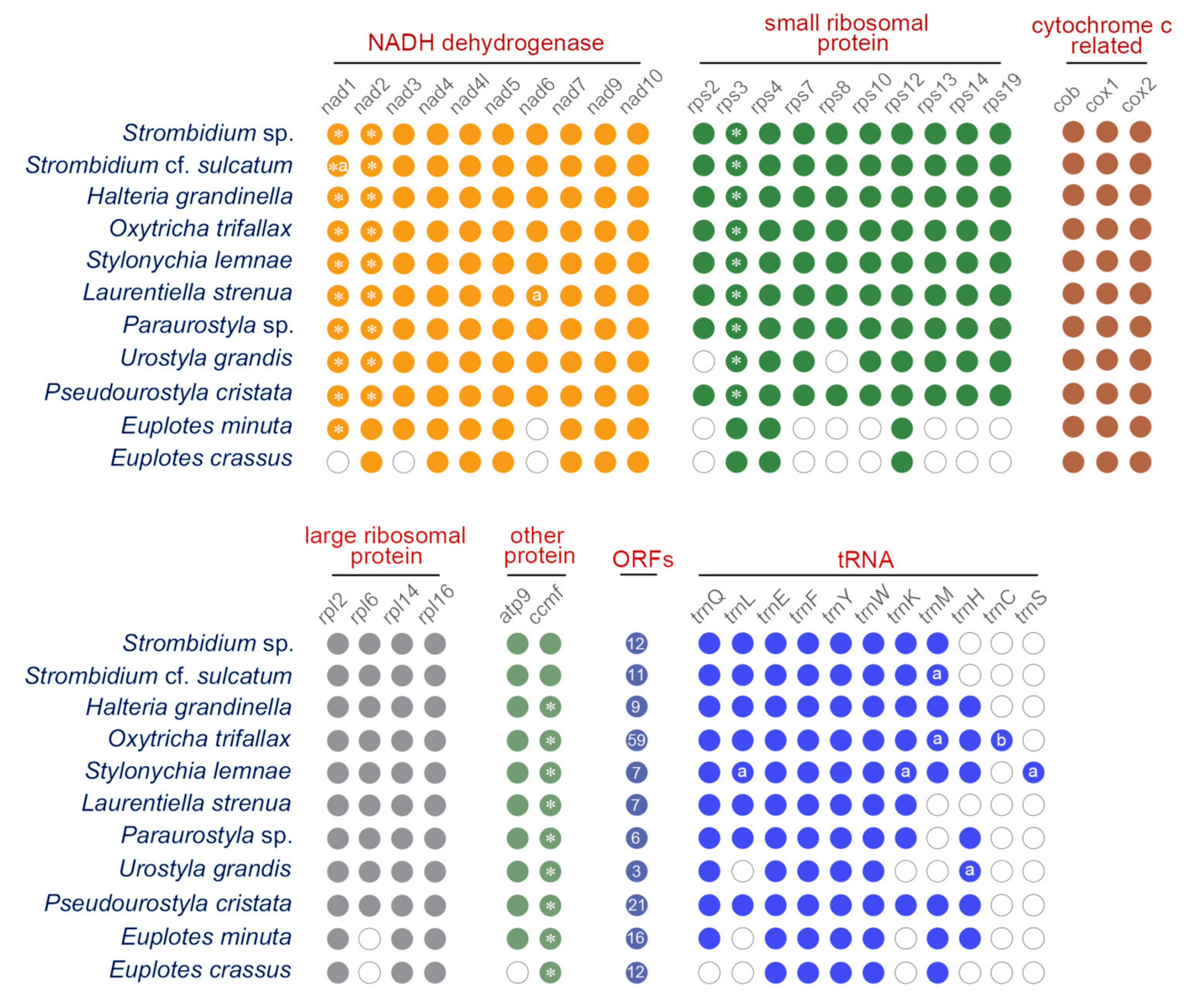
2.1. Mitogenome Overview
2.2. Mitogenome Comparison among Species in the Class Spirotrichea
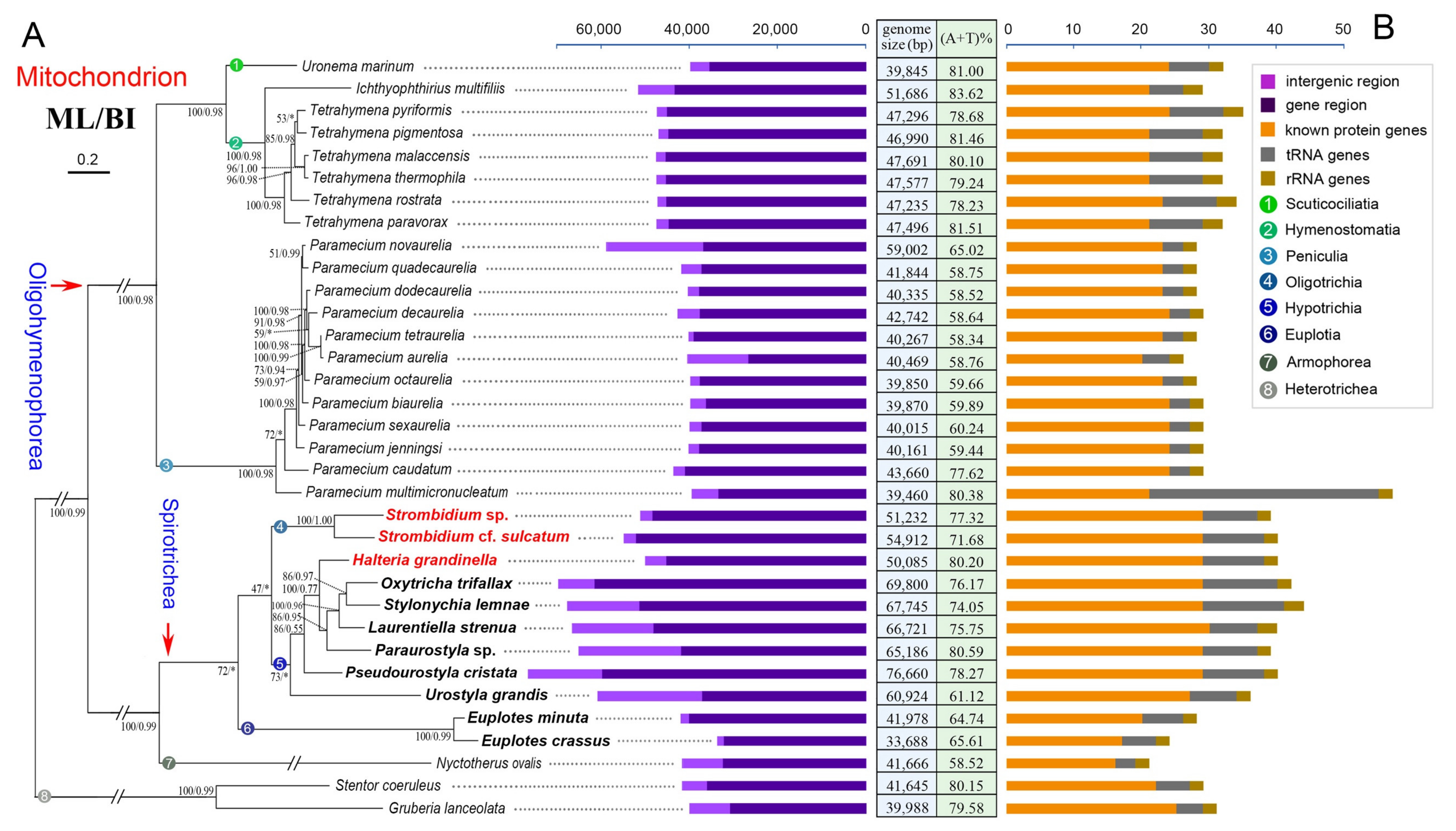
2.3. Phylogenetic Analyses Based on Mitochondrial Proteins
3. Discussion
3.1. Ccmf and Other Split Protein-Coding Genes in Mitogenome
3.2. ORFs with Unknown Functions in Mitogenome
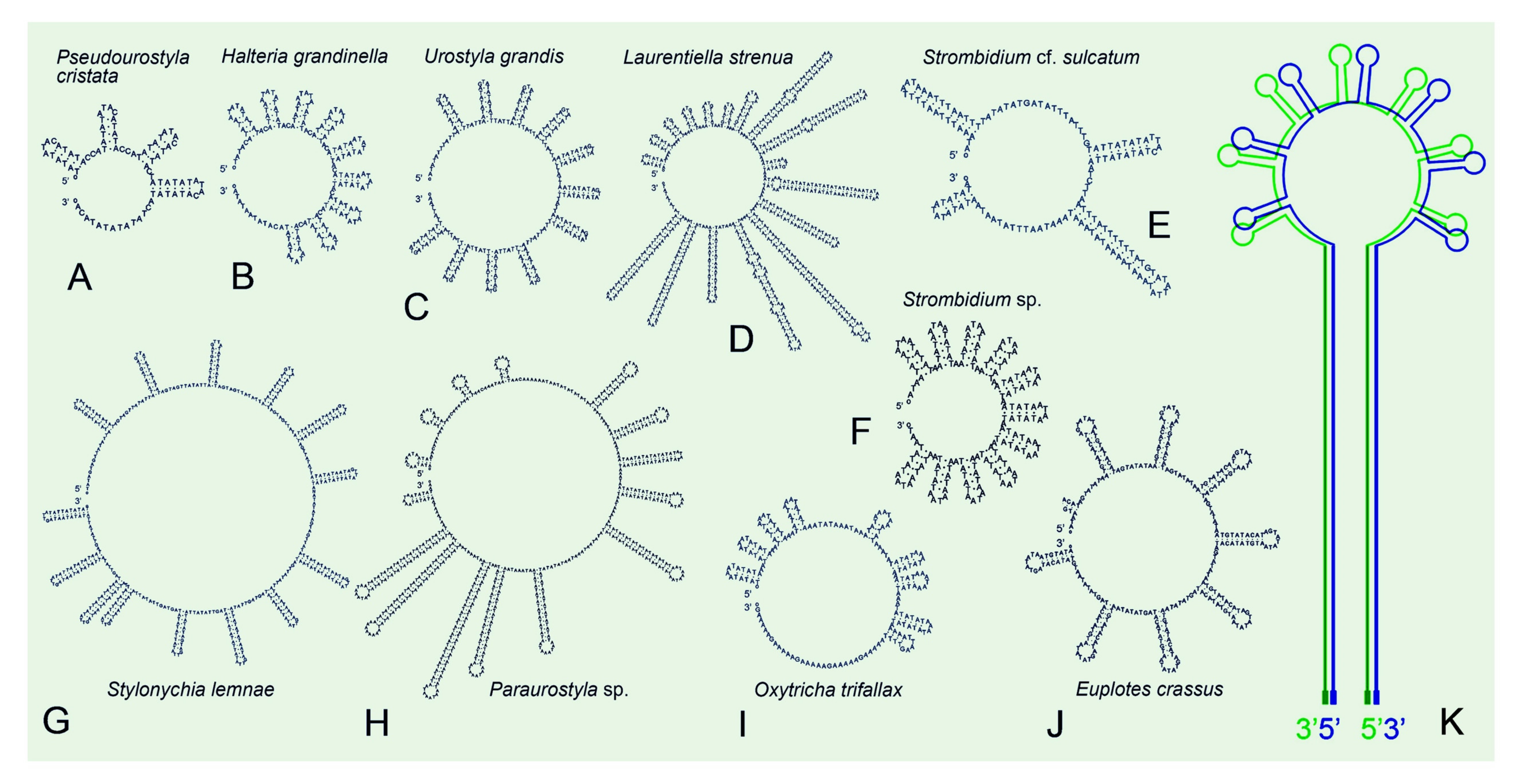
3.3. Repeat Regions in Mitogenomes
3.4. Phylogenetic Position of Halteria grandinella
4. Materials and Methods
4.1. Cell Cultures
4.2. Mitochondrial DNA, Genomic DNA Extraction, and High-Throughput Sequencing
4.3. Mitogenome Assembly and Annotation
4.4. Phylogenetic Analyses and Topology Testing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The origin and diversification of mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampl, V.; Čepička, I.; Eliáš, M. Was the mitochondrion necessary to start eukaryogenesis? Trends Microbiol. 2019, 27, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Rickettsia, typhus and the mitochondrial connection. Nature 1998, 396, 109–110. [Google Scholar] [CrossRef] [PubMed]
- Mottis, A.; Herzig, S.; Auwerx, J. Mitocellular communication: Shaping health and disease. Science 2019, 366, 827–832. [Google Scholar] [CrossRef]
- Hämäläinen, R.H.; Landoni, J.C.; Ahlqvist, K.J.; Goffart, S.; Ryytty, S.; Rahman, M.O.; Brilhante, V.; Icay, K.; Hautaniemi, S.; Wang, L. Defects in mtDNA replication challenge nuclear genome stability through nucleotide depletion and provide a unifying mechanism for mouse progerias. Nat. Metab. 2019, 1, 958–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Rand, D.M. The units of selection on mitochondrial DNA. Annu. Rev. Ecol. Syst. 2003, 32, 415–448. [Google Scholar] [CrossRef]
- Cole, L.W.; Guo, W.; Mower, J.P.; Palmer, J.D. High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evol. 2018, 35, 2773–2785. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, E.S.; Chabbert, C.D.; Klaus, B.; Steinmetz, L.M. A genome-wide map of mitochondrial DNA recombination in yeast. Genetics 2014, 198, 755–771. [Google Scholar] [CrossRef] [Green Version]
- Ladoukakis, E.D.; Zouros, E. Direct evidence for homologous recombination in mussel (Mytilus galloprovincialis) mitochondrial DNA. Mol. Biol. Evol. 2001, 18, 1168–1175. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.W.; Lang, B.F.; Burger, G. Mitochondria of protists. Annu. Rev. Genet. 2004, 38, 477–524. [Google Scholar] [CrossRef]
- Li, X.; Huang, J.; Filker, S.; Stoeck, T.; Bi, Y.; Yu, Y.; Song, W. Spatio-temporal patterns of zooplankton in a main-stem dam affected tributary: A case study in the Xiangxi River of the Three Gorges Reservoir, China. Sci. China Life Sci. 2019, 62, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jiang, Y.; Gao, F.; Zheng, W.; Krock, T.J.; Stover, N.A.; Lu, C.; Katz, L.A.; Song, W. Genome analyses of the new model protist Euplotes vannus focusing on genome rearrangement and resistance to environmental stressors. Mol. Ecol. Resour. 2019, 19, 1292–1308. [Google Scholar] [CrossRef]
- Cheng, T.; Wang, Y.; Huang, J.; Chen, X.; Zhao, X.; Gao, S.; Song, W. Our recent progress in epigenetic research using the model ciliate, Tetrahymena thermophila. Mar. Life Sci. Technol. 2019, 1, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Sheng, Y.; Liu, Y.; Zhang, W.; Cheng, T.; Duan, L.; Pan, B.; Qiao, Y.; Liu, Y.; Gao, S. A distinct class of eukaryotic MT-A70 methyltransferases maintain symmetric DNA N6-adenine methylation at the ApT dinucleotides as an epigenetic mark associated with transcription. Nucleic Acids Res. 2019, 47, 11771–11789. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.R.; Wang, C.; Jiang, Y.; Katz, A.L.; Gao, F.; Yan, Y. Further analyses of variation of ribosome DNA copy number and polymorphism in ciliates provide insights relevant to studies of both molecular ecology and phylogeny. Sci. China Life Sci. 2019, 62, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Maurer-Alcalá, X.X.; Knight, R.; Pond, S.L.K.; Katz, L.A. Single-cell transcriptomics reveal a correlation between genome architecture and gene family evolution in ciliates. mBio 2019, 10, e02524-19. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Xiong, J.; Mao, F.; Sheng, Y.; Chen, X.; Feng, L.; Dui, W.; Yang, W.; Kapusta, A.; Feschotte, C. RNAi-dependent Polycomb repression controls transposable elements in Tetrahymena. Genes Dev. 2019, 33, 348–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, D.H.; Kolisko, M. Molecules illuminate morphology: Phylogenomics confirms convergent evolution among ‘oligotrichous’ ciliates. Int. J. Syst. Evol. Microbiol. 2017, 67, 3676–3682. [Google Scholar] [CrossRef] [PubMed]
- Slabodnick, M.M.; Ruby, J.G.; Reiff, S.B.; Swart, E.C.; Gosai, S.; Prabakaran, S.; Witkowska, E.; Larue, G.E.; Fisher, S.; Freeman, R.M. The macronuclear genome of Stentor coeruleus reveals tiny introns in a giant cell. Curr. Biol. 2017, 27, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Y.; Duan, L.; Cheng, T.; Qiao, Y.; Stover, N.A.; Gao, S. The completed macronuclear genome of a model ciliate Tetrahymena thermophila and its application in genome scrambling and copy number analyses. Sci. China Life Sci. 2020, 63, 1534–1542. [Google Scholar] [CrossRef]
- Wang, Y.R.; Jiang, Y.; Liu, Y.; Li, Y.; Katz, L.A.; Gao, F.; Yan, Y. Comparative studies on the polymorphism and copy number variation of mtSSU rDNA in ciliates (Protista, Ciliophora): Implications for phylogenetic, environmental, and ecological research. Microorganisms 2020, 8, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.; Li, Y.; Ma, H.; Al-Rasheid, K.A.; Warren, A.; Wang, Y.; Yan, Y. Morphology and molecular phylogeny of four trachelocercid ciliates (Protozoa, Ciliophora, Karyorelictea) found in marine coastal habitats of northern China, with description of a new genus, two new species and a new combination. Front. Mar. Sci. 2021, 7, 615903. [Google Scholar] [CrossRef]
- Gray, M.W.; Lang, B.F.; Cedergren, R.; Golding, G.B.; Lemieux, C.; Sankoff, D.; Turmel, M.; Brossard, N.; Delage, E.; Littlejohn, T.G. Genome structure and gene content in protist mitochondrial DNAs. Nucleic Acids Res. 1998, 26, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Suyama, Y.; Miura, K. Size and structural variations of mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1968, 60, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Morin, G.B.; Cech, T.R. The telomeres of the linear mitochondrial DNA of Tetrahymena thermophila consist of 53 bp tandem repeats. Cell 1986, 46, 873–883. [Google Scholar] [CrossRef]
- Pritchard, A.E.; Cummings, D.J. Replication of linear mitochondrial DNA from Paramecium: Sequence and structure of the initiation-end crosslink. Proc. Natl. Acad. Sci. USA 1981, 78, 7341–7345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradian, M.M.; Beglaryan, D.; Skozylas, J.M.; Kerikorian, V. Complete mitochondrial genome sequence of three Tetrahymena species reveals mutation hot spots and accelerated nonsynonymous substitutions in Ymf genes. PLoS ONE 2007, 2, e650. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Zhu, Y.; Littlejohn, T.G.; Greenwood, S.J.; Schnare, M.N.; Lang, B.F.; Gray, M.W. Complete sequence of the mitochondrial genome of Tetrahymena pyriformis and comparison with Paramecium aurelia mitochondrial DNA. J. Mol. Biol. 2000, 297, 365–380. [Google Scholar] [CrossRef] [Green Version]
- Brunk, C.F.; Lee, L.C.; Tran, A.B.; Li, J. Complete sequence of the mitochondrial genome of Tetrahymena thermophila and comparative methods for identifying highly divergent genes. Nucleic Acids Res. 2003, 31, 1673–1682. [Google Scholar] [CrossRef] [Green Version]
- Barth, D.; Berendonk, T.U. The mitochondrial genome sequence of the ciliate Paramecium caudatum reveals a shift in nucleotide composition and codon usage within the genus Paramecium. BMC Genom. 2011, 12, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valverde, J.R.; Marco, R.; Garesse, R. A conserved heptamer motif for ribosomal RNA transcription termination in animal mitochondria. Proc. Natl. Acad. Sci. USA 1994, 91, 5368–5371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johri, P.; Marinov, G.K.; Doak, T.G.; Lynch, M. Population genetics of Paramecium mitochondrial genomes: Recombination, mutation spectrum, and efficacy of selection. Genome Biol. Evol. 2019, 11, 1398–1416. [Google Scholar] [CrossRef] [Green Version]
