
A Genomic Blueprint of Flax Fungal Parasite Fusarium oxysporum f. sp. lini

 ,
,
Abstract
:1. Introduction
2. Results
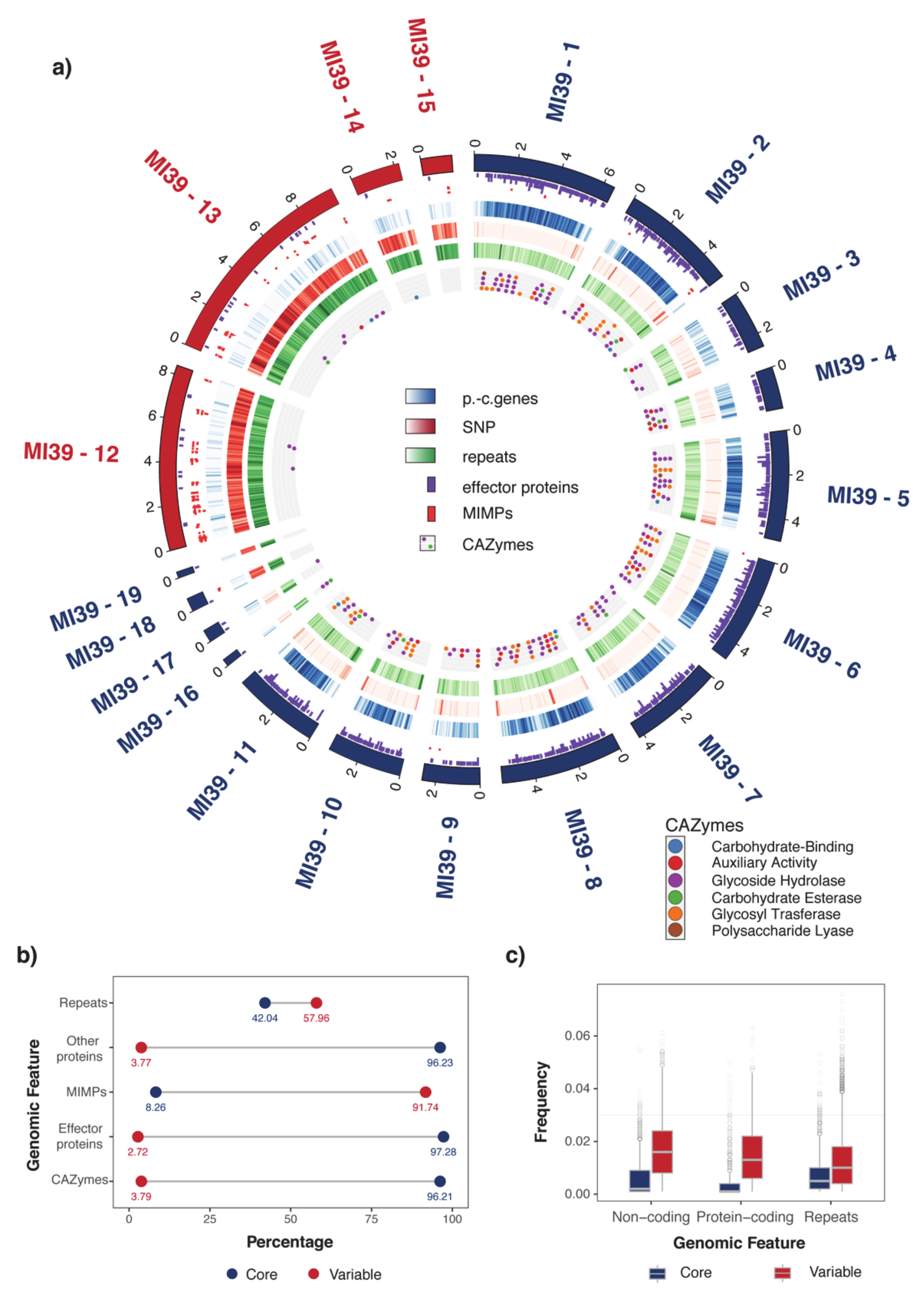
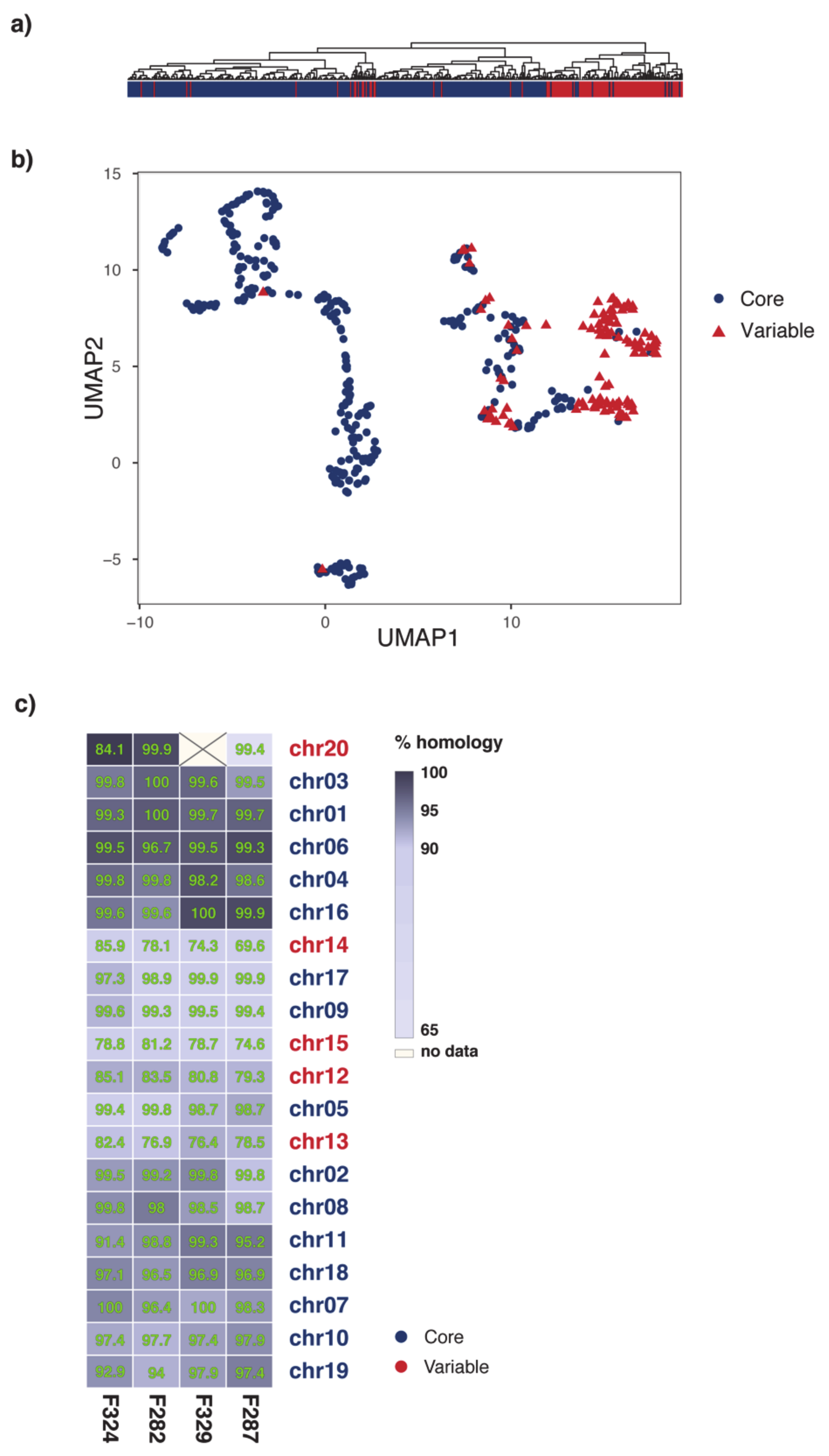
2.1. Organization of MI39 Reference Genome
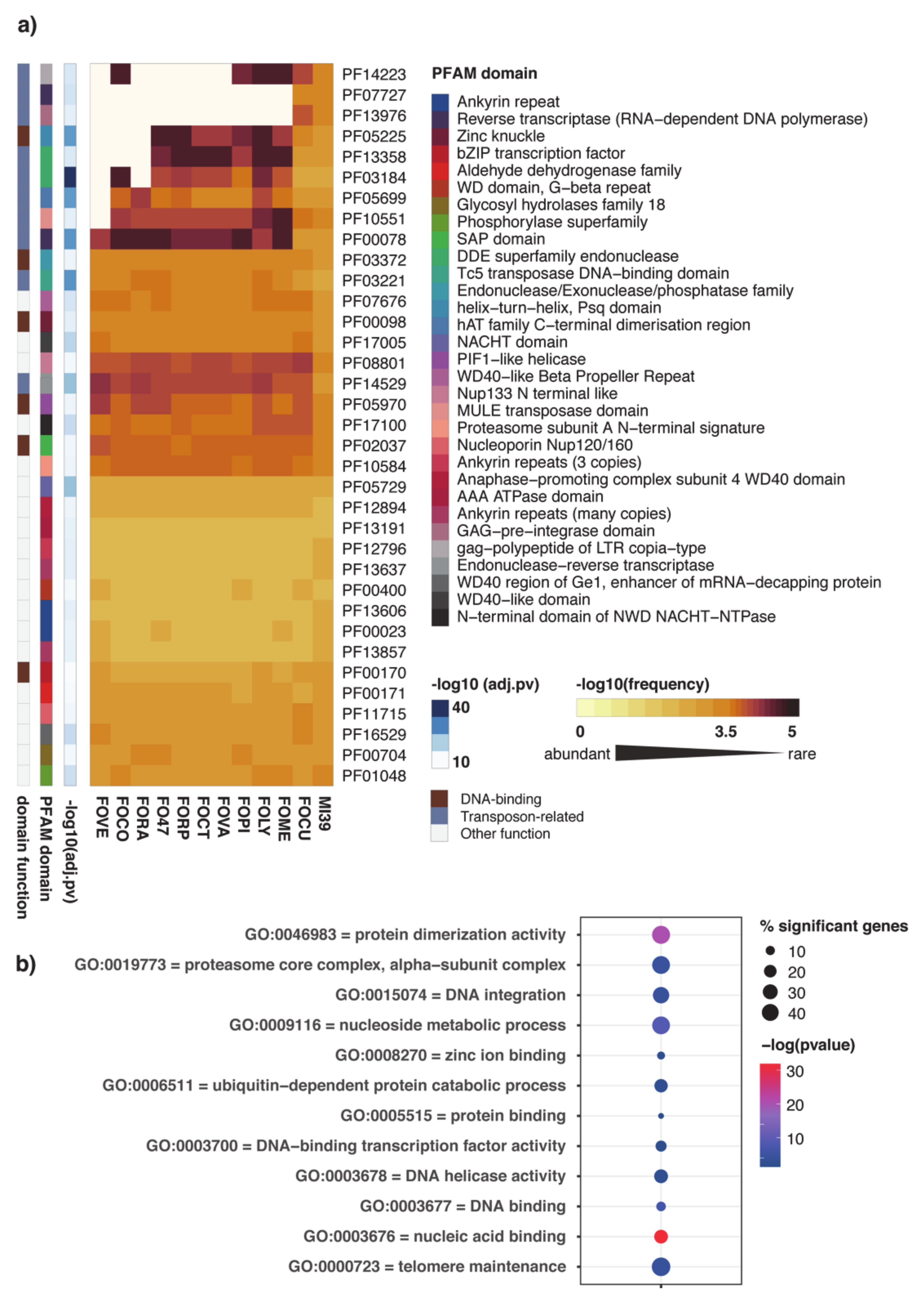
2.2. Functional Annotation of FOLINI Proteome
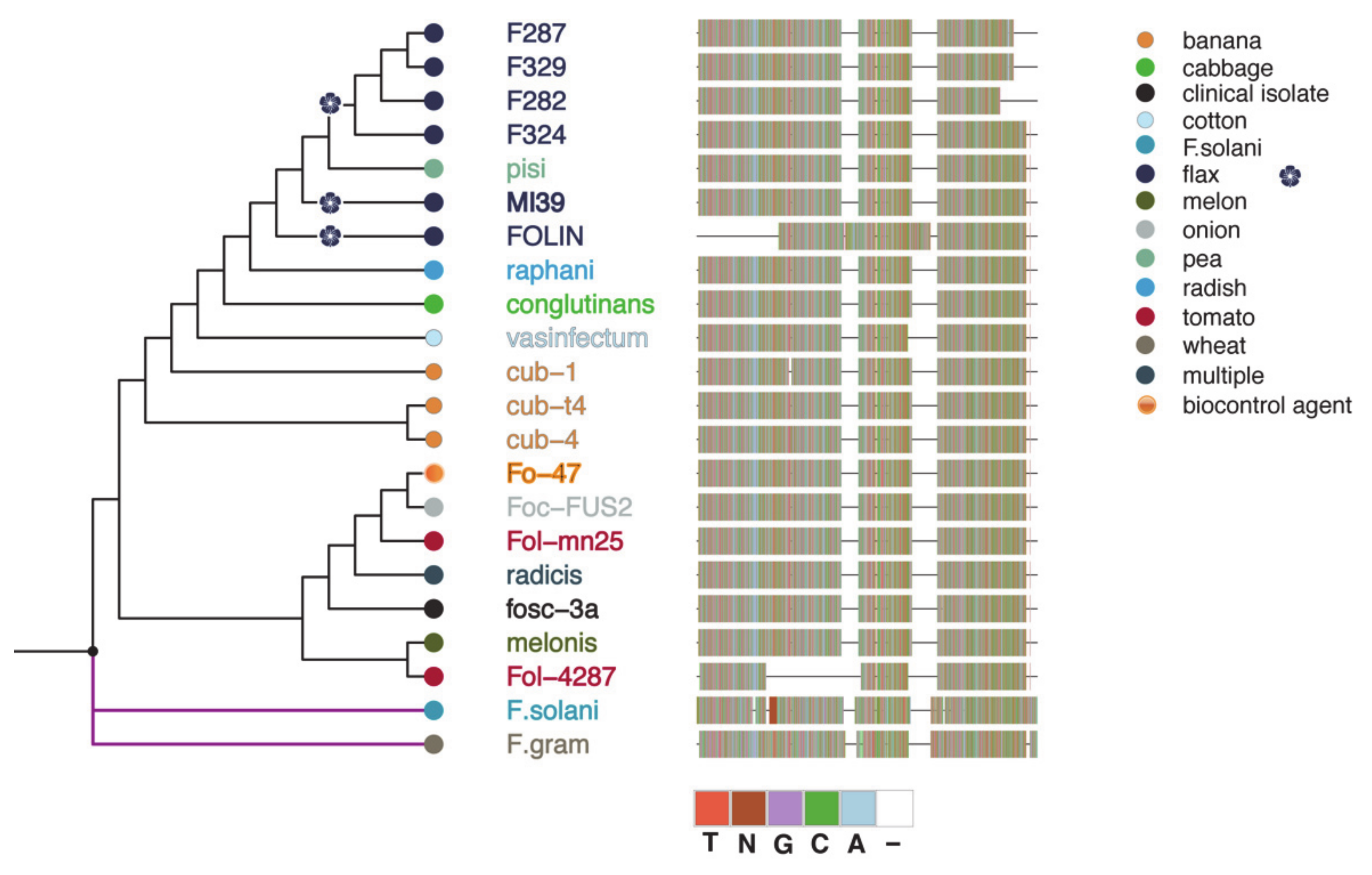
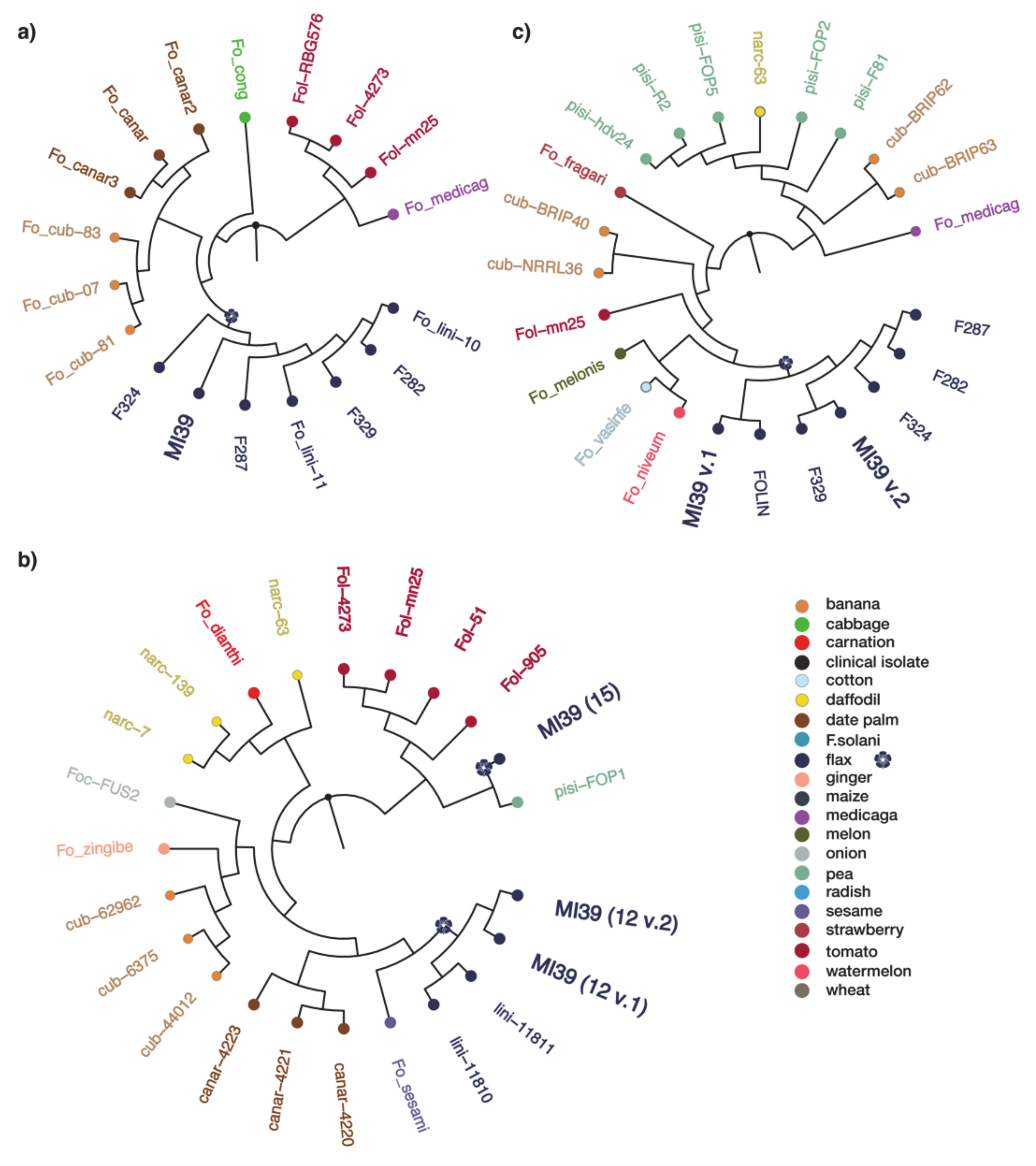
2.3. Phylogeny
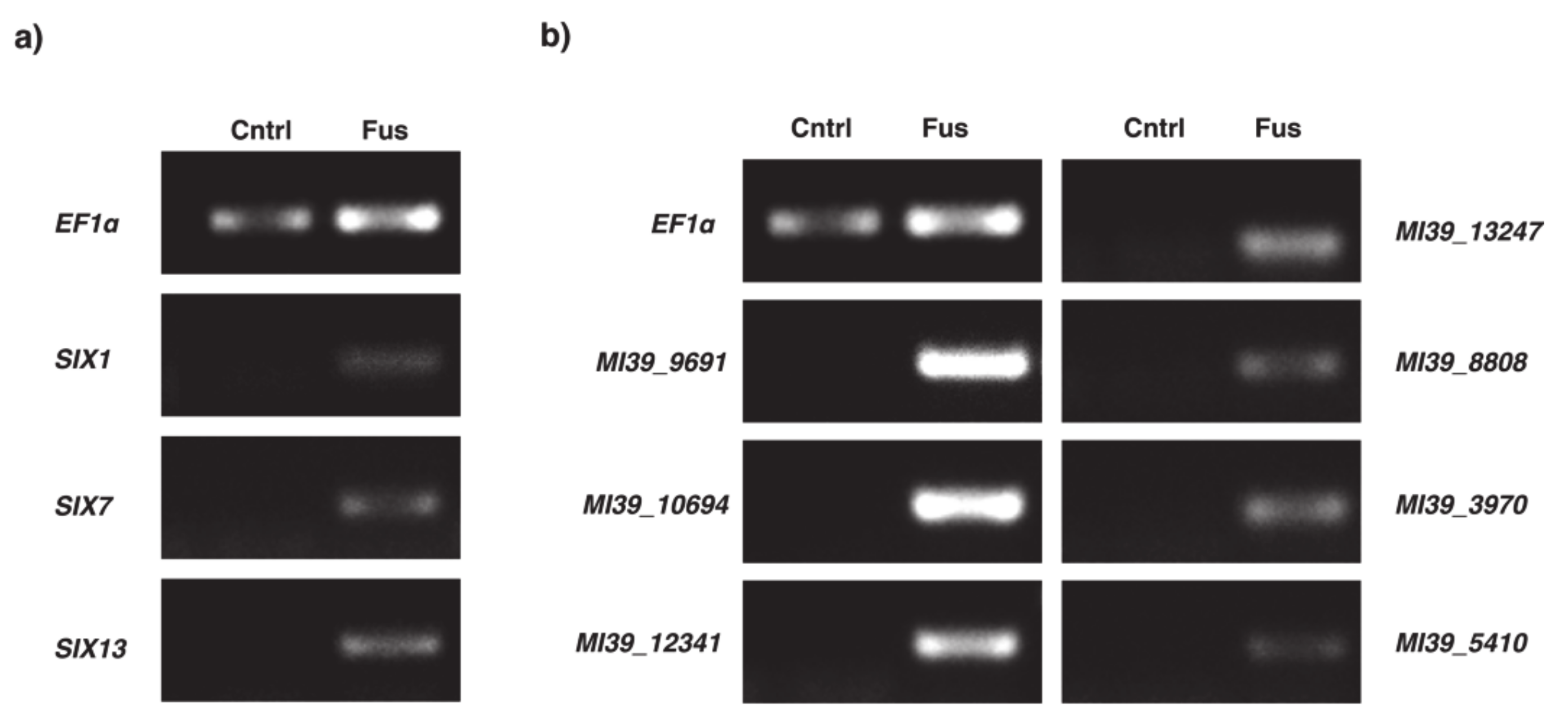
2.4. Expression Analysis of Selected Genes within Variable Compartment
3. Discussion
4. Materials and Methods
4.1. Data Sources and Sequence Analysis
4.2. Proteins Annotation
4.3. Phylogenetic Analysis
4.4. RNA Isolation, Reverse Transcript, and PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Ma, L.-J. Deciphering Pathogenicity of Fusarium oxysporum From a Phylogenomics Perspective. Adv. Genet. 2017, 100, 179–209. [Google Scholar]
- Ma, L.-J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.-J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef]
- Williams, A.H.; Sharma, M.; Thatcher, L.F.; Azam, S.; Hane, J.K.; Sperschneider, J.; Kidd, B.N.; Anderson, J.P.; Ghosh, R.; Garg, G.; et al. Comparative genomics and prediction of conditionally dispensable sequences in legume–infecting Fusarium oxysporum formae speciales facilitates identification of candidate effectors. BMC Genom. 2016, 17, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Armitage, A.D.; Taylor, A.; Sobczyk, M.K.; Baxter, L.; Greenfield, B.P.J.; Bates, H.J.; Wilson, F.; Jackson, A.C.; Ott, S.; Harrison, R.J.; et al. Characterisation of pathogen-specific regions and novel effector candidates in Fusarium oxysporum f. sp. cepae. Sci. Rep. 2018, 8, 13530. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Raffaele, S.; Kamoun, S. The two-speed genomes of filamentous pathogens: Waltz with plants. Curr. Opin. Genet. Dev. 2015, 35, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Frantzeskakis, L.; Kusch, S.; Panstruga, R. The need for speed: Compartmentalized genome evolution in filamentous phytopathogens. Mol. Plant Pathol. 2018, 20, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Torres, D.E.; Oggenfuss, U.; Croll, D.; Seidl, M.F. Genome evolution in fungal plant pathogens: Looking beyond the two-speed genome model. Fungal Biol. Rev. 2020, 34, 136–143. [Google Scholar] [CrossRef]
- Vlaardingerbroek, I.; Beerens, B.; Rose, L.; Fokkens, L.; Cornelissen, B.J.C.; Rep, M. Exchange of core chromosomes and horizontal transfer of lineage-specific chromosomes in Fusarium oxysporum. Environ. Microbiol. 2016, 18, 3702–3713. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fokkens, L.; Conneely, L.J.; Rep, M. Partial pathogenicity chromosomes in Fusarium oxysporumare sufficient to cause disease and can be horizontally transferred. Environ. Microbiol. 2020, 12, 2033. [Google Scholar]
- Li, J.; Fokkens, L.; van Dam, P.; Rep, M. Related mobile pathogenicity chromosomes in Fusarium oxysporumdetermine host range on cucurbits. Mol. Plant Pathol. 2020, 21, 761–776. [Google Scholar] [CrossRef] [Green Version]
