
Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis

 ,
,  and
and 
Abstract
:1. Introduction
2. Epigenetic Modifications Regulate Epithelial to Mesenchymal Transition (EMT)
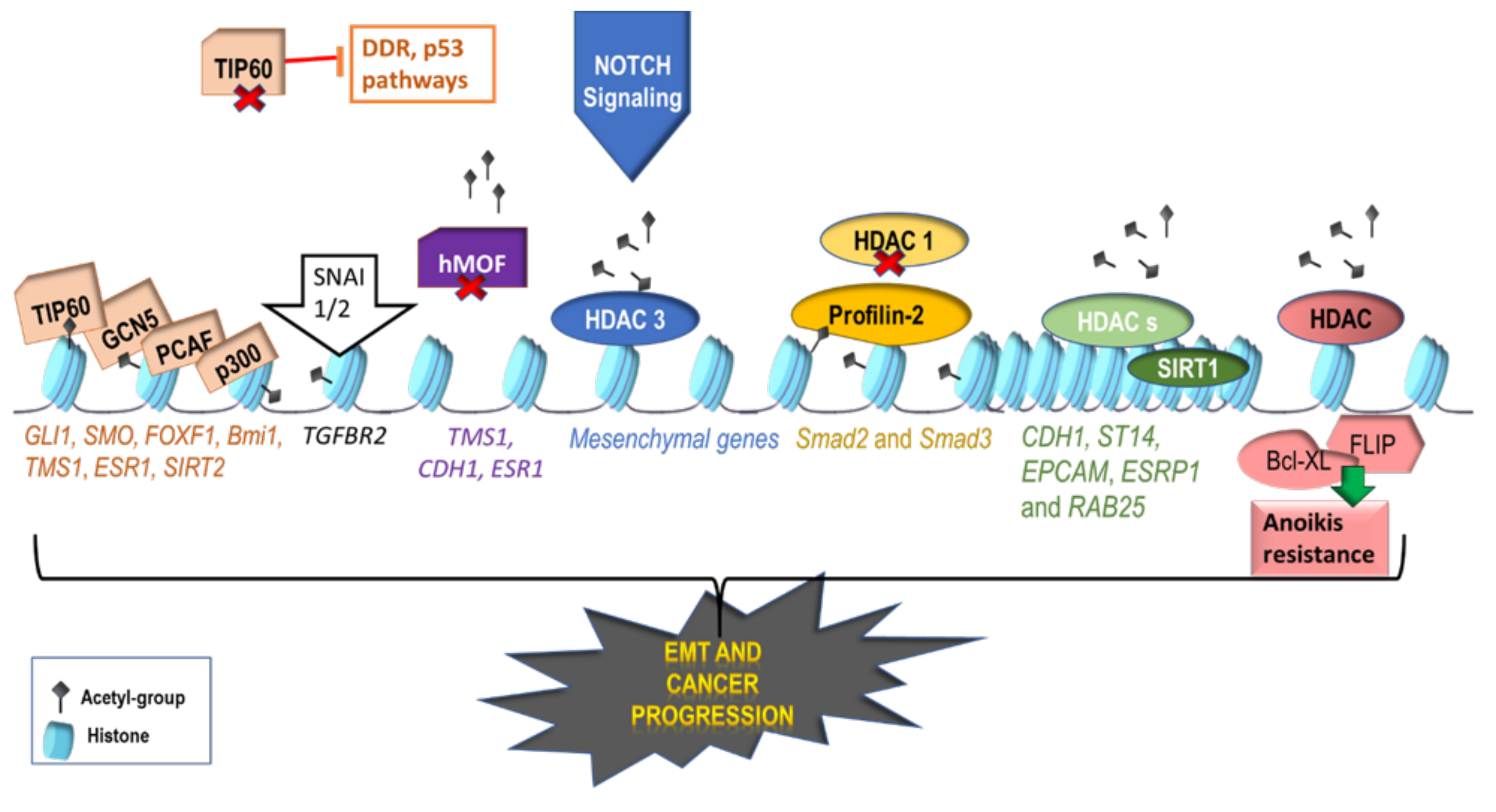
2.1. Activating Histone Modifications Involved in EMT
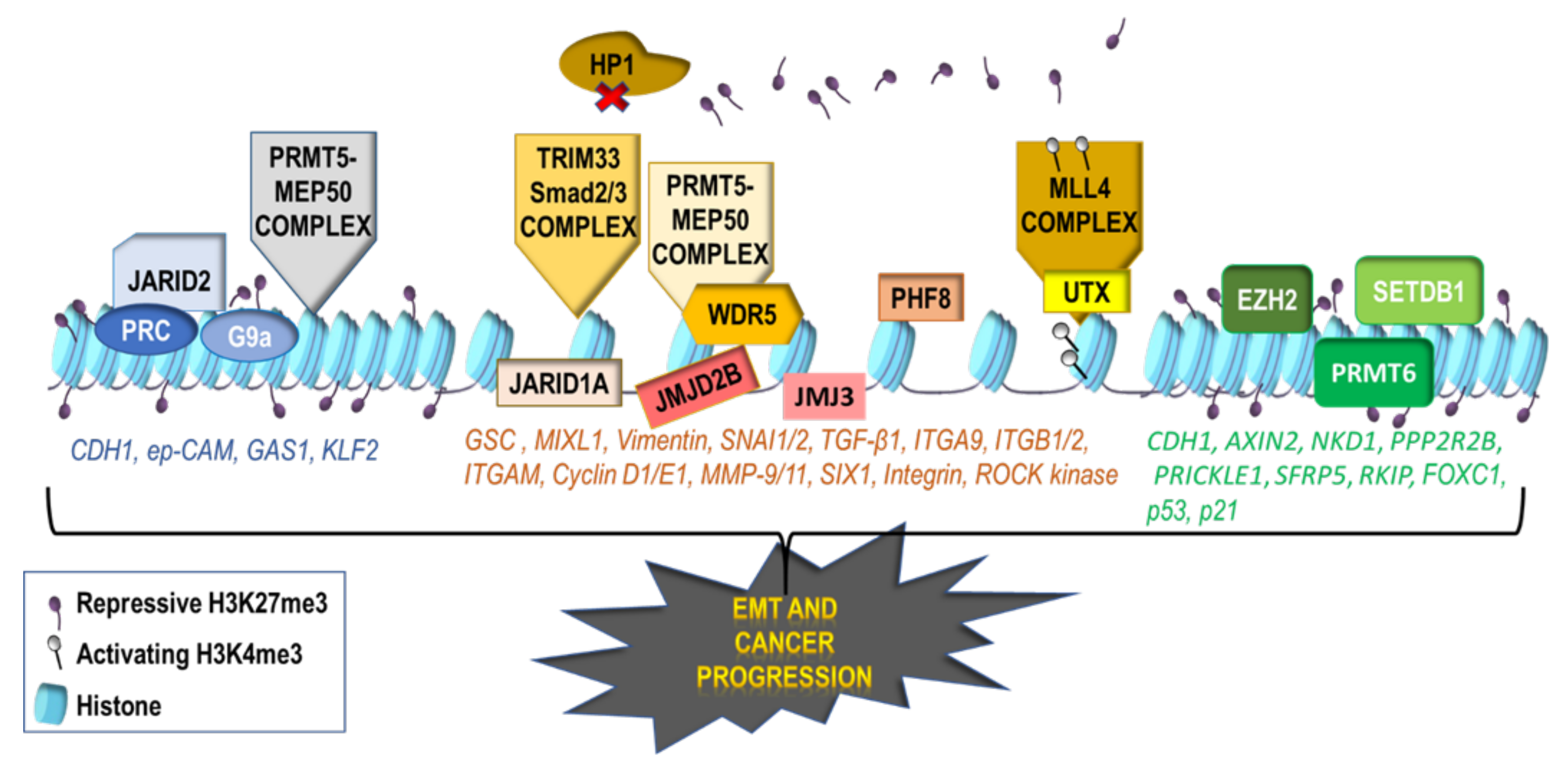
2.2. Repressive Histone Modifications Involved in EMT
2.3. DNA Methylation Involved in EMT
2.4. Non-Coding RNAs (Nc-RNAs) Involved in EMT
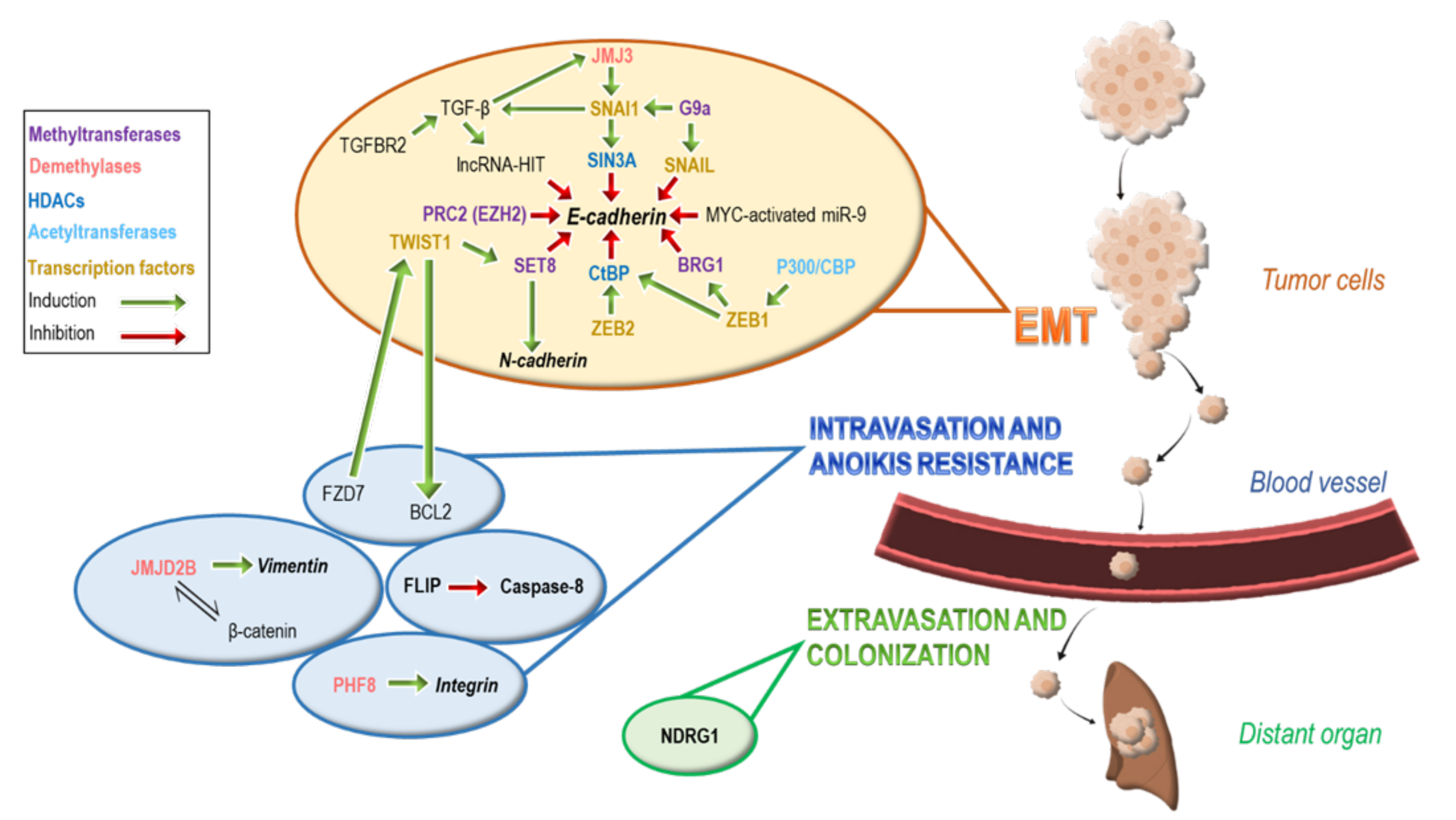
3. Epigenetic Modifications Involved in Intravasation and Anoikis Resistance
4. Epigenetic Modifications Involved in Extravasation and Colonization
5. Cancer-Type Specific Histone Modifications Regulating Tumor Metastasis
5.1. Head and Neck Cancer
5.2. Lung Cancer
5.3. Breast Cancer
5.4. Gastrointestinal Cancers
5.5. Prostate Cancer
6. Targeting Options
6.1. Low Molecular Weight EMT Inhibitors
6.2. Histone-Modifying Drugs
6.3. DNA Methylation Inhibitors
6.4. NcRNAs

{kind=link}
{kind=link}
{kind=link}
| Drug Category | Drug Name | Cancer Type | Effect | Reference | |
|---|---|---|---|---|---|
| LMW EMT inhibitors | BRD4 Inhibitors | MS417 | Colorectal | Inhibits metastasis via induction of E-cadherin and inhibition of Vimentin | [121] |
| JQ1 | Breast | Blocks BRD4 binding to K73/K76Ac2 on the TWIST transcription factor, resulting in decreased WNT5A expression and inhibition of EMT and metastasis | [122] | ||
| Histone binder inhibitor | UNC3866 | Prostate | Blocks the binding of CBX4/7 to methyl-lysine; unclarified role in EMT reversal | [124] | |
| CSC targeting drugs | Salinomycin | Breast, GI, Leukemia, Lung, Prostate | Influences the Twist-Snail/ZEB-E-cadherin axis and Wnt-TGF-β-BMP to inhibit EMT | [126,127,128,129,130] | |
| Histone-modifying drugs | DNMT Inhibitors | Chaetocin | AML | Restores E-cadherin and p15INK4B expression by reducing SUV39H1- mediated H3K9me3 on their promoters | [154] |
| BIX01294 | HeLa cells | Induces E-cadherin expression by inhibiting G9a and GLP-mediated H3K9me2 deposition on its promoter. | [155,156] | ||
| UNC0638 | Pancreatic | Induces E-cadherin expression by inhibiting G9a and GLP-mediated H3K9me2 deposition on its promoter. | [157] | ||
| Zebularine | Pancreatic | Used in combination with SAHA to promote the differentiation of cancer cells | [137] | ||
| HDAC Inhibitors | SAHA | Pancreatic | Combined with Zebularine in order to promote differentiation of cancer cells | [137] | |
| Panobinostat | Hepatocellular | Inhibits cancer proliferation and induces apoptosis of cancer cells. Also increases differentiation markers in vivo | [135] | ||
| Mocetinostat | Pancreatic | Specific antagonism of ZEB1-mediated silencing of miR-203, which is associated with tumor recurrence | [139] | ||
| Entinostat | Breast | In phase III clinical trial | [143] | ||
| PRMT5 Inhibitors | shRNA- mediated inhibition | Breast, Lung | Reduces metastatic potential, EMT and proliferation rate of cancer cells | [80,146] | |
| EZH2 Inhibitors | Tazemetostat | Advanced solid tumors and lymphomas | Under clinical trial investigation | [152] | |
| GSK2816126 | Lymphomas, solid tumors and MM | Clinical study terminated due to little effect in treatment efficiency | [152] | ||
| Erk Inhibitors | AZD6244 | Breast | Combination with EZH2 inhibitors suppresses TGF-β-induced EMT | [150] | |
| LSD1 Inhibitors | Parnate | Myelogenous leukemia | Decreases motility and invasiveness, increases E-cadherin | [158] | |
| Pargyline | Lung | Suppresses proliferation, migration and invasion of cancer cells | [159,160] | ||
| GSK2879552 | Lung | Clinical study discontinued | [161] | ||