- Johri, P.; Krenek, S.; Marinov, G.K.; Doak, T.G.; Berendonk, T.U.; Lynch, M. Population genomics of Paramecium species. Mol. Biol. Evol. 2017, 34, 1194–1216. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gao, Y.; Hou, Y.; Ye, S.; Wang, L.; Sun, J.; Li, Q. Mitochondrial genome sequencing and analysis of scuticociliates (Uronema marinum) isolated from Takifugu rubripes. Mitochondrial DNA Part B 2018, 3, 736–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, A.; Haites, R.; Young, N.; Billman-Jacobe, H. The mitochondrial genome of Tetrahymena rostrata. Mitochondrial DNA Part B 2020, 5, 53–54. [Google Scholar] [CrossRef]
- Chen, X. Genome Architecture, Rearrangement and Evolution in the Ciliate Oxytricha Trifallax. Ph.D. Thesis, Princeton University, Princeton, NJ, USA, 2015. [Google Scholar]
- De Graaf, R.M.; Van Alen, T.A.; Dutilh, B.E.; Kuiper, J.W.; Van Zoggel, H.J.; Huynh, M.B.; Görtz, H.-D.; Huynen, M.A.; Hackstein, J.H. The mitochondrial genomes of the ciliates Euplotes minuta and Euplotes crassus. BMC Genom. 2009, 10, 514. [Google Scholar] [CrossRef] [Green Version]
- Swart, E.C.; Nowacki, M.; Shum, J.; Stiles, H.; Higgins, B.P.; Doak, T.G.; Schotanus, K.; Magrini, V.J.; Minx, P.; Mardis, E.R. The Oxytricha trifallax mitochondrial genome. Genome Biol. Evol. 2011, 4, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Park, M.-H.; Min, G.-S. The complete mitochondrial genome of Gruberia lanceolata (Gruber, 1884) Kahl, 1932 (Ciliophora: Heterotrichea). Mitochondrial DNA Part B 2019, 4, 3443–3445. [Google Scholar] [CrossRef]
- De Graaf, R.M.; Ricard, G.; Van Alen, T.A.; Duarte, I.; Dutilh, B.E.; Burgtorf, C.; Kuiper, J.W.; Van Der Staay, G.W.; Tielens, A.G.; Huynen, M.A. The organellar genome and metabolic potential of the hydrogen-producing mitochondrion of Nyctotherus ovalis. Mol. Biol. Evol. 2011, 28, 2379–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotterová, J.; Salomaki, E.; Pánek, T.; Bourland, W.; Žihala, D.; Táborský, P.; Edgcomb, V.P.; Beinart, R.A.; Kolísko, M.; Čepička, I. Genomics of new ciliate lineages provides insight into the evolution of obligate anaerobiosis. Curr. Biol. 2020, 30, 2037–2050. [Google Scholar] [CrossRef]
- Lewis, W.H.; Lind, A.E.; Sendra, K.M.; Onsbring, H.; Williams, T.A.; Esteban, G.F.; Hirt, R.P.; Ettema, T.J.; Embley, T.M. Convergent evolution of hydrogenosomes from mitochondria by gene transfer and loss. Mol. Biol. Evol. 2020, 37, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Coyne, R.S.; Hannick, L.; Shanmugam, D.; Hostetler, J.B.; Brami, D.; Joardar, V.S.; Johnson, J.; Radune, D.; Singh, I.; Badger, J.H. Comparative genomics of the pathogenic ciliate Ichthyophthirius multifiliis, its free-living relatives and a host species provide insights into adoption of a parasitic lifestyle and prospects for disease control. Genome Biol. 2011, 12, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Fan, X.; Gao, F.; Al-Farraj, S.A.; El-Serehy, H.A.; Song, W. Further analyses on the phylogeny of the subclass Scuticociliatia (Protozoa, Ciliophora) based on both nuclear and mitochondrial data. Mol. Phylogenet. Evol. 2019, 139, 106565. [Google Scholar] [CrossRef]
- Agatha, S. A cladistic approach for the classification of oligotrichid ciliates (Ciliophora: Spirotricha). Acta Protozool. 2004, 43, 201–217. [Google Scholar]
- Foissner, W.; Muller, H.; Agatha, S. A comparative fine structural and phylogenetic analysis of resting cysts in oligotrich and hypotrich Spirotrichea (Ciliophora). Eur. J. Protistol. 2007, 43, 295–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Yan, Y.; Chen, X.; Al-Farraj, S.A.; El-Serehy, H.A.; Gao, F. Further analyses on the evolutionary “key-protist” Halteria (Protista, Ciliophora) based on transcriptomic data. Zool. Scr. 2019, 48, 813–825. [Google Scholar] [CrossRef]
- Gao, F.; Warren, A.; Zhang, Q.; Gong, J.; Miao, M.; Sun, P.; Xu, D.; Huang, J.; Yi, Z.; Song, W. The all-data-based evolutionary hypothesis of ciliated protists with a revised classification of the phylum Ciliophora (Eukaryota, Alveolata). Sci. Rep. 2016, 6, 24874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, E.A.; Muller, K.M.; Cannone, J.; Hogan, D.J.; Gutell, R.; Prescott, D.M. Phylogenetic relationships among 28 spirotrichous ciliates documented by rDNA. Mol. Phylogenet. Evol. 2003, 29, 258–267. [Google Scholar] [CrossRef]
- Paiva, T.S.; Borges, B.N.; Harada, M.L.; Silva-Neto, I.D. Comparative phylogenetic study of Stichotrichia (Alveolata: Ciliophora: Spirotrichea) based on 18S-rDNA sequences. Genet. Mol. Res. 2009, 8, 223–246. [Google Scholar] [CrossRef]
- Zheng, W.; Wang, C.; Lynch, M.; Gao, S. The compact macronuclear genome of the ciliate Halteria grandinella: A transcriptome-like genome with 23,000 nanochromosomes. mBio 2021, 12, e01964-20. [Google Scholar] [CrossRef]
- Nishimura, Y.; Tanifuji, G.; Kamikawa, R.; Yabuki, A.; Hashimoto, T.; Inagaki, Y. Mitochondrial genome of Palpitomonas bilix: Derived genome structure and ancestral system for cytochrome c maturation. Genome Biol. Evol. 2016, 8, 3090–3098. [Google Scholar] [CrossRef] [Green Version]
- Park, K.M.; Min, G.S.; Kim, S. The mitochondrial genome of the ciliate Pseudourostyla cristata (Ciliophora, Urostylida). Mitochondrial DNA Part B 2019, 4, 66–67. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.W.; Jackson, A.P.; Rigden, D.J.; Willis, A.C.; Ferguson, S.J.; Ginger, M.L. Order within a mosaic distribution of mitochondrial c-type cytochrome biogenesis systems? FEBS J. 2008, 275, 2385–2402. [Google Scholar] [CrossRef] [PubMed]
- Nosek, J.; Tomáška, Ľ. Mitochondrial genome diversity: Evolution of the molecular architecture and replication strategy. Curr. Genet. 2003, 44, 73–84. [Google Scholar] [CrossRef]
- Hauth, A.M.; Maier, U.G.; Lang, B.F.; Burger, G. The Rhodomonas salina mitochondrial genome: Bacteria-like operons, compact gene arrangement and complex repeat region. Nucleic Acids Res. 2005, 33, 4433–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Lane, C.E.; Curtis, B.A.; Kozera, C.; Bowman, S.; Archibald, J.M. Complete sequence and analysis of the mitochondrial genome of Hemiselmis andersenii CCMP644 (Cryptophyceae). BMC Genom. 2008, 9, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, O.; Bonnard, G.; Grienenberger, J.M.; Kloareg, B.; Boyen, C. Transcription initiation and RNA processing in the mitochondria of the red alga Chondrus crispus: Convergence in the evolution of transcription mechanisms in mitochondria. J. Mol. Biol. 1998, 283, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Saint-Louis, D.; Gray, M.W.; Lang, B.F. Complete sequence of the mitochondrial DNA of the red alga Porphyra purpurea: Cyanobacterial introns and shared ancestry of red and green algae. Plant Cell 1999, 11, 1675–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- L’abbé, D.; Duhaime, J.F.; Lang, B.F.; Morais, R. The transcription of DNA in chicken mitochondria initiates from one major bidirectional promoter. J. Biol. Chem. 1991, 266, 10844–10850. [Google Scholar] [CrossRef]
- Casane, D.; Dennebouy, N.; De Rochambeau, H.; Mounolou, J.; Monnerot, M. Nonneutral evolution of tandem repeats in the mitochondrial DNA control region of lagomorphs. Mol. Biol. Evol. 1997, 14, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, A.; Laping, J.; Seilhamer, J.; Cummings, D. Inter-species sequence diversity in the replication initiation region of Paramecium mitochondrial DNA. J. Mol. Biol. 1983, 164, 1–15. [Google Scholar] [CrossRef]
- Lynn, D.H. The Ciliated Protozoa: Characterization, Classification and Guide to the Literature, 3rd ed.; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Wilbert, N. Eine verbesserte Technik der Protargolimprä gnation für Ciliaten. Mikrokosmos 1975, 64, 171–179. [Google Scholar]
- Gordon, A.; Hannon, G. Fastx-toolkit. FASTQ/A short-reads preprocessing tools. 2010, unpublished. [Google Scholar]
- Chen, X.; Wang, C.; Pan, B.; Lu, B.; Li, C.; Shen, Z.; Warren, A.; Li, L. Single-cell genomic sequencing of three peritrichs (Protista, Ciliophora) reveals less biased stop codon usage and more prevalent programmed ribosomal frameshifting than in other ciliates. Front. Mar. Sci. 2020, 7, 602323. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant review with the integrative genomics viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valach, M.; Burger, G.; Gray, M.W.; Lang, B.F. Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res. 2014, 42, 13764–13777. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- De Rijk, P.; De Wachter, R. RnaViz, a program for the visualisation of RNA secondary structure. Nucleic Acids Res. 1997, 25, 4679–4684. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [Green Version]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Accession No. | Genome Size (bp) | Gene Region (bp) | Intergenic Region (bp) | Overall A+T Content (%) | Known Protein Genes | tRNA Genes | rRNA Genes |
|---|---|---|---|---|---|---|---|---|
| Strombidium sp. | MT471315 | 51,232 | 48,614 | 2618 | 77.32 | 29 | 8 | 2 |
| Strombidium cf. sulcatum | MT471316 | 54,912 | 52,218 | 2694 | 71.68 | 29 | 9 | 2 |
| Halteria grandinella | MT471317 | 50,085 | 45,401 | 4684 | 80.20 | 29 | 9 | 2 |
| Oxytricha trifallax | JN383843 | 69,800 | 61,685 | 8115 | 76.17 | 29 | 11 | 2 |
| Stylonychia lemnae | KX524144 | 67,745 | 51,501 | 16,244 | 74.05 | 29 | 12 | 3 |
| Laurentiella strenua | KX529838 | 66,721 | 48,266 | 18,455 | 75.75 | 30 | 7 | 3 |
| Paraurostyla sp. | KX524143 | 65,186 | 42,149 | 23,037 | 80.59 | 29 | 8 | 2 |
| Urostyla grandis | KX494929 | 60,924 | 37,327 | 23,597 | 61.12 | 27 | 7 | 2 |
| Pseudourostyla cristata | MH888186 | 76,660 | 59,952 | 16,708 | 78.27 | 29 | 9 | 2 |
| Euplotes minuta | GQ903130 | 41,978 | 40,257 | 1721 | 64.74 | 20 | 6 | 2 |
| Euplotes crassus | GQ903131 | 33,688 | 32,273 | 1415 | 65.61 | 17 | 5 | 2 |
| Tetrahymena pyriformis | AF160864 | 47,296 | 45,285 | 2011 | 78.68 | 24 | 8 | 3 |
| Tetrahymena thermophila | AF396436 | 47,577 | 45,619 | 1958 | 79.24 | 21 | 8 | 3 |
| Tetrahymena malaccensis | DQ927303 | 47,691 | 45,528 | 2163 | 80.10 | 21 | 8 | 3 |
| Tetrahymena paravorax | DQ927304 | 47,496 | 44,812 | 2684 | 81.51 | 21 | 8 | 3 |
| Tetrahymena pigmentosa | DQ927305 | 46,990 | 44,889 | 2101 | 81.46 | 21 | 8 | 3 |
| Tetrahymena rostrata | MN025427 | 47,235 | 45,336 | 1899 | 78.23 | 23 | 8 | 3 |
| Ichthyophthirius multifiliis | JN227086 | 51,686 | 43,469 | 8217 | 83.62 | 21 | 5 | 3 |
| Uronema marinum | MG272262 | 39,845 | 35,544 | 4301 | 81.00 | 24 | 6 | 2 |
| Paramecium caudatum | FN424190 | 43,660 | 41,091 | 2569 | 77.62 | 24 | 3 | 2 |
| Paramecium aurelia | NC001324 | 40,469 | 26,808 | 13,661 | 58.76 | 20 | 4 | 2 |
| Paramecium tetraurelia | - | 40,267 | 39,204 | 1063 | 58.34 | 23 | 3 | 2 |
| Paramecium sexaurelia | - | 40,015 | 37,474 | 2541 | 60.24 | 24 | 3 | 2 |
| Paramecium multimicronucleatum | - | 39,460 | 33,539 | 5921 | 80.38 | 21 | 34 | 2 |
| Paramecium biaurelia | - | 39,870 | 36,297 | 3573 | 59.89 | 24 | 3 | 2 |
| Paramecium octaurelia | - | 39,850 | 37,784 | 2066 | 59.66 | 23 | 3 | 2 |
| Paramecium novaurelia | - | 59,002 | 36,996 | 22,006 | 65.02 | 23 | 3 | 2 |
| Paramecium decaurelia | - | 42,742 | 37,779 | 4963 | 58.64 | 24 | 3 | 2 |
| Paramecium dodecaurelia | - | 40,335 | 37,947 | 2388 | 58.52 | 23 | 3 | 2 |
| Paramecium quadecaurelia | - | 41,844 | 37,450 | 4394 | 58.75 | 23 | 3 | 2 |
| Paramecium jenningsi | - | 40,161 | 38,010 | 2151 | 59.44 | 24 | 3 | 2 |
| Nyctotherus ovalis | GU057832 | 41,666 | 32,511 | 9155 | 58.52 | 16 | 3 | 2 |
| Stentor coeruleus | MPUH01000652 | 41,645 | 36,154 | 5491 | 80.15 | 22 | 5 | 2 |
| Gruberia lanceolata | MK301177 | 39,988 | 30,910 | 9078 | 79.58 | 25 | 4 | 2 |
| Species | Central Repeat | Telomeric Repeat (5′–3′) | Terminal Inverted Repeat | ||
|---|---|---|---|---|---|
| Length (bp) | A+T Content | Repeat Unit (Number of Repeats) | |||
| Strombidium sp. | 170 | 100.00% | ATAATATAATAATAT (11) | CTCCCTTATCTAGTCTTT (both ends) | * |
| Strombidium cf. sulcatum | 142 | 96.48% | ATAAATTTAATTTTA (2) + irregular sequence for the rest | TTATATCCTTTCTCCCCTATATCTCTATAGTACT (both ends) | * |
| Halteria grandinella | 168 | 94.05% | TATACATATAATATATA (9) | AAAACAGCTCCGTTCCAATACTACTAACTAA (both ends) | * |
| Oxytricha trifallax | ~285 | 96.76% | TATATAAA (11) + TATAAATAAA (3) + AAAAAG (5) | CGACTCCTCTATCCTCATCCTAGACTCCGCTTACT (both ends) | ~1800 bp |
| Stylonychia lemnae | ~607 | 92.29% | TATARTAGTTATATTATA (27) | TTCATACCTTTACTAGATACCCGCCTCCGGCTCTCC (3′ end) | ~3100 bp |
| Laurentiella strenua | 733 | 98.91% | ATATAAATGTATATAA (7) + ATAAA(TA)nT (49) + TTT(AT)n (4), n = 0–8 | CCTACTACGCTTCATACGCTAAA (partial) (both ends) | ~2400 bp |
| Paraurostyla sp. | ~802 | 98.86% | ATATAACAAATA (7) + AAATAA(TA)nAT (20), n = 2–29 | * | * |
| Urostyla grandis | ~279 | 95.91% | ATATATTTATTAATATATAGTAT (10) | GTAGCACATGTAG (3′ end) | * |
| Pseudourostyla cristata | 80 | 86.25% | TATATATACATATAC (3) + (TA)nC (3), n = 3 or 5 | * | * |
| Euplotes minuta | ~1596 | 83.36% | ATAGTATATAATGTATAC (63) + ATAGTATATAATGTTAC (1) + ATAGTATATAATTGTTAC (18) | * | * |
| Euplotes crassus | ~416 | 83.54% | ATAGTATATAATGTATAC (15) | * | * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Li, C.; Zhang, X.; Wang, C.; Roger, A.J.; Gao, F. Characterization and Comparative Analyses of Mitochondrial Genomes in Single-Celled Eukaryotes to Shed Light on the Diversity and Evolution of Linear Molecular Architecture. Int. J. Mol. Sci. 2021, 22, 2546. https://doi.org/10.3390/ijms22052546
Zhang T, Li C, Zhang X, Wang C, Roger AJ, Gao F. Characterization and Comparative Analyses of Mitochondrial Genomes in Single-Celled Eukaryotes to Shed Light on the Diversity and Evolution of Linear Molecular Architecture. International Journal of Molecular Sciences. 2021; 22(5):2546. https://doi.org/10.3390/ijms22052546
Chicago/Turabian StyleZhang, Tengteng, Chao Li, Xue Zhang, Chundi Wang, Andrew J. Roger, and Feng Gao. 2021. "Characterization and Comparative Analyses of Mitochondrial Genomes in Single-Celled Eukaryotes to Shed Light on the Diversity and Evolution of Linear Molecular Architecture" International Journal of Molecular Sciences 22, no. 5: 2546. https://doi.org/10.3390/ijms22052546
APA StyleZhang, T., Li, C., Zhang, X., Wang, C., Roger, A. J., & Gao, F. (2021). Characterization and Comparative Analyses of Mitochondrial Genomes in Single-Celled Eukaryotes to Shed Light on the Diversity and Evolution of Linear Molecular Architecture. International Journal of Molecular Sciences, 22(5), 2546. https://doi.org/10.3390/ijms22052546