- Kistler, H.C. Genetic Diversity in the Plant-Pathogenic Fungus. Phytopathology 1997, 87, 474–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, M.L.; Jimenez, D.R.C.; Leandro, L.F.; Munkvold, G.P. Genotypic and Phenotypic Characterization of Fungi in the Fusarium oxysporum Species Complex from Soybean Roots. Phytopathology 2014, 104, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Nirmaladevi, D.; Venkataramana, M.; Srivastava, R.K.; Uppalapati, S.R.; Gupta, V.K.; Yli-Mattila, T.; Clement Tsui, K.M.; Srinivas, C.; Niranjana, S.R.; Chandra, N.S. Molecular phylogeny, pathogenicity and toxigenicity of Fusarium oxysporum f. sp. lycopersici. Sci. Rep. 2016, 6, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokkens, L.; Shahi, S.; Connolly, L.R.; Stam, R.; Schmidt, S.M.; Smith, K.M.; Freitag, M.; Rep, M. The multi-speed genome of Fusarium oxysporumreveals association of histone modifications with sequence divergence and footprints of past horizontal chromosome transfer events. BioRxiv 2020, 701, 315–362. [Google Scholar]
- Husaini, A.M.; Sakina, A.; Cambay, S.R. Host–Pathogen Interaction in Fusarium oxysporum Infections: Where Do We Stand? Mol. Plant Microbe Interact. 2018, 31, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Houterman, P.M.; Cornelissen, B.J.C.; Rep, M. Suppression of Plant Resistance Gene-Based Immunity by a Fungal Effector. PLoS Pathog. 2008, 4, e1000061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.M.; Lukasiewicz, J.; Farrer, R.; van Dam, P.; Bertoldo, C.; Rep, M. Comparative genomics of Fusarium oxysporum f. sp. melonisreveals the secreted protein recognized by the Fom-2resistance gene in melon. New Phytol. 2015, 209, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Houterman, P.M.; Ma, L.; van Ooijen, G.; de Vroomen, M.J.; Cornelissen, B.J.C.; Takken, F.L.W.; Rep, M. The effector protein Avr2 of the xylem-colonizing fungus Fusarium oxysporum activates the tomato resistance protein I-2 intracellularly. Plant J. 2009, 58, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Rep, M.; van der Does, H.C.; Meijer, M.; van Wijk, R.; Houterman, P.M.; Dekker, H.L.; de Koster, C.G.; Cornelissen, B.J.C. A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 2004, 53, 1373–1383. [Google Scholar] [CrossRef]
- Kanapin, A.; Samsonova, A.; Rozhmina, T.; Bankin, M.; Logachev, A.; Samsonova, M. The Genome Sequence of Five Highly Pathogenic Isolates of Fusarium oxysporum f. sp. lini. Mol. Plant Microbe Interact. 2020, 33, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Baayen, R.P.; O’Donnell, K.; Bonants, P.J.; Cigelnik, E.; Kroon, L.P.; Roebroeck, E.J.; Waalwijk, C. Gene Genealogies and AFLP Analyses in the Fusarium oxysporum Complex Identify Monophyletic and Nonmonophyletic Formae Speciales Causing Wilt and Rot Disease. Phytopathology 2000, 90, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yu, H.; Ma, L.-J. Accessory Chromosomes in Fusarium oxysporum. Phytopathology 2020, 110, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, B.; Yang, J.; Bi, F.; Dong, T.; Yang, Q.; Hu, C.; Xiang, D.; Chen, H.; Huang, H.; et al. A Cerato-Platanin Family Protein FocCP1 Is Essential for the Penetration and Virulence of Fusarium oxysporum f. sp. cubense Tropical Race 4. Int. J. Mol. Sci. 2019, 20, 3785. [Google Scholar] [CrossRef] [Green Version]