| DNA methylation inhibitors | Decitabine | MDS, AML | Changes cancer cell morphology, differentiation markers and inhibits proliferation | [163] | |
| 5-aza-2′-deoxycytidine | Gastric | Improves sensitivity to chemotherapy, restores epithelial phenotypes by promoting E-cadherin re-expression and inhibits EMT | [167,168] | ||
7. Conclusions—Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EMT | Epithelial to Mesenchymal Transition |
| P300/CBP | E1A Binding Protein P300/CREB Binding Protein |
| ZEB1 | Zinc Finger E-Box Binding Homeobox 1 |
| BRG1 | BRM/SWI2-Related Gene 1 |
| CtBP | C-terminal-binding protein |
| HDAC | Histone deacetylase |
| SNAI1 | Snail Family of zinc finger proteins 1 |
| SNAI2 | Slug |
| JMJ3 | Jumonji Domain Containing 3 Histone Lysine Demethylase |
| G9a | Euchromatic Histone Lysine Methyltransferase 2 |
| SIN3A | SIN3 Transcription Regulator Family Member A |
| SET8 | Lysine Methyltransferase 5A |
| H | Histone |
| K | Lysine |
| me | methylation |
| ac | acetylation |
| R | Arginine |
| TWIST1 | Twist Family BHLH Transcription Factor 1 |
| EZH2 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit |
| PRC2 | Polycomb Repressive Complex 2 |
| FZD7 | Frizzled Class Receptor 7 |
| FLIP | CASP8 and FADD Like Apoptosis Regulator |
| JMJD2B | Jumonji Domain-Containing Protein 2B |
| PHF8 | PHD Finger Protein 8 |
| NDRG1 | N-Myc Downstream Regulated 1 |
| GLI1 | Glioma-Associated Oncogene Homolog 1 |
| SMO | Smoothened, Frizzled Class Receptor |
| FOXF1 | Forkhead Box F1 |
| Bmi1 | Polycomb Complex Protein BMI-1 |
| SIRT2 | Sirtuin 2 |
| TGFBR2 | TGF-β Receptor type 2 |
| GCN5 | General Control of Amino Acid Synthesis Protein 5-Like 2 |
| PCAF | P300/CBP-Associated Factor |
| TIP60 | Tat Interacting Protein, 60 kDa |
| DDR | DNA Damage Response |
| hMOF | Males-absent-on-the-first histone acetyltransferase |
| TMS1 | Target of Methylation-induced Silencing |
| ESR1 | Estrogen Receptor 1 |
| Smad2 | Mothers Against Decapentaplegic Homolog 2 |
| EPCAM | Epithelial Cell Adhesion Molecule |
| ST14 | Suppression of Tumorigenicity 14 |
| ESRP1 | Epithelial Splicing Regulatory Protein 1 |
| RAB25 | RAB25, Member RAS Oncogene Family |
| FLIP | CASP8 And FADD Like Apoptosis Regulator |
| PRMT | Protein Arginine Methyltransferase |
| MEP50 | Methylosome Protein 50 |
| GAS1 | Growth Arrest Specific 1 |
| PRC | Polycomb Repressive Complex |
| KLF2 | Kruppel Like Factor 2 |
| JARID | Jumonji, AT Rich Interactive Domain |
| TRIM33 | Tripartite Motif Containing 33 |
| Smad2/3 | Mothers Against Decapentaplegic Homolog 2/3 |
| HP1 | Heterochromatin Protein 1-Alpha |
| GSC | Goosecoid Homeobox |
| MIXL1 | Mix Paired-Like Homeobox |
| WDR5 | WD Repeat-Containing Protein 5 |
| KDM6B | Lysine (K)-Specific Demethylase 6B |
| ITG | Integrin |
| UTX | Ubiquitously- Transcribed X Chromosome Tetratricopeptide Repeat Protein |
| MMP-9/11 | Metalloproteinase-9/11 |
| SIX1 | Sine Oculis Homeobox Homolog 1 |
| MLL4 | Mixed-Lineage Leukemia Protein 4 |
| ROCK kinase | Rho-Associated Protein Kinase 1 |
| FOXC1 | Forkhead Box C1 |
| SETDB1 | SET Domain Bifurcated Histone Lysine Methyltransferase 1 |
| CDH1 | Cadherin 1 |
| AXIN2 | Axis Inhibition Protein 2 |
| NKD1 | Naked1 |
| PPP2R2B | Protein Phosphatase 2 Regulatory Subunit 2 beta |
| PRICKLE1 | Prickle Planar Cell Polarity Protein |
| SFRP5 | Secreted Frizzled Related Protein 5 |
| RKIP | Raf Kinase Inhibitory Protein |
References
- Lin, T.F.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J.L.D.P. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Sadanandam, A.; Varney, M.L.; Kinarsky, L.; Ali, H.; Mosley, R.L.; Singh, R.K. Identification of Functional Cell Adhesion Molecules with a Potential Role in Metastasis by a Combination of in vivo Phage Display and in silico Analysis. OMICS A J. Integr. Biol. 2007, 11, 41–57. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Y.; Kriska, A.; Chen, H. Chapter 28—Epigenetic Regulation in Cancer Metastasis; Tollefsbol, T.O.B.T.-M.E., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 499–514. ISBN 978-0-12-803239-8. [Google Scholar]
- Costa, F.F. Epigenomics in cancer management. Cancer Manag. Res. 2010, 2, 255–265. [Google Scholar] [CrossRef]
- Hake, S.B.; Xiao, A.; Allis, C.D. Linking the epigenetic “language” of covalent histone modifications to cancer. Br. J. Cancer 2004, 90, 761–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone methyltransferase SETDB1: A common denominator of tumorigenesis with therapeutic potential. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions between β-catenin and transforming growth factor-β signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.-Z.; Tsai, Y.-P.; Yang, M.-H.; Huang, C.-H.; Chang, S.-Y.; Chang, C.-C.; Teng, S.-C.; Wu, K.-J. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef]
- Hao, S.; He, W.; Li, Y.; Ding, H.; Hou, Y.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Targeted inhibition of β-catenin/CBP signaling ameliorates renal interstitial fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, Y.; Specht, S.; Lunz, J.G.; Isse, K.; Corbitt, N.; Takizawa, T.; Demetris, A.J. Cooperation of p300 and PCAF in the control of microRNA 200c/141 transcription and epithelial characteristics. PLoS ONE 2012, 7, e32449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-Q.; Yan, F.-Q.; Wang, L.-H.; Yin, W.-J.; Chang, T.-Y.; Liu, J.-P.; Wu, K.-J. Identification of new hypoxia-regulated epithelial-mesenchymal transition marker genes labeled by H3K4 acetylation. Genes. Chromosomes Cancer 2020, 59, 73–83. [Google Scholar] [CrossRef]
- Yang, M.-H.; Hsu, D.S.-S.; Wang, H.-W.; Wang, H.-J.; Lan, H.-Y.; Yang, W.-H.; Huang, C.-H.; Kao, S.-Y.; Tzeng, C.-H.; Tai, S.-K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef]
- Dhasarathy, A.; Phadke, D.; Mav, D.; Shah, R.R.; Wade, P.A. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS ONE 2011, 6, e26514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor-Vazirani, P.; Kagey, J.D.; Powell, D.R.; Vertino, P.M. Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer Res. 2008, 68, 6810–6821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Højfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.E.; Ling, D.; Nelson, C.; Atmadibrata, B.; Dinger, M.E.; Xu, N.; Mizukami, T.; Liu, P.Y.; Liu, B.; Cheung, B.; et al. The histone demethylase JMJD1A induces cell migration and invasion by up-regulating the expression of the long noncoding RNA MALAT1. Oncotarget 2014, 5, 1793–1804. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Li, W.; Zang, W.; Liu, Z.; Xu, X.; Yu, H.; Yang, Q.; Jia, J. JMJD2B Promotes Epithelial—Mesenchymal Transition by Cooperating with β-Catenin and Enhances Gastric Cancer Metastasis. Clin. Cancer Res. 2013, 19, 6419–6429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkman, M.; Östling, P.; Härmä, V.; Virtanen, J.; Mpindi, J.-P.; Rantala, J.; Mirtti, T.; Vesterinen, T.; Lundin, M.; Sankila, A.; et al. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene 2012, 31, 3444–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadoss, S.; Chen, X.; Wang, C.-Y. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 44508–44517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, A.; Mourtzoukou, D.; Kosmidou, V.; Avlonitis, S.; Kontogeorgos, G.; Zografos, G.; Pintzas, A. EZH2 is regulated by ERK/AKT and targets integrin alpha2 gene to control Epithelial—Mesenchymal Transition and anoikis in colon cancer cells. Int. J. Biochem. Cell Biol. 2013, 45, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.-S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef] [Green Version]
- Vanharanta, S.; Shu, W.; Brenet, F.; Hakimi, A.A.; Heguy, A.; Viale, A.; Reuter, V.E.; Hsieh, J.J.-D.; Scandura, J.M.; Massagué, J. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat. Med. 2013, 19, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.-I.; Evers, B.M.; Zhou, B.P. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene 2013, 32, 1351–1362. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Kim, Y.; Bhin, J.; Shin, H.-J.R.; Nam, H.J.; Lee, S.H.; Yoon, J.-B.; Binda, O.; Gozani, O.; Hwang, D.; et al. Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc. Natl. Acad. Sci. USA 2011, 108, 13510–13515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, Inhibited Indirectly by LincRNA HOTAIR, Directly Inhibits SETDB1 and Reverses the EMT of Breast Cancer Stem Cells by Downregulating the STAT3 Pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Regina, C.; Compagnone, M.; Peschiaroli, A.; Lena, A.; Annicchiarico-Petruzzelli, M.; Piro, M.C.; Melino, G.; Candi, E. Setdb1, a novel interactor of ΔNp63, is involved in breast tumorigenesis. Oncotarget 2016, 7, 28836–28848. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.-C.; Lu, J.-W.; Yang, J.-Y.; Lin, I.-H.; Ou, D.-L.; Lin, Y.-H.; Chou, K.-H.; Huang, W.-F.; Wang, W.-P.; Huang, Y.-L.; et al. H3K9 Histone Methyltransferase, KMT1E/SETDB1, Cooperates with the SMAD2/3 Pathway to Suppress Lung Cancer Metastasis. Cancer Res. 2014, 74, 7333–7343. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Sun, L.; Li, Q.; Han, X.; Lei, L.; Zhang, H.; Shang, Y. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012, 31, 110–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Mizzen, C.A. The multiple facets of histone H4-lysine 20 methylation. Biochem. Cell Biol. 2009, 87, 151–161. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Shinchi, Y.; Hieda, M.; Nishioka, Y.; Matsumoto, A.; Yokoyama, Y.; Kimura, H.; Matsuura, S.; Matsuura, N. SUV420H2 suppresses breast cancer cell invasion through down regulation of the SH2 domain-containing focal adhesion protein tensin-3. Exp. Cell Res. 2015, 334, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tilló, E.; Lázaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [Green Version]
- Jordan, N.V.; Prat, A.; Abell, A.N.; Zawistowski, J.S.; Sciaky, N.; Karginova, O.A.; Zhou, B.; Golitz, B.T.; Perou, C.M.; Johnson, G.L. SWI/SNF chromatin-remodeling factor Smarcd3/Baf60c controls epithelial-mesenchymal transition by inducing Wnt5a signaling. Mol. Cell. Biol. 2013, 33, 3011–3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, G.; Chiba, T.; Aoba, T.; Imai, K. Epigenetic inactivation of E-cadherin by promoter hypermethylation in oral carcinoma cells. Odontology 2007, 95, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, M.K.; Misra, S.; Khedkar, S.V.; Hamilton, N.; Irvin-Wilson, C.; Sharan, C.; Sealy, L.; Chaudhuri, G. Regulation of BRCA2 Gene Expression by the SLUG Repressor Protein in Human Breast Cells. J. Biol. Chem. 2005, 280, 17163–17171. [Google Scholar] [CrossRef] [Green Version]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.-O.; Heidecke, C.-D.; Friess, H.; Büchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.-J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef]
- Carmona, F.J.; Davalos, V.; Vidal, E.; Gomez, A.; Heyn, H.; Hashimoto, Y.; Vizoso, M.; Martinez-Cardus, A.; Sayols, S.; Ferreira, H.J.; et al. A comprehensive DNA methylation profile of epithelial-to-mesenchymal transition. Cancer Res. 2014, 74, 5608–5619. [Google Scholar] [CrossRef] [Green Version]
- Fukagawa, A.; Ishii, H.; Miyazawa, K.; Saitoh, M. δEF1 associates with DNMT1 and maintains DNA methylation of the E-cadherin promoter in breast cancer cells. Cancer Med. 2015, 4, 125–135. [Google Scholar] [CrossRef]
- Papageorgis, P.; Lambert, A.W.; Ozturk, S.; Gao, F.; Pan, H.; Manne, U.; Alekseyev, Y.O.; Thiagalingam, A.; Abdolmaleky, H.M.; Lenburg, M.; et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res. 2010, 70, 968–978. [Google Scholar] [CrossRef] [Green Version]
- Davalos, V.; Moutinho, C.; Villanueva, A.; Boque, R.; Silva, P.; Carneiro, F.; Esteller, M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 2012, 31, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Vrba, L.; Jensen, T.J.; Garbe, J.C.; Heimark, R.L.; Cress, A.E.; Dickinson, S.; Stampfer, M.R.; Futscher, B.W. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS ONE 2010, 5, e8697. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, E.D.; Bramsen, J.B.; Hulf, T.; Dyrskjøt, L.; Ramanathan, R.; Hansen, T.B.; Villadsen, S.B.; Gao, S.; Ostenfeld, M.S.; Borre, M.; et al. Coordinated epigenetic repression of the miR-200 family and miR-205 in invasive bladder cancer. Int. J. Cancer 2011, 128, 1327–1334. [Google Scholar] [CrossRef]
- Sansom, O.J.; Maddison, K.; Clarke, A.R. Mechanisms of disease: Methyl-binding domain proteins as potential therapeutic targets in cancer. Nat. Clin. Pract. Oncol. 2007, 4, 305–315. [Google Scholar] [CrossRef]
- McCabe, M.T.; Brandes, J.C.; Vertino, P.M. Cancer DNA methylation: Molecular mechanisms and clinical implications. Clin. Cancer Res. 2009, 15, 3927–3937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Xia, H.; Ng, S.S.; Jiang, S.; Cheung, W.K.C.; Sze, J.; Bian, X.-W.; Kung, H.-F.; Lin, M.C. miR-200a-mediated downregulation of ZEB2 and CTNNB1 differentially inhibits nasopharyngeal carcinoma cell growth, migration and invasion. Biochem. Biophys. Res. Commun. 2010, 391, 535–541. [Google Scholar] [CrossRef]
- Varambally, S.; Cao, Q.; Mani, R.-S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef] [Green Version]
- Abba, M.L.; Patil, N.; Leupold, J.H.; Allgayer, H. MicroRNA Regulation of Epithelial to Mesenchymal Transition. J. Clin. Med. 2016, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Richards, E.J.; Zhang, G.; Li, Z.-P.; Permuth-Wey, J.; Challa, S.; Li, Y.; Kong, W.; Dan, S.; Bui, M.M.; Coppola, D.; et al. Long non-coding RNAs (LncRNA) regulated by transforming growth factor (TGF) β: LncRNA-hit-mediated TGFβ-induced epithelial to mesenchymal transition in mammary epithelia. J. Biol. Chem. 2015, 290, 6857–6867. [Google Scholar] [CrossRef] [Green Version]
- Marconi, A.; Atzei, P.; Panza, C.; Fila, C.; Tiberio, R.; Truzzi, F.; Wachter, T.; Leverkus, M.; Pincelli, C. FLICE/caspase-8 activation triggers anoikis induced by beta1-integrin blockade in human keratinocytes. J. Cell Sci. 2004, 117, 5815–5823. [Google Scholar] [CrossRef] [Green Version]
- Mawji, I.A.; Simpson, C.D.; Hurren, R.; Gronda, M.; Williams, M.A.; Filmus, J.; Jonkman, J.; Da Costa, R.S.; Wilson, B.C.; Thomas, M.P.; et al. Critical role for Fas-associated death domain-like interleukin-1-converting enzyme-like inhibitory protein in anoikis resistance and distant tumor formation. J. Natl. Cancer Inst. 2007, 99, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Asad, M.; Heong, V.; Wong, M.K.; Tan, T.Z.; Ye, J.; Kuay, K.T.; Thiery, J.P.; Scott, C.; Huang, R.Y.-J. The FZD7-TWIST1 axis is responsible for anoikis resistance and tumorigenesis in ovarian carcinoma. Mol. Oncol. 2019, 13, 757–780. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.A.; Kang, Y. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. 2013, 91, 411–429. [Google Scholar] [CrossRef]
- McDonald, B.; Spicer, J.; Giannais, B.; Fallavollita, L.; Brodt, P.; Ferri, L.E. Systemic inflammation increases cancer cell adhesion to hepatic sinusoids by neutrophil mediated mechanisms. Int. J. Cancer 2009, 125, 1298–1305. [Google Scholar] [CrossRef]
- Khatib, A.M.; Kontogiannea, M.; Fallavollita, L.; Jamison, B.; Meterissian, S.; Brodt, P. Rapid induction of cytokine and E-selectin expression in the liver in response to metastatic tumor cells. Cancer Res. 1999, 59, 1356–1361. [Google Scholar]
- Läubli, H.; Stevenson, J.L.; Varki, A.; Varki, N.M.; Borsig, L. L-Selectin Facilitation of Metastasis Involves Temporal Induction of Fut7-Dependent Ligands at Sites of Tumor Cell Arrest. Cancer Res. 2006, 66, 1536–1542. [Google Scholar] [CrossRef] [Green Version]
- Guan, R.J.; Ford, H.L.; Fu, Y.; Li, Y.; Shaw, L.M.; Pardee, A.B. Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Res. 2000, 60, 749–755. [Google Scholar]
- Bandyopadhyay, S.; Pai, S.K.; Hirota, S.; Hosobe, S.; Takano, Y.; Saito, K.; Piquemal, D.; Commes, T.; Watabe, M.; Gross, S.C.; et al. Role of the putative tumor metastasis suppressor gene Drg-1 in breast cancer progression. Oncogene 2004, 23, 5675–5681. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.-N.; Ding, W.-Q.; Guo, X.-J.; Yuan, X.-W.; Wang, D.-M.; Song, J.-G. Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat. Commun. 2015, 6, 8230. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Katsuno, Y.; Meyer, D.; Budi, E.H.; Chen, S.-H.; Koeppen, H.; Wang, H.; Akhurst, R.J.; Derynck, R. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition. EMBO Rep. 2018, 19, 135–155. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, Y.; Tamiya, T.; Takada, I.; Fukaya, T.; Sugiyama, Y.; Inoue, N.; Kimura, A.; Morita, R.; Kashiwagi, I.; Takimoto, T.; et al. Histone 3 lysine 9 (H3K9) methyltransferase recruitment to the interleukin-2 (IL-2) promoter is a mechanism of suppression of IL-2 transcription by the transforming growth factor-β-smad pathway. J. Biol. Chem. 2011, 286, 35456–35465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Sun, H.; Sun, W.-J.; Bao, H.-B.; Si, S.-H.; Fan, J.-L.; Lin, P.; Cui, R.-J.; Pan, Y.-J.; Wen, S.-M.; et al. Role of RbBP5 and H3K4me3 in the vicinity of Snail transcription start site during epithelial-mesenchymal transition in prostate cancer cell. Oncotarget 2016, 7, 65553–65567. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-W.; Hua, K.-T.; Kao, H.-J.; Chi, C.-C.; Wei, L.-H.; Johansson, G.; Shiah, S.-G.; Chen, P.-S.; Jeng, Y.-M.; Cheng, T.-Y.; et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lorton, B.; Gupta, V.; Shechter, D. A TGFβ-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 2017, 36, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.-C.; Lee, C.-F.; Li, Y.-S.; Chen, Y.-R.; Hsiao, P.-W.; Chan, M.-Y.; Lin, F.-M.; Huang, H.-D.; Chen, Y.-T.; Jeng, Y.-M.; et al. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013, 73, 4711–4721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M.; Galaine, J.; Guenat, D.; Mougey, V.; Perrard, J.; et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2018, 7, e1423170. [Google Scholar] [CrossRef] [Green Version]
- Messier, T.L.; Gordon, J.A.R.; Boyd, J.R.; Tye, C.E.; Browne, G.; Stein, J.L.; Lian, J.B.; Stein, G.S. Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 2016, 7, 5094–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judes, G.; Dubois, L.; Rifaï, K.; Idrissou, M.; Mishellany, F.; Pajon, A.; Besse, S.; Daures, M.; Degoul, F.; Bignon, Y.-J.; et al. TIP60: An actor in acetylation of H3K4 and tumor development in breast cancer. Epigenomics 2018, 10, 1415–1430. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-T.; Liu, K.-Y.; Jeng, W.-Y.; Chiang, C.-M.; Chai, C.-Y.; Chiou, S.-S.; Huang, M.-S.; Yokoyama, K.K.; Wang, S.-N.; Huang, S.-K.; et al. PCAF-mediated acetylation of ISX recruits BRD4 to promote epithelial-mesenchymal transition. EMBO Rep. 2020, 21, e48795. [Google Scholar] [CrossRef]
- Cho, M.-H.; Park, J.-H.; Choi, H.-J.; Park, M.-K.; Won, H.-Y.; Park, Y.-J.; Lee, C.H.; Oh, S.-H.; Song, Y.-S.; Kim, H.S.; et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun. 2015, 6, 7821. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Ding, S.; Huang, H.; Luo, P.; Qing, B.; Zhang, S.; Tang, R. HDAC1 triggers the proliferation and migration of breast cancer cells via upregulation of interleukin-8. Biol. Chem. 2017, 398, 1347–1356. [Google Scholar] [CrossRef]
- Karamouzis, M.V.; Badra, F.A.; Papavassiliou, A.G. Breast cancer: The upgraded role of HER-3 and HER-4. Int. J. Biochem. Cell Biol. 2007, 39, 851–856. [Google Scholar] [CrossRef]
- Kwak, S.-M.; Seo, J.; Hwang, J.-T.; Sung, G.-J.; Song, J.-H.; Jeong, J.-H.; Lee, S.-H.; Yoon, H.-G.; Choi, H.-K.; Choi, K.-C. EGFR-c-Src-Mediated HDAC3 Phosphorylation Exacerbates Invasion of Breast Cancer Cells. Cells 2019, 8, 930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Li, G.; Su, F.; Cai, Y.; Shi, L.; Meng, Y.; Liu, Z.; Sun, J.; Wang, M.; Qian, M.; et al. HDAC8 cooperates with SMAD3/4 complex to suppress SIRT7 and promote cell survival and migration. Nucleic Acids Res. 2020, 48, 2912–2923. [Google Scholar] [CrossRef]
- Li, Z.; Hou, P.; Fan, D.; Dong, M.; Ma, M.; Li, H.; Yao, R.; Li, Y.; Wang, G.; Geng, P.; et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2017, 24, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Geng, P.; Zhang, Y.; Liu, X.; Zhang, N.; Liu, Y.; Liu, X.; Lin, C.; Yan, X.; Li, Z.; Wang, G.; et al. Automethylation of protein arginine methyltransferase 7 and its impact on breast cancer progression. FASEB J. 2017, 31, 2287–2300. [Google Scholar] [CrossRef] [Green Version]
- Nakakido, M.; Deng, Z.; Suzuki, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. PRMT6 increases cytoplasmic localization of p21CDKN1A in cancer cells through arginine methylation and makes more resistant to cytotoxic agents. Oncotarget 2015, 6, 30957–30967. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhou, Z.; Jin, S.; Xu, K.; Zhang, H.; Xu, J.; Sun, Q.; Wang, J.; Xu, J. PRMT9 promotes hepatocellular carcinoma invasion and metastasis via activating PI3K/Akt/GSK-3β/Snail signaling. Cancer Sci. 2018, 109, 1414–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Liu, Z.; Cheung, W.K.C.; Zhao, M.; Chen, S.Y.; Chan, S.W.; Booth, C.J.; Nguyen, D.X.; Yan, Q. Histone demethylase RBP2 is critical for breast cancer progression and metastasis. Cell Rep. 2014, 6, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Enkhbaatar, Z.; Terashima, M.; Oktyabri, D.; Tange, S.; Ishimura, A.; Yano, S.; Suzuki, T. KDM5B histone demethylase controls epithelial-mesenchymal transition of cancer cells by regulating the expression of the microRNA-200 family. Cell Cycle 2013, 12, 2100–2112. [Google Scholar] [CrossRef]
- Tang, B.; Qi, G.; Tang, F.; Yuan, S.; Wang, Z.; Liang, X.; Li, B.; Yu, S.; Liu, J.; Huang, Q.; et al. JARID1B promotes metastasis and epithelial-mesenchymal transition via PTEN/AKT signaling in hepatocellular carcinoma cells. Oncotarget 2015, 6, 12723–12739. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Shi, L.; Gui, B.; Yu, W.; Wang, J.; Zhang, D.; Han, X.; Yao, Z.; Shang, Y. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 2011, 71, 6899–6908. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Sharma, A.; Dhar, S.S.; Lee, S.-H.; Gu, B.; Chan, C.-H.; Lin, H.-K.; Lee, M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014, 74, 1705–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.-J.; Park, J.-H.; Park, M.; Won, H.-Y.; Joo, H.-S.; Lee, C.H.; Lee, J.-Y.; Kong, G. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO Rep. 2015, 16, 1288–1298. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Shi, J.; Li, W.; Tanaka, K.; Allton, K.L.; Richardson, D.; Li, J.; Franco, H.L.; Nagari, A.; Malladi, V.S.; et al. Histone modification profiling in breast cancer cell lines highlights commonalities and differences among subtypes. BMC Genom. 2018, 19, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsli-Ceppioglu, S.; Dagdemir, A.; Judes, G.; Lebert, A.; Penault-Llorca, F.; Bignon, Y.-J.; Bernard-Gallon, D. The Epigenetic Landscape of Promoter Genome-wide Analysis in Breast Cancer. Sci. Rep. 2017, 7, 6597. [Google Scholar] [CrossRef]
- Healey, M.A.; Hu, R.; Beck, A.H.; Collins, L.C.; Schnitt, S.J.; Tamimi, R.M.; Hazra, A. Association of H3K9me3 and H3K27me3 repressive histone marks with breast cancer subtypes in the Nurses’ Health Study. Breast Cancer Res. Treat. 2014, 147, 639–651. [Google Scholar] [CrossRef]
- Hirukawa, A.; Smith, H.W.; Zuo, D.; Dufour, C.R.; Savage, P.; Bertos, N.; Johnson, R.M.; Bui, T.; Bourque, G.; Basik, M.; et al. Targeting EZH2 reactivates a breast cancer subtype-specific anti-metastatic transcriptional program. Nat. Commun. 2018, 9, 2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifaï, K.; Judes, G.; Idrissou, M.; Daures, M.; Bignon, Y.-J.; Penault-Llorca, F.; Bernard-Gallon, D. SIRT1-dependent epigenetic regulation of H3 and H4 histone acetylation in human breast cancer. Oncotarget 2018, 9, 30661–30678. [Google Scholar] [CrossRef]
- Hu, Y.; Zheng, Y.; Dai, M.; Wang, X.; Wu, J.; Yu, B.; Zhang, H.; Cui, Y.; Kong, W.; Wu, H.; et al. G9a and histone deacetylases are crucial for Snail2-mediated E-cadherin repression and metastasis in hepatocellular carcinoma. Cancer Sci. 2019, 110, 3442–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natoni, F.; Diolordi, L.; Santoni, C.; Montani, M.S.G. Sodium butyrate sensitises human pancreatic cancer cells to both the intrinsic and the extrinsic apoptotic pathways. Biochim. Biophys. Acta Mol. Cell Res. 2005, 1745, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, Q.; Shang, K.; Zhang, J.; Chuai, S.; Kong, D.; Zhou, T.; Fu, S.; Liang, Y.; Li, C.; Chen, Z.; et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat. Commun. 2015, 6, 8651. [Google Scholar] [CrossRef] [Green Version]
- Xia, R.; Jin, F.; Lu, K.; Wan, L.; Xie, M.; Xu, T.; De, W.; Wang, Z. SUZ12 promotes gastric cancer cell proliferation and metastasis by regulating KLF2 and E-cadherin. Tumor Biol. 2015, 36, 5341–5351. [Google Scholar] [CrossRef]
- Malouf, G.G.; Taube, J.H.; Lu, Y.; Roysarkar, T.; Panjarian, S.; Estecio, M.R.; Jelinek, J.; Yamazaki, J.; Raynal, N.J.-M.; Long, H.; et al. Architecture of epigenetic reprogramming following Twist1-mediated epithelial-mesenchymal transition. Genome Biol. 2013, 14, R144. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Thompson, E.W.; Williams, E.D. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs 2007, 185, 7–19. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neureiter, D.; Herold, C.; Ocker, M. Gastrointestