
Assessing Plasmin Generation in Health and Disease

 , and
, and
Abstract
:1. Introduction
2. Plasmin and the Molecular Mechanism of Plasmin Generation
3. Diseases Associated with Plasmin Generation
4. Fibrinolytic Assays Are Overshadowed by Clotting Tests
5. Assays That Measure Fibrinolytic System Components in Blood
6. Functional Assays That Measure Fibrinolytic Potential
7. Assays That Measure Plasmin Generation Kinetics
8. Plasmin Generation in Health and Disease
9. Summary and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cesarman-Maus, G.; Hajjar, K.A. Molecular mechanisms of fibrinolysis. Br. J. Haematol. 2005, 129, 307–321. [Google Scholar] [CrossRef]
- Forsgren, M.; Råden, B.; Israelsson, M.; Larsson, K.; Hedén, L.O. Molecular cloning and characterization of a full-length cDNA clone for human plasminogen. FEBS Lett. 1987, 213, 254–260. [Google Scholar] [CrossRef] [Green Version]
- van der Vorm, L.N.; Remijn, J.A.; de Laat, B.; Huskens, D. Effects of Plasmin on von Willebrand Factor and Platelets: A Narrative Review. TH Open 2018, 2, e218–e228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaller, J.; Gerber, S.S. The plasmin-antiplasmin system: Structural and functional aspects. Cell. Mol. Life Sci. 2011, 68, 785–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holvoet, P.; Lijnen, H.R.; Collen, D. A monoclonal antibody specific for Lys-plasminogen. Application to the study of the activation pathways of plasminogen in vivo. J. Biol. Chem. 1985, 260, 12106–12111. [Google Scholar] [CrossRef]
- Ny, T.; Sawdey, M.; Lawrence, D.; Millan, J.L.; Loskutoff, D.J. Cloning and sequence of a cDNA coding for the human beta-migrating endothelial-cell-type plasminogen activator inhibitor. Proc. Natl. Acad. Sci. USA 1986, 83, 6776–6780. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.D.; Wun, T.C.; Sadler, J.E. cDNA cloning and expression in Escherichia coli of a plasminogen activator inhibitor from human placenta. J. Biol. Chem. 1987, 262, 3718–3725. [Google Scholar] [CrossRef]
- Whyte, C.S.; Mutch, N.J. uPA-mediated plasminogen activation is enhanced by polyphosphate. Haematologica 2020, 106, 522. [Google Scholar] [CrossRef] [Green Version]
- Stoppelli, M.P.; Corti, A.; Soffientini, A.; Cassani, G.; Blasi, F.; Assoian, R.K. Differentiation-enhanced binding of the amino-terminal fragment of human urokinase plasminogen activator to a specific receptor on U937 monocytes. Proc. Natl. Acad. Sci. USA 1985, 82, 4939–4943. [Google Scholar] [CrossRef] [Green Version]
- Nieuwenhuizen, W. Fibrin-mediated plasminogen activation. Ann. N. Y. Acad. Sci. 2001, 936, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.W.; Ravindran, S.; Patston, P.A. The reaction between plasmin and C1-inhibitor results in plasmin inhibition by the serpin mechanism. Blood Coagul. Fibrinolysis 2002, 13, 711–714. [Google Scholar] [CrossRef]
- Holmes, W.E.; Nelles, L.; Lijnen, H.R.; Collen, D. Primary structure of human alpha 2-antiplasmin, a serine protease inhibitor (serpin). J. Biol. Chem. 1987, 262, 1659–1664. [Google Scholar] [CrossRef]
- Plow, E.F.; Collen, D. The presence and release of alpha 2-antiplasmin from human platelets. Blood 1981, 58, 1069–1074. [Google Scholar] [CrossRef] [Green Version]
- Ratnoff, O.D.; Pensky, J.; Ogston, D.; Naff, G.B. The inhibition of plasmin, plasma kallikrein, plasma permeability factor, and the C’1r subcomponent of the first component of complement by serum C’1 esterase inhibitor. J. Exp. Med. 1969, 129, 315–331. [Google Scholar] [CrossRef]
- Thorsen, S.; Philips, M. Isolation of tissue-type plasminogen activator-inhibitor complexes from human plasma. Evidence for a rapid plasminogen activator inhibitor. Biochim. Et Biophys. Acta 1984, 802, 111–118. [Google Scholar] [CrossRef]
- Harpel, P.C.; Cooper, N.R. Studies on human plasma C1 inactivator-enzyme interactions. I. Mechanisms of interaction with C1s, plasmin, and trypsin. J. Clin. Investig. 1975, 55, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.M.; Perkins, S.J.; Sim, R.B.; Willis, A.C.; Feighery, C.; Jackson, J. Degradation of C1-inhibitor by plasmin: Implications for the control of inflammatory processes. Mol. Med. 1997, 3, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Sulikowski, T.; Patston, P.A. The inhibition of TNK-t-PA by C1-inhibitor. Blood Coagul. Fibrinolysis 2001, 12, 75–77. [Google Scholar] [CrossRef]
- Nesheim, M. Thrombin and fibrinolysis. Chest 2003, 124 (Suppl. S3), 33s–39s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Alemany, R.; Suelves, M.; Muñoz-Cánoves, P. Plasmin generation dependent on alpha-enolase-type plasminogen receptor is required for myogenesis. Thromb. Haemost. 2003, 90, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Alemany, R.; Suelves, M.; Diaz-Ramos, A.; Vidal, B.; Munoz-Canoves, P. Alpha-enolase plasminogen receptor in myogenesis. Front Biosci 2005, 10, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Pancholi, V.; Fischetti, V.A. α-Enolase, a Novel Strong Plasmin(ogen) Binding Protein on the Surface of Pathogenic Streptococci. J. Biol. Chem. 1998, 273, 14503–14515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madureira, P.A.; O’Connell, P.A.; Surette, A.P.; Miller, V.A.; Waisman, D.M. The Biochemistry and Regulation of S100A10: A Multifunctional Plasminogen Receptor Involved in Oncogenesis. J. Biomed. Biotechnol. 2012, 2012, 353687. [Google Scholar] [CrossRef] [Green Version]