- Faino, L.; Seidl, M.F.; Shi-Kunne, X.; Pauper, M.; van den Berg, G.C.M.; Wittenberg, A.H.J.; Thomma, B.P.H.J. Transposons passively and actively contribute to evolution of the two-speed genome of a fungal pathogen. Genome Res. 2016, 26, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. 1), i351–i358. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Tello, D.; Gil, J.; Loaiza, C.D.; Riascos, J.J.; Cardozo, N.; Duitama, J. NGSEP3: Accurate variant calling across species and sequencing protocols. Bioinformatics 2019, 35, 4716–4723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Russell, P.; Meyer, M.; Mauceli, E.; Alföldi, J.; Di Palma, F.; Lindblad-Toh, K. Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics 2010, 26, 1145–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Sperschneider, J.; Dodds, P.N.; Gardiner, D.M.; Singh, K.B.; Taylor, J.M. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol. Plant Pathol. 2018, 19, 2094–2110. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inf. 2009, 23, 205–211. [Google Scholar]
- Fischer, S.; Brunk, B.P.; Chen, F.; Gao, X.; Harb, O.S.; Iodice, J.B.; Shanmugam, D.; Roos, D.S.; Stoeckert, C.J. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinform. 2011, 35, 6–12. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Haeseler von, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Elements | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Length (Mb) | Proteins | MIMPs | Secondary Metabolites Clusters | CAZYmes | Effector Genes | Repeats | SNPs | TFs |
| MI39-1 | 6.5 | 372.93 | 0.615 | 0.307 | 6.76 | 36.00 | 263.39 | 186.00 | 3.076 |
| MI39-2 | 5.28 | 343.92 | 1.704 | 0.946 | 5.87 | 29.54 | 254.34 | 505.65 | 2.651 |
| MI39-3 | 2.64 | 93.33 | 0 | 0.379 | 2.65 | 9.86 | 257.61 | 335.01 | 0 |
| MI39-4 | 1.9 | 149.64 | 0 | 0 | 8.95 | 18.96 | 186.53 | 245.01 | 0 |
| MI39-5 | 4.99 | 315.10 | 0 | 0.400 | 7.80 | 34.61 | 279.09 | 328.50 | 0.601 |
| MI39-6 | 4.82 | 389.17 | 0.414 | 0.622 | 8.50 | 40.22 | 244.24 | 101.59 | 2.489 |
| MI39-7 | 4.23 | 301.70 | 0 | 0.472 | 4.24 | 27.38 | 269.36 | 147.78 | 2.364 |
| MI39-8 | 5.51 | 316.39 | 0 | 0.181 | 8.89 | 31.23 | 221.22 | 475.86 | 1.633 |
| MI39-9 | 2.62 | 101.66 | 0.764 | 0.382 | 3.82 | 13.37 | 218.22 | 491.87 | 0 |
| MI39-10 | 3.36 | 273.75 | 0 | 0 | 5.93 | 25.23 | 295.12 | 404.39 | 1.785 |
| MI39-11 | 3.9 | 312.49 | 0 | 0 | 6.40 | 28.94 | 323.51 | 404.71 | 1.282 |
| MI39-12 | 8.4 | 19.86 | 17.84 | 0 | 0.35 | 1.784 | 830.75 | 9296.25 | 0.238 |
| MI39-13 | 9.7 | 23.18 | 4.84 | 0 | 0.82 | 1.957 | 828.96 | 9924.00 | 0.103 |
| MI39-14 | 2.21 | 35.66 | 0.902 | 0 | 0.45 | 0.902 | 803.50 | 6367.08 | 0 |
| MI39-15 | 1.41 | 19.81 | 8.49 | 0 | 0 | 0.707 | 834.91 | 5314.45 | 0.709 |
| MI39-16 | 0.33 | 45.33 | 0 | 0 | 48.35 | 9.06 | 507.72 | 456.34 | 0 |
| MI39-17 | 0.56 | 51.52 | 0 | 0 | 30.20 | 10.66 | 483.25 | 577.41 | 0 |
| MI39-18 | 0.61 | 29.48 | 3.27 | 1.637 | 0 | 1.637 | 804.19 | 5929.12 | 0 |
| MI39-19 | 0.28 | 25.34 | 0 | 0 | 0 | 3.620 | 1035.39 | 7247.74 | 0 |
| MI39-20 | 0.035 | 0 | 0 | 0 | 0 | 0 | 688.17 | 200.71 | 0 |
| p-value (core/variable) | 0.002 | 0.003 | 0.058 | 0.024 | 0.002 | 0.009 | 0.003 | 0.677 | |
| Gene ID | Chromosome | Start | End | Type | Most Similar Known Cluster | Similarity |
|---|---|---|---|---|---|---|
| MI39_11795 | MI39-1 | 396,772 | 398,794 | NRPS | ||
| MI39_7258 | MI39-1 | 592,293 | 593,294 | saccharide | ||
| MI39_7862 | MI39-1 | 967,185 | 970,941 | NRPS-like | ||
| MI39_1628 | MI39-1 | 2,347,825 | 2,349,019 | saccharide | ||
| MI39_10036 | MI39-2 | 298,534 | 305,960 | T1PKS | ||
| MI39_231 | MI39-2 | 2,684,091 | 2,688,260 | NRPS-like | ||
| MI39_13258 | MI39-2 | 2,684,091 | 2,688,694 | NRPS-like | ||
| MI39_2278 | MI39-2 | 3,261,368 | 3,262,567 | saccharide | ||
| MI39_14038 | MI39-2 | 3,765,374 | 3,773,009 | fatty_acid | ||
| MI39_2573 | MI39-2 | 3,765,377 | 3,768,012 | fatty_acid | ||
| MI39_9444 | MI39-2 | 3,765,849 | 3,768,012 | fatty_acid | ||
| MI39_12401 | MI39-2 | 4,574,044 | 4,577,898 | NRPS-like | ||
| MI39_11789 | MI39-2 | 4,575,793 | 4,577,898 | NRPS-like | ||
| MI39_1311 | MI39-3 | 1,671,638 | 1,674,973 | NRPS-like | ||
| MI39_9164 | MI39-5 | 232,718 | 235,765 | NRPS-like | ||
| MI39_1436 | MI39-5 | 2,035,026 | 2,036,282 | terpene | ||
| MI39_10721 | MI39-6 | 699,152 | 703,444 | T1PKS | naphthopyrone | Polyketide 100% |
| MI39_2845 | MI39-6 | 1,385,216 | 1,386,634 | terpene | ||
| MI39_12234 | MI39-6 | 1,385,216 | 1,386,765 | terpene | ||
| MI39_7678 | MI39-6 | 2,432,577 | 2,438,251 | fatty_acid | ||
| MI39_10493 | MI39-6 | 2,432,577 | 2,435,482 | fatty_acid | ||
| MI39_10754 | MI39-6 | 2,432,577 | 2,436,631 | fatty_acid | ||
| MI39_13479 | MI39-7 | 383,162 | 391,130 | T1PKS | ||
| MI39_5620 | MI39-7 | 2,604,581 | 2,606,040 | T3PKS | ||
| MI39_8099 | MI39-8 | 3,062,963 | 3,066,332 | NRPS-like | ||
| MI39_5810 | MI39-9 | 1,725,235 | 1,737,100 | T1PKS, NRPS | ACT-Toxin II | Polyketide 100% |
| MI39_602 | MI39-10 | 1,433,101 | 1,435,614 | saccharide | ||
| MI39_9909 | MI39-10 | 1,433,101 | 1,434,782 | saccharide | ||
| MI39_4863 | MI39-11 | 2,099,210 | 2,100,758 | saccharide | ||
| MI39_8184 | MI39-18 | 373,737 | 383,484 | T1PKS, NRPS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samsonova, A.; Kanapin, A.; Bankin, M.; Logachev, A.; Gretsova, M.; Rozhmina, T.; Samsonova, M. A Genomic Blueprint of Flax Fungal Parasite Fusarium oxysporum f. sp. lini. Int. J. Mol. Sci. 2021, 22, 2665. https://doi.org/10.3390/ijms22052665
Samsonova A, Kanapin A, Bankin M, Logachev A, Gretsova M, Rozhmina T, Samsonova M. A Genomic Blueprint of Flax Fungal Parasite Fusarium oxysporum f. sp. lini. International Journal of Molecular Sciences. 2021; 22(5):2665. https://doi.org/10.3390/ijms22052665
Chicago/Turabian StyleSamsonova, Anastasia, Alexander Kanapin, Michael Bankin, Anton Logachev, Maria Gretsova, Tatyana Rozhmina, and Maria Samsonova. 2021. "A Genomic Blueprint of Flax Fungal Parasite Fusarium oxysporum f. sp. lini" International Journal of Molecular Sciences 22, no. 5: 2665. https://doi.org/10.3390/ijms22052665
APA StyleSamsonova, A., Kanapin, A., Bankin, M., Logachev, A., Gretsova, M., Rozhmina, T., & Samsonova, M. (2021). A Genomic Blueprint of Flax Fungal Parasite Fusarium oxysporum f. sp. lini. International Journal of Molecular Sciences, 22(5), 2665. https://doi.org/10.3390/ijms22052665