- Sochaj-Gregorczyk, A.; Ksiazek, M.; Waligorska, I.; Straczek, A.; Benedyk, M.; Mizgalska, D.; Thøgersen, I.B.; Enghild, J.J.; Potempa, J. Plasmin inhibition by bacterial serpin: Implications in gum disease. Faseb J. 2020, 34, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Ogiwara, K.; Nogami, K.; Nishiya, K.; Shima, M. Plasmin-induced procoagulant effects in the blood coagulation: A crucial role of coagulation factors V and VIII. Blood Coagul. Fibrinolysis 2010, 21, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Pryzdial, E.L.G.; Lavigne, N.; Dupuis, N.; Kessler, G.E. Plasmin Converts Factor X from Coagulation Zymogen to Fibrinolysis Cofactor*. J. Biol. Chem. 1999, 274, 8500–8505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, D.; Schindler, S.; Zipfel, P.F. Plasminogen Is a Complement Inhibitor. J. Biol. Chem. 2012, 287, 18831–18842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kost, C.; Benner, K.; Stockmann, A.; Linder, D.; Preissner, K.T. Limited plasmin proteolysis of vitronectin. Characterization of the adhesion protein as morpho-regulatory and angiostatin-binding factor. Eur. J. Biochem. 1996, 236, 682–688. [Google Scholar] [CrossRef]
- Miles, L.A.; Parmer, R.J. Plasminogen receptors: The first quarter century. Semin. Thromb. Hemost. 2013, 39, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolev, K.; Longstaff, C. Bleeding related to disturbed fibrinolysis. Br. J. Haematol. 2016, 175, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Draxler, D.F.; Medcalf, R.L. The Fibrinolytic System—More Than Fibrinolysis? Transfus. Med. Rev. 2015, 29, 102–109. [Google Scholar] [CrossRef]
- Medcalf, R.L. Fibrinolysis, inflammation, and regulation of the plasminogen activating system. J. Thromb. Haemost. 2007, 5, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Medcalf, R.L. Fibrinolysis: From blood to the brain. J. Thromb. Haemost. 2017, 15, 2089–2098. [Google Scholar] [CrossRef] [PubMed]
- Kwaan, H.; Lisman, T.; Medcalf, R.L. Fibrinolysis: Biochemistry, Clinical Aspects, and Therapeutic Potential. Semin Thromb Hemost 2017, 43, 113–114. [Google Scholar] [CrossRef] [Green Version]
- Hunt, B.J.; Segal, H. Hyperfibrinolysis. J. Clin. Pathol. 1996, 49, 958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rømer, J.; Bugge, T.H.; Fyke, C.; Lund, L.R.; Flick, M.J.; Degen, J.L.; Danø, K. Impaired wound healing in mice with a disrupted plasminogen gene. Nat. Med. 1996, 2, 287–292. [Google Scholar] [CrossRef]
- Creemers, E.; Cleutjens, J.; Smits, J.; Heymans, S.; Moons, L.; Collen, D.; Daemen, M.; Carmeliet, P. Disruption of the plasminogen gene in mice abolishes wound healing after myocardial infarction. Am. J. Pathol. 2000, 156, 1865–1873. [Google Scholar] [CrossRef] [Green Version]
- Madureira, P.A.; Hill, R.; Miller, V.A.; Giacomantonio, C.; Lee, P.W.K.; Waisman, D.M. Annexin A2 is a novel cellular redox regulatory protein involved in tumorigenesis. Oncotarget 2011, 2, 1075–1093. [Google Scholar] [CrossRef] [PubMed]
- Ranson, M.; Andronicos, N.M.; O’Mullane, M.J.; Baker, M.S. Increased plasminogen binding is associated with metastatic breast cancer cells: Differential expression of plasminogen binding proteins. Br. J. Cancer 1998, 77, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Suelves, M.; López-Alemany, R.; Lluís, F.; Aniorte, G.; Serrano, E.; Parra, M.; Carmeliet, P.; Muñoz-Cánoves, P. Plasmin activity is required for myogenesis in vitro and skeletal muscle regeneration in vivo. Blood 2002, 99, 2835–2844. [Google Scholar] [CrossRef] [Green Version]
- Tsirka, S.E.; Bugge, T.H.; Degen, J.L.; Strickland, S. Neuronal death in the central nervous system demonstrates a non-fibrin substrate for plasmin. Proc. Natl. Acad. Sci. USA 1997, 94, 9779–9781. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.L.; Strickland, S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 1997, 91, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Leebeek, F.W.; Rijken, D.C. The Fibrinolytic Status in Liver Diseases. Semin. Thromb. Hemost. 2015, 41, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Miszta, A.; Kopec, A.K.; Pant, A.; Holle, L.A.; Byrnes, J.R.; Lawrence, D.A.; Hansen, K.C.; Flick, M.J.; Luyendyk, J.P.; de Laat, B.; et al. A high-fat diet delays plasmin generation in a thrombomodulin-dependent manner in mice. Blood 2020, 135, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Tersteeg, C.; Joly, B.S.; Gils, A.; Lijnen, R.; Deckmyn, H.; Declerck, P.J.; Plaimauer, B.; Coppo, P.; Veyradier, A.; Maas, C.; et al. Amplified endogenous plasmin activity resolves acute thrombotic thrombocytopenic purpura in mice. J. Thromb. Haemost. 2017, 15, 2432–2442. [Google Scholar] [CrossRef]
- Thiebaut, A.M.; Gauberti, M.; Ali, C.; De Lizarrondo, S.M.; Vivien, D.; Yepes, M.; Roussel, B.D. The role of plasminogen activators in stroke treatment: Fibrinolysis and beyond. Lancet. Neurol. 2018, 17, 1121–1132. [Google Scholar] [CrossRef]
- Sakkinen, P.A.; Cushman, M.; Psaty, B.M.; Rodriguez, B.; Boineau, R.; Kuller, L.H.; Tracy, R.P. Relationship of Plasmin Generation to Cardiovascular Disease Risk Factors in Elderly Men and Women. Arter. Thromb. Vasc. Biol. 1999, 19, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Moore, H.B.; Moore, E.E.; Neal, M.D.; Sheppard, F.R.; Kornblith, L.Z.; Draxler, D.F.; Walsh, M.; Medcalf, R.L.; Cohen, M.J.; Cotton, B.A.; et al. Fibrinolysis Shutdown in Trauma: Historical Review and Clinical Implications. Anesth. Analg. 2019, 129, 762–773. [Google Scholar] [CrossRef]
- Yang, C.D.; Hwang, K.K.; Yan, W.; Gallagher, K.; FitzGerald, J.; Grossman, J.M.; Hahn, B.H.; Chen, P.P. Identification of anti-plasmin antibodies in the antiphospholipid syndrome that inhibit degradation of fibrin. J. Immunol. 2004, 172, 5765–5773. [Google Scholar] [CrossRef] [Green Version]
- Krone, K.A.; Allen, K.L.; McCrae, K.R. Impaired fibrinolysis in the antiphospholipid syndrome. Curr. Rheumatol. Rep. 2010, 12, 53–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, J.S.; Comery, T.A.; Martone, R.L.; Elokdah, H.; Crandall, D.L.; Oganesian, A.; Aschmies, S.; Kirksey, Y.; Gonzales, C.; Xu, J.; et al. Enhanced clearance of Aβ in brain by sustaining the plasmin proteolysis cascade. Proc. Natl. Acad. Sci. USA 2008, 105, 8754–8759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, A.L.; Medcalf, R.L. Tissue-Type Plasminogen Activator: A Multifaceted Modulator of Neurotransmission and Synaptic Plasticity. Neuron 2006, 50, 673–678. [Google Scholar] [CrossRef] [Green Version]
- Yepes, M.; Lawrence, D.A. New functions for an old enzyme: Nonhemostatic roles for tissue-type plasminogen activator in the central nervous system. Exp. Biol. Med. 2004, 229, 1097–1104. [Google Scholar] [CrossRef]
- Melchor, J.P.; Strickland, S. Tissue plasminogen activator in central nervous system physiology and pathology. Thromb. Haemost. 2005, 93, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Coccheri, S. COVID-19: The crucial role of blood coagulation and fibrinolysis. Intern. Emerg. Med. 2020, 15, 1369–1373. [Google Scholar] [CrossRef] [PubMed]
- de Maat, S.; Björkqvist, J.; Suffritti, C.; Wiesenekker, C.P.; Nagtegaal, W.; Koekman, A.; van Dooremalen, S.; Pasterkamp, G.; de Groot, P.G.; Cicardi, M.; et al. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J. Allergy Clin. Immunol. 2016, 138, 1414–1423. [Google Scholar] [CrossRef] [Green Version]
- Chandler, W.L.; Velan, T. Plasmin generation and D-dimer formation during cardiopulmonary bypass. Blood Coagul. Fibrinolysis 2004, 15, 583–591. [Google Scholar] [CrossRef]
- Ye, Y.; Vattai, A.; Zhang, X.; Zhu, J.; Thaler, C.J.; Mahner, S.; Jeschke, U.; von Schönfeldt, V. Role of Plasminogen Activator Inhibitor Type 1 in Pathologies of Female Reproductive Diseases. Int. J. Mol. Sci. 2017, 18, 1651. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Zhang, Y.; Yang, H.; Hou, W.; Jin, B.; Hou, J.; Li, H.; Zhao, H.; Zhou, J. Characteristics of fibrinolytic disorders in acute promyelocytic leukemia. Hematolgy 2018, 23, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Ploplis, V.A.; French, E.L.; Carmeliet, P.; Collen, D.; Plow, E.F. Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood 1998, 91, 2005–2009. [Google Scholar] [CrossRef] [Green Version]
- Busuttil, S.J.; Ploplis, V.A.; Castellino, F.J.; Tang, L.; Eaton, J.W.; Plow, E.F. A central role for plasminogen in the inflammatory response to biomaterials. J. Thromb. Haemost. 2004, 2, 1798–1805. [Google Scholar] [CrossRef]
- Chapman, H.A., Jr.; Vavrin, Z.; Hibbs, J.B., Jr. Macrophage fibrinolytic activity: Identification of two pathways of plasmin formation by intact cells and of a plasminogen activator inhibitor. Cell 1982, 28, 653–662. [Google Scholar] [CrossRef]
- Winter, W.E.; Flax, S.D.; Harris, N.S. Coagulation Testing in the Core Laboratory. Lab. Med. 2017, 48, 295–313. [Google Scholar] [CrossRef]
- de Laat-Kremers, R.M.W.; Ninivaggi, M.; Devreese, K.M.J.; de Laat, B. Towards standardization of thrombin generation assays: Inventory of thrombin generation methods based on results of an International Society of Thrombosis and Haemostasis Scientific Standardization Committee survey. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kintigh, J.; Monagle, P.; Ignjatovic, V. A review of commercially available thrombin generation assays. Res. Pract. Thromb. Haemost. 2018, 2, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Berntorp, E.; Salvagno, G.L. Standardization and clinical utility of thrombin-generation assays. Semin. Thromb. Hemost. 2008, 34, 670–682. [Google Scholar] [CrossRef]
- Sivapalaratnam, S.; Collins, J.; Gomez, K. Diagnosis of inherited bleeding disorders in the genomic era. Br. J. Haematol. 2017, 179, 363–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhart, J.; Kepa, S.; Hofer, S.; Koder, S.; Kaider, A.; Wolberg, A.S.; Haslacher, H.; Quehenberger, P.; Eigenbauer, E.; Panzer, S.; et al. Fibrinolysis in patients with a mild-to-moderate bleeding tendency of unknown cause. Ann. Hematol. 2017, 96, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Al Dieri, R.; de Laat, B.; Hemker, H.C. Thrombin generation: What have we learned? Blood Rev. 2012, 26, 197–203. [Google Scholar] [CrossRef]
- Samad, F.; Ruf, W. Inflammation, obesity, and thrombosis. Blood 2013, 122, 3415–3422. [Google Scholar] [CrossRef]
- Lisman, T.; Ariëns, R.A. Alterations in Fibrin Structure in Patients with Liver Diseases. Semin. Thromb. Hemost. 2016, 42, 389–396. [Google Scholar] [CrossRef]
- Kelchtermans, H.; Pelkmans, L.; Bouwhuis, A.; Schurgers, E.; Lindhout, T.; Huskens, D.; Miszta, A.; Hemker, H.C.; Lance, M.D.; de Laat, B. Simultaneous measurement of thrombin generation and fibrin formation in whole blood under flow conditions. Thromb. Haemost. 2016, 116, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Longstaff, C. Measuring fibrinolysis: From research to routine diagnostic assays. J. Thromb. Haemost. 2018, 16, 652–662. [Google Scholar] [CrossRef]
- Whiting, D.; DiNardo, J.A. TEG and ROTEM: Technology and clinical applications. Am. J. Hematol. 2014, 89, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T. Comparison between thromboelastography and thromboelastometry. Minerva Anestesiol. 2019, 85, 1346–1356. [Google Scholar] [CrossRef]
- Gonzalez, E.; Moore, E.E.; Moore, H.B. Management of Trauma-Induced Coagulopathy with Thrombelastography. Crit. Care Clin. 2017, 33, 119–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitlur, M.; Sorensen, B.; Rivard, G.E.; Young, G.; Ingerslev, J.; Othman, M.; Nugent, D.; Kenet, G.; Escobar, M.; Lusher, J. Standardization of thromboelastography: A report from the TEG-ROTEM working group. Haemophilia 2011, 17, 532–537. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, C.U.; Tynngård, N.; Reinstrup, P.; Engström, M. Monitoring fibrinolysis in whole blood by viscoelastic instruments: A comparison of ROTEM and ReoRox. Scand. J. Clin. Lab. Investig. 2013, 73, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Solomon, C.; Schöchl, H.; Ranucci, M.; Schött, U.; Schlimp, C.J. Comparison of fibrin-based clot elasticity parameters measured by free oscillation rheometry (ReoRox®) versus thromboelastometry (ROTEM®). Scand. J. Clin. Lab. Investig. 2015, 75, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Van Geffen, M.; Loof, A.; Lap, P.; Boezeman, J.; van Gorkom, B.A.P.L.; Brons, P.; Verbruggen, B.; Kraaij, M.V.; Van Heerde, W.L. A novel hemostasis assay for the simultaneous measurement of coagulation and fibrinolysis. Hematology 2011, 16, 327–336. [Google Scholar] [CrossRef]
- Van Geffen, M.; Menegatti, M.; Loof, A.; Lap, P.; Karimi, M.; Laros-van Gorkom, B.A.P.; Brons, P.; Van Heerde, W.L. Retrospective evaluation of bleeding tendency and simultaneous thrombin and plasmin generation in patients with rare bleeding disorders. Haemophilia 2012, 18, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Van Geffen, M.; Cugno, M.; Lap, P.; Loof, A.; Cicardi, M.; Van Heerde, W. Alterations of coagulation and fibrinolysis in patients with angioedema due to C1-inhibitor deficiency. Clin. Exp. Immunol. 2012, 167, 472–478. [Google Scholar] [CrossRef]
- Simpson, M.L.; Goldenberg, N.A.; Jacobson, L.J.; Bombardier, C.G.; Hathaway, W.E.; Manco-Johnson, M.J. Simultaneous thrombin and plasmin generation capacities in normal and abnormal states of coagulation and fibrinolysis in children and adults. Thromb. Res. 2011, 127, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Nogami, K.; Shima, M. Simultaneous measurement of thrombin and plasmin generation to assess the interplay between coagulation and fibrinolysis. Thromb. Haemost. 2013, 110, 761–768. [Google Scholar] [CrossRef]
- Tarandovskiy, I.D.; Rajabi, A.A.; Karnaukhova, E.; Buehler, P.W. Contradictory to its effects on thrombin, C1-inhibitor reduces plasmin generation in the presence of thrombomodulin. J. Thromb. Thrombolysis 2019, 48, 81–87. [Google Scholar] [CrossRef]
- Tarandovskiy, I.D.; Buehler, P.W.; Ataullakhanov, F.I.; Karnaukhova, E. C1-esterase inhibitor enhances thrombin generation and spatial fibrin clot propagation in the presence of thrombomodulin. Thromb. Res. 2019, 176, 54–60. [Google Scholar] [CrossRef]
- Tarandovskiy, I.D.; Shin, H.K.H.; Baek, J.H.; Karnaukhova, E.; Buehler, P.W. Interspecies comparison of simultaneous thrombin and plasmin generation. Sci. Rep. 2020, 10, 3885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miszta, A.; Ahmadzia, H.K.; Luban, N.L.C.; Li, S.; Guo, D.; Holle, L.A.; Berger, J.S.; James, A.H.; Gobburu, J.V.S.; van den Anker, J.; et al. Application of a plasmin generation assay to define pharmacodynamic effects of tranexamic acid in women undergoing cesarean delivery. J. Thromb. Haemost. 2020, 19, 221–232. [Google Scholar] [CrossRef]
- De Smedt, E.; Al Dieri, R.; Spronk, H.M.H.; Hamulyak, K.; ten Cate, H.; Hemker, H.C. The technique of measuring thrombin generation with fluorogenic substrates: 1. Necessity of adequate calibration. Thromb. Haemost. 2008, 100, 343–349. [Google Scholar] [CrossRef]
- Hemker, H.C.; Kremers, R. Data management in thrombin generation. Thromb. Res. 2013, 131, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intagliata, N.M.; Argo, C.K.; Stine, J.G.; Lisman, T.; Caldwell, S.H.; Violi, F. Concepts and Controversies in Haemostasis and Thrombosis Associated with Liver Disease: Proceedings of the 7th International Coagulation in Liver Disease Conference. Thromb. Haemost. 2018, 118, 1491–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saes, J.L.; Schols, S.E.M.; Betbadal, K.F.; van Geffen, M.; Verbeek-Knobbe, K.; Gupta, S.; Hardesty, B.M.; Shapiro, A.D.; van Heerde, W.L. Thrombin and plasmin generation in patients with plasminogen or plasminogen activator inhibitor type 1 deficiency. Haemophilia 2019, 25, 1073–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valke, L.; Bukkems, L.H.; Barteling, W.; van Gorkom, B.A.P.L.; Blijlevens, N.M.A.; Mathôt, R.A.A.; van Heerde, W.L.; Schols, S.E.M. Pharmacodynamic monitoring of factor VIII replacement therapy in hemophilia A: Combining thrombin and plasmin generation. J. Thromb. Haemost. 2020, 18, 3222–3231. [Google Scholar] [CrossRef]
- Bouck, E.G.; Denorme, F.; Holle, L.A.; Middelton, E.A.; Blair, A.; de Laat, B.; Schiffman, J.D.; Yost, C.C.; Rondina, M.T.; Wolberg, A.S.; et al. COVID-19 and Sepsis Are Associated With Different Abnormalities in Plasma Procoagulant and Fibrinolytic Activity. Arter. Thromb. Vasc. Biol. 2020, 41, 401–414. [Google Scholar] [CrossRef]
- De Jongh, R.; Ninivaggi, M.; Mesotten, D.; Bai, C.; Marcus, B.; Huskens, D.; Stragier, H.; Miszta, A.; Verbruggen, J.; de Laat-Kremers, R.M.W.; et al. Vascular activation is a strong predictor of mortality in coronavirus disease 2019 patients on the ICU. Blood Coagul. Fibrinolysis 2021. Publish Ahead of Print. [Google Scholar] [CrossRef]
- Maino, A.; Garagiola, I.; Artoni, A.; Al-Humood, S.; Peyvandi, F. A novel mutation of alpha2-plasmin inhibitor gene causes an inherited deficiency and a bleeding tendency. Haemoph. Off. J. World Fed. Hemoph. 2008, 14, 166. [Google Scholar] [CrossRef]
- Mehta, R.; Shapiro, A.D. Plasminogen activator inhibitor type 1 deficiency. Haemoph. Off. J. World Fed. Hemoph. 2008, 14, 1255–1260. [Google Scholar] [CrossRef]
- Lisman, T. Decreased Plasma Fibrinolytic Potential As a Risk for Venous and Arterial Thrombosis. Semin. Thromb. Hemost. 2017, 43, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Vaughan, D.E.; Stampfer, M.J.; Manson, J.E.; Shen, C.; Newcomer, L.M.; Goldhaber, S.Z.; Hennekens, C.H. Baseline fibrinolytic state and the risk of future venous thrombosis. A prospective study of endogenous tissue-type plasminogen activator and plasminogen activator inhibitor. Circulation 1992, 85, 1822–1827. [Google Scholar] [CrossRef] [Green Version]
- Crowther, M.A.; Roberts, J.; Roberts, R.; Johnston, M.; Stevens, P.; Skingley, P.; Patrassi, G.M.; Sartori, M.T.; Hirsh, J.; Prandoni, P.; et al. Fibrinolytic variables in patients with recurrent venous thrombosis: A prospective cohort study. Thromb. Haemost. 2001, 85, 390–394. [Google Scholar]
- Reed, G.L.; Houng, A.K.; Singh, S.; Wang, D. alpha2-Antiplasmin: New Insights and Opportunities for Ischemic Stroke. Semin. Thromb. Hemost. 2017, 43, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tilburg, N.H.; Rosendaal, F.R.; Bertina, R.M. Thrombin activatable fibrinolysis inhibitor and the risk for deep vein thrombosis. Blood 2000, 95, 2855–2859. [Google Scholar] [CrossRef] [PubMed]
- Folsom, A.R.; Cushman, M.; Heckbert, S.R.; Rosamond, W.D.; Aleksic, N. Prospective study of fibrinolytic markers and venous thromboembolism. J. Clin. Epidemiol. 2003, 56, 598–603. [Google Scholar] [CrossRef]
- Matsuo, O.; Lijnen, H.R.; Ueshima, S.; Kojima, S.; Smyth, S.S. A guide to murine fibrinolytic factor structure, function, assays, and genetic alterations. J. Thromb. Haemost. 2007, 5, 680–689. [Google Scholar] [CrossRef]
- Casanave, E.B.; Tentoni, J. Comparative fibrinolysis. Fibrinolysis Thrombolysis 2014. Available online: https://www.intechopen.com/books/fibrinolysis-and-thrombolysis/comparative-fibrinolysis (accessed on 20 February 2021).
- Mackman, N. Role of Tissue Factor in Hemostasis, Thrombosis, and Vascular Development. Arter. Thromb. Vasc. Biol. 2004, 24, 1015–1022. [Google Scholar] [CrossRef]
- Bugge, T.H.; Kombrinck, K.W.; Flick, M.J.; Daugherty, C.C.; Danton, M.J.; Degen, J.L. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 1996, 87, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Bollen, L.; Peetermans, M.; Peeters, M.; Van Steen, K.; Hoylaerts, M.F.; Declerck, P.J.; Verhamme, P.; Gils, A. Active PAI-1 as marker for venous thromboembolism: Case–control study using a comprehensive panel of PAI-1 and TAFI assays. Thromb. Res. 2014, 134, 1097–1102. [Google Scholar] [CrossRef]
- Meltzer, M.E.; Lisman, T.; de Groot, P.G.; Meijers, J.C.M.; le Cessie, S.; Doggen, C.J.M.; Rosendaal, F.R. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood 2010, 116, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Sperzel, M.; Huetter, J. Evaluation of aprotinin and tranexamic acid in different in vitro and in vivo models of fibrinolysis, coagulation and thrombus formation. J. Thromb. Haemost. 2007, 5, 2113–2118. [Google Scholar] [CrossRef]
- Silva, M.M.; Thelwell, C.; Williams, S.C.; Longstaff, C. Regulation of fibrinolysis by C-terminal lysines operates through plasminogen and plasmin but not tissue-type plasminogen activator. J. Thromb. Haemost. 2012, 10, 2354–2360. [Google Scholar] [CrossRef]
- Picetti, R.; Shakur-Still, H.; Medcalf, R.L.; Standing, J.F.; Roberts, I. What concentration of tranexamic acid is needed to inhibit fibrinolysis? A systematic review of pharmacodynamics studies. Blood Coagul. Fibrinolysis 2019, 30, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotton, B.A.; Harvin, J.A.; Kostousouv, V.; Minei, K.M.; Radwan, Z.A.; Schochl, H.; Wade, C.E.; Holcomb, J.B.; Matijevic, N. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J. Trauma Acute Care Surg. 2012, 73, 365–370. [Google Scholar] [CrossRef]
- Gall, L.S.; Brohi, K.; Davenport, R.A. Diagnosis and Treatment of Hyperfibrinolysis in Trauma (A European Perspective). Semin. Thromb. Hemost. 2017, 43, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Stibbe, J.; Kluft, C.; Brommer, E.J.; Gomes, M.; de Jong, D.S.; Nauta, J. Enhanced fibrinolytic activity during cardiopulmonary bypass in open-heart surgery in man is caused by extrinsic (tissue-type) plasminogen activator. Eur. J. Clin. Investig. 1984, 14, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Solomon, C.; Collis, R.E.; Collins, P.W. Haemostatic monitoring during postpartum haemorrhage and implications for management. Br. J. Anaesth. 2012, 109, 851–863. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Soff, G.A.; Kampel, L.J. Disseminated intravascular coagulation with excessive fibrinolysis in prostate cancer: A case series and review of the literature. Oncology 2011, 81, 119–125. [Google Scholar] [CrossRef]
- collaborators, C.-T.; Shakur, H.; Roberts, I.; Bautista, R.; Caballero, J.; Coats, T.; Dewan, Y.; El-Sayed, H.; Gogichaishvili, T.; Gupta, S.; et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): A randomised, placebo-controlled trial. Lancet 2010, 376, 23–32. [Google Scholar] [CrossRef]
- Forbes, C.D.; Barr, R.D.; Reid, G.; Thomson, C.; Prentice, C.R.; McNicol, G.P.; Douglas, A.S. Tranexamic acid in control of haemorrhage after dental extraction in haemophilia and Christmas disease. Br. Med. J. 1972, 2, 311–313. [Google Scholar] [CrossRef] [Green Version]
- Strickland, D.K. A new plasminogen receptor. Blood 2010, 115, 1315–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, H.; Baik, N.; Kiosses, W.B.; Krajewski, S.; Miles, L.A.; Parmer, R.J. The Novel Plasminogen Receptor, Plasminogen ReceptorKT (Plg-RKT), Regulates Catecholamine Release. J. Biol. Chem. 2011, 286, 33125–33133. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Gong, Y.; Grella, D.K.; Castellino, F.J.; Miles, L.A. Endogenous plasmin converts Glu-plasminogen to Lys-plasminogen on the monocytoid cell surface. J. Thromb. Haemost. 2003, 1, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Castellino, F.J.; Gong, Y. Critical Role for Conversion of Glu-Plasminogen to Lys-Plasminogen for Optimal Stimulation of Plasminogen Activation on Cell Surfaces. Trends Cardiovasc. Med. 2003, 13, 21–30. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Friedlander, R.J., Jr.; Nicholas, R.L.; Nachman, R.L. Binding of Lys-plasminogen to monocytes/macrophages. J. Clin. Investig. 1988, 82, 1948–1955. [Google Scholar] [CrossRef] [Green Version]
- Whyte, C.S.; Morrow, G.B.; Baik, N.; Booth, N.A.; Jalal, M.M.; Parmer, R.J.; Miles, L.A.; Mutch, N.J. Exposure of plasminogen and a novel plasminogen receptor, Plg-RKT, on activated human and murine platelets. Blood 2021, 137, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Yamamoto, K.; Loskutoff, D.J. Distribution and regulation of plasminogen activator inhibitor-1 in murine adipose tissue in vivo. Induction by tumor necrosis factor-alpha and lipopolysaccharide. J. Clin. Investig. 1996, 97, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Pluskota, E.; Soloviev, D.A.; Bdeir, K.; Cines, D.B.; Plow, E.F. Integrin αMβ2 Orchestrates and Accelerates Plasminogen Activation and Fibrinolysis by Neutrophils. J. Biol. Chem. 2004, 279, 18063–18072. [Google Scholar] [CrossRef] [Green Version]
- Dudani, A.K.; Ganz, P.R. Endothelial cell surface actin serves as a binding site for plasminogen, tissue plasminogen activator and lipoprotein(a). Br. J. Haematol. 1996, 95, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Ginsberg, M.H.; White, J.G.; Plow, E.F. Plasminogen interacts with human platelets through two distinct mechanisms. J. Clin. Investig. 1986, 77, 2001–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lishko, V.K.; Novokhatny, V.V.; Yakubenko, V.P.; Skomorovska-Prokvolit, H.V.; Ugarova, T.P. Characterization of plasminogen as an adhesive ligand for integrins alphaMbeta2 (Mac-1) and alpha5beta1 (VLA-5). Blood 2004, 104, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Hembrough, T.A.; Vasudevan, J.; Allietta, M.M.; Glass, W.F., 2nd; Gonias, S.L. A cytokeratin 8-like protein with plasminogen-binding activity is present on the external surfaces of hepatocytes, HepG2 cells and breast carcinoma cell lines. J. Cell Sci. 1995, 108 Pt 3, 1071–1082. [Google Scholar]
- Hawley, S.B.; Tamura, T.; Miles, L.A. Purification, cloning, and characterization of a profibrinolytic plasminogen-binding protein, TIP49a. J. Biol. Chem. 2001, 276, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Herren, T.; Burke, T.A.; Das, R.; Plow, E.F. Identification of histone H2B as a regulated plasminogen receptor. Biochemistry 2006, 45, 9463–9474. [Google Scholar] [CrossRef] [PubMed]
- Parkkinen, J.; Raulo, E.; Merenmies, J.; Nolo, R.; Kajander, E.O.; Baumann, M.; Rauvala, H. Amphoterin, the 30-kDa protein in a family of HMG1-type polypeptides. Enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J. Biol. Chem. 1993, 268, 19726–19738. [Google Scholar] [CrossRef]
- Kanalas, J.J.; Makker, S.P. Identification of the rat Heymann nephritis autoantigen (GP330) as a receptor site for plasminogen. J. Biol. Chem. 1991, 266, 10825–10829. [Google Scholar] [CrossRef]
- Davis, G.E.; Pintar Allen, K.A.; Salazar, R.; Maxwell, S.A. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. J. Cell Sci. 2001, 114 Pt 5, 917–930. [Google Scholar]
- Vallier, L.; Cointe, S.; Lacroix, R.; Bonifay, A.; Judicone, C.; Dignat-George, F.; Kwaan, H.C. Microparticles and Fibrinolysis. Semin. Thromb. Hemost. 2017, 43, 129–134. [Google Scholar] [CrossRef]
- Ginestra, A.; Monea, S.; Seghezzi, G.; Dolo, V.; Nagase, H.; Mignatti, P.; Vittorelli, M.L. Urokinase Plasminogen Activator and Gelatinases Are Associated with Membrane Vesicles Shed by Human HT1080 Fibrosarcoma Cells. J. Biol. Chem. 1997, 272, 17216–17222. [Google Scholar] [CrossRef] [Green Version]
- Dejouvencel, T.; Doeuvre, L.; Lacroix, R.; Plawinski, L.; Dignat-George, F.; Lijnen, H.R.; Anglés-Cano, E. Fibrinolytic cross-talk: A new mechanism for plasmin formation. Blood 2010, 115, 2048–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lijnen, H.R. Elements of the Fibrinolytic System. Ann. N. Y. Acad. Sci. 2001, 936, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Koster, A.; Quinones, Q.J.; Milling, T.J.; Key, N.S. Antifibrinolytic Therapy and Perioperative Considerations. Anesthesiology 2018, 128, 657–670. [Google Scholar] [CrossRef]
- Berntorp, E.; Follrud, C.; Lethagen, S. No increased risk of venous thrombosis in women taking tranexamic acid. Thromb. Haemost. 2001, 86, 714–715. [Google Scholar] [PubMed]
- Aronson, D.L.; Chang, P.; Kessler, C.M. Platelet-dependent thrombin generation after in vitro fibrinolytic treatment. Circulation 1992, 85, 1706–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bijnen, S.T.; Østerud, B.; Barteling, W.; Verbeek-Knobbe, K.; Willemsen, M.; van Heerde, W.L.; Muus, P. Alterations in markers of coagulation and fibrinolysis in patients with Paroxysmal Nocturnal Hemoglobinuria before and during treatment with eculizumab. Thromb. Res. 2015, 136, 274–281. [Google Scholar] [CrossRef] [PubMed]



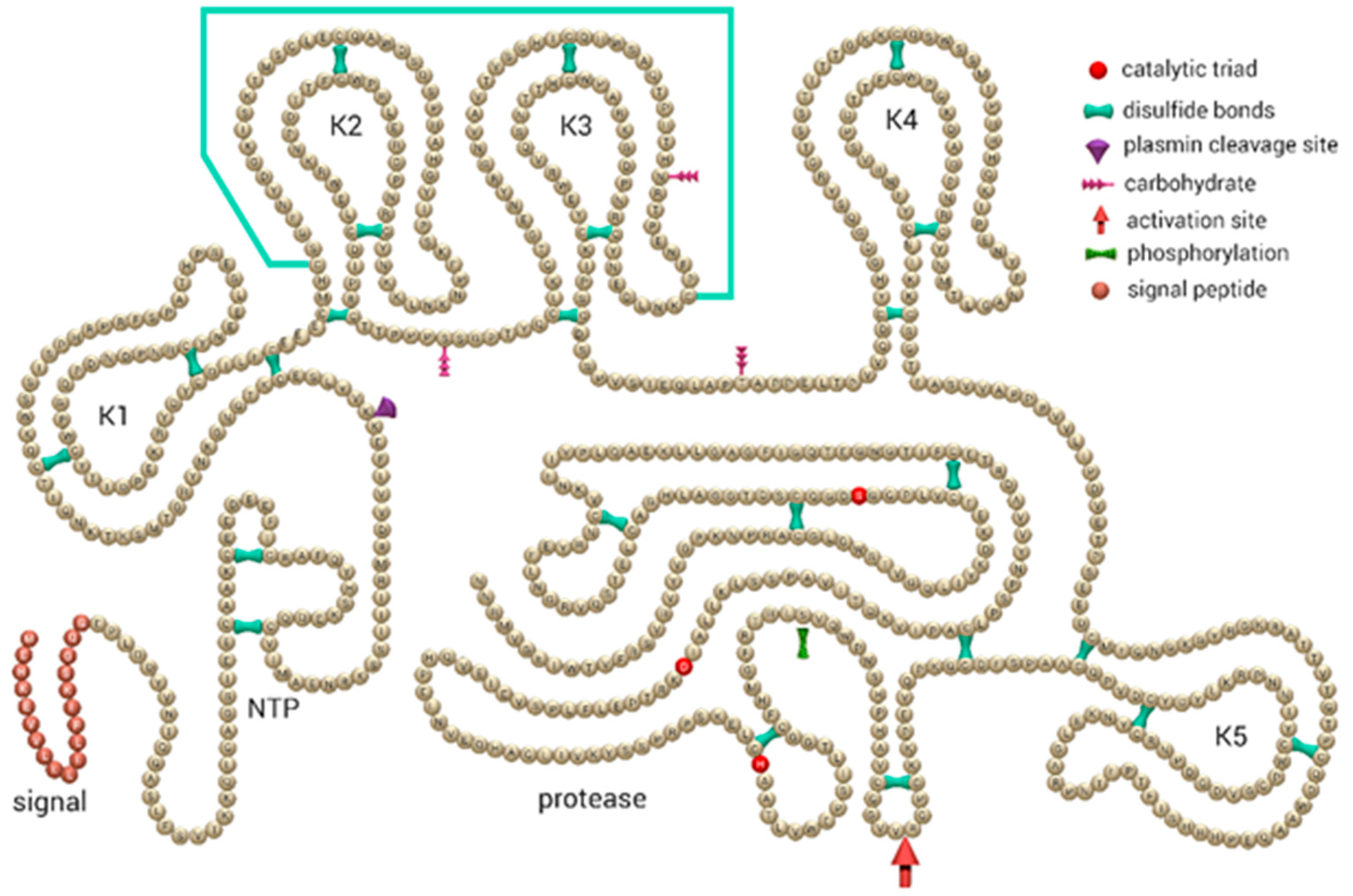{kind=link}
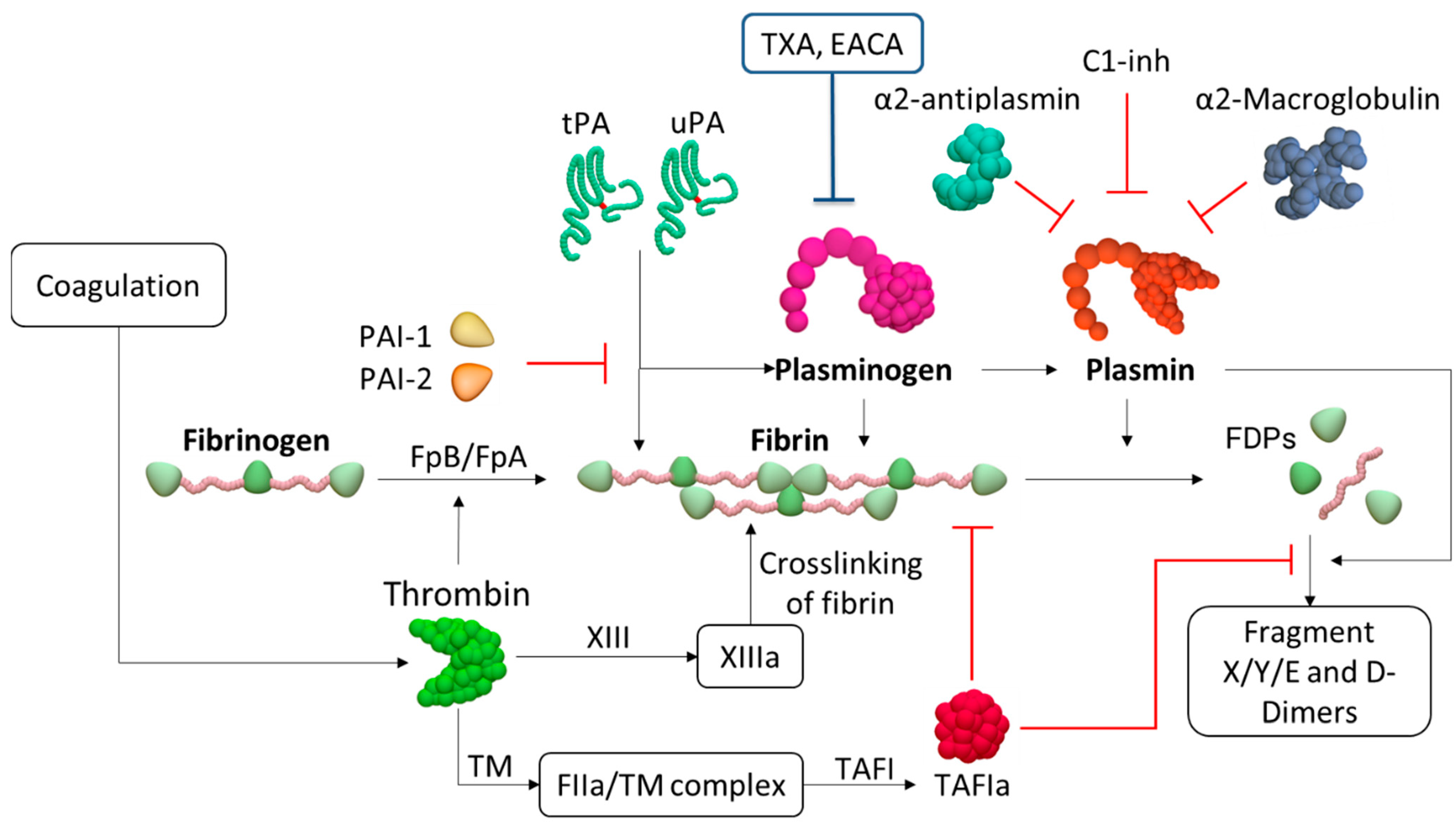{kind=link}
| Processes and Diseases | Key Finding(s) | Reference |
|---|---|---|
| Wound healing | Plasminogen is required for the repair of skin wounds in mice; plasmin-mediated proteolysis plays a central role in cardiac wound healing after myocardial infarction in mice | [36,37] |
| Oncogenesis and metastasis | S100A10 is a key regulator of cellular plasmin production; plasminogen-binding proteins were detected in the plasma membranes of the human breast cancer cell line MDA-MB-231 | [23,38,39] |
| Degradation of extracellular matrix, muscle regeneration | Inhibition of plasmin activity with α2-antiplasmin results in decreased myoblast fusion and differentiation in vitro | [40] |
| Cell migration or tissue remodeling | α-enolase constitutes a receptor for plasminogen on several leukocyte cell types, serving to localize and promote plasminogen activation | [20,21] |
| Apoptosis | Disruption of neuron–ECM interaction via tPA/plasmin degradation of laminin sensitizes hippocampal neurons to cell death | [41,42] |
| Liver diseases, mainly cirrhosis | Changes in tPA, PAI-1, and active PAI-1, leading to hyperfibrinolysis | [43] |
| Obesity | Thrombomodulin-dependent activation of TAFI results in delayed PG | [44] |
| Thrombotic thrombocytopenic purpura | Plasmin levels are increased during acute TTP, although limited via suppression by α2-antiplasmin and PAI-1 | [45] |
| Von Willebrand factor disease | Plasmin is able to proteolyze von Willebrand factor | [3] |
| Stroke | Plasminogen has a protective effect on the ischemic brain by improving the clearance of macrovascular thrombi and restoring reperfusion | [46] |
| Cardiovascular diseases | Plasmin-α2-antiplasmin complex levels predict acute myocardial infarction in the elderly | [47] |
| Trauma | Changes after severe injury lead to trauma-induced coagulopathy and coagulation changes that cause hyperfibrinolysis | [48] |
| Antiphospholipid syndrome | Elevated levels of PAI-1; presence of antibodies inhibiting plasmin and tPA | [49,50] |
| Gum disease | Miropin expressed by Tannerella forsythia is an efficient inhibitor of plasmin | [24] |
| Alzheimer disease | Plasmin contributes to the catabolism and clearance of neurotoxic β-amyloid (Aβ) | [51] |
| Central nervous system | tPA is a modulator of neurotransmission and the synaptic plasticity process; tPA/Plasminogen axis contributes to excitotoxic neuronal degeneration | [52,53,54] |
| Coronavirus disease-2019 | Enhanced plasmin generation potential in plasma | [55] |
| Hereditary angioedema | Plasmin cleaves and activates factor XII associated with hereditary angioedema | [56] |
| Cardiopulmonary bypass | Increased D-dimer and PG, measured by plasmin-α2-antiplasmin complexes and antiplasmin level | [57] |
| Placenta disorders | PAI-1 is responsible for inhibiting extracellular matrix degradation, thereby causing an inhibition of trophoblasts invasion | [58] |
| Acute promyelocytic leukemia | Decreased PAI-1 and plasminogen, and increased tPA, uPA, and uPAR leads to hyperfibrinolysis | [59] |
| Inflammation | Plasminogen, plasminogen activators, and inhibitors have been identified in exudates and extracts of inflamed tissue; endotoxin-stimulated macrophages hydrolyze fibrin by a plasmin-mediated process in the absence of plasminogen activator | [60,61,62] |
| NHA | STA | STPGA | PGA | |
|---|---|---|---|---|
| Substrates (thrombin and plasmin) | Bz-AGR-AMC and bis-(CBZ-FR)-Rho | Boc-VPR-AMC and Boc-EKK-AMC | Z-GGR-AMC and Boc-EKK-AMC | Boc-EKK-AMC |
| Specificity of the plasmin substrate | Plasmin, thrombin, factor Xa | Plasmin | Plasmin | Plasmin |
| Calibration method | Calibration with plasmin | - | Michaelis–Menten equation using the obtained values of AMC production rate | Calibration with α2M-plasmin |
| Method to obtain plasmin curve | Comparing the arbitrary fluorescence values to a calibration curve | Subtracting the fluorescence reading of the blank well | Plasmin is calculated using the obtained values of AMC production rate | Comparing the arbitrary fluorescence values to a calibration curve |
| Correction for substrate consumption/inner filter effect | +/+ | −/− | −/+ | +/+ |
| tPA (μg/mL) | 0.38 | 0.45 | 0.7 | 0.31 or 1.25 |
| TF (pM) | 0.3 | 5 | 4.5 | 0.5 or 1 |
| Plasma volume | 80 | 90 | 80 | 30 |
| Published applications in health and diseases | -PG in patients with plasminogen and PAI-1 deficiency [92] -PG in hemophilia A patients [93] -PG in patients with rare bleeding disorders [81] -PG in patients with angioedema [82] | -Abnormalities in fibrinolysis in adults and children [83] -PG in human plasma with fibrinolytic deficiency [84] | -Inhibitory effect of C-1 inhibitor [85] | -PG in mice model of diet-induced obesity [44] -PG in mice with fibrin(ogen) deficiency [44] -Pharmacodynamics of TXA [88] -PG in coronavirus disease-2019 patients [94,95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miszta, A.; Huskens, D.; Donkervoort, D.; Roberts, M.J.M.; Wolberg, A.S.; de Laat, B. Assessing Plasmin Generation in Health and Disease. Int. J. Mol. Sci. 2021, 22, 2758. https://doi.org/10.3390/ijms22052758
Miszta A, Huskens D, Donkervoort D, Roberts MJM, Wolberg AS, de Laat B. Assessing Plasmin Generation in Health and Disease. International Journal of Molecular Sciences. 2021; 22(5):2758. https://doi.org/10.3390/ijms22052758
Chicago/Turabian StyleMiszta, Adam, Dana Huskens, Demy Donkervoort, Molly J. M. Roberts, Alisa S. Wolberg, and Bas de Laat. 2021. "Assessing Plasmin Generation in Health and Disease" International Journal of Molecular Sciences 22, no. 5: 2758. https://doi.org/10.3390/ijms22052758
APA StyleMiszta, A., Huskens, D., Donkervoort, D., Roberts, M. J. M., Wolberg, A. S., & de Laat, B. (2021). Assessing Plasmin Generation in Health and Disease. International Journal of Molecular Sciences, 22(5), 2758. https://doi.org/10.3390/ijms22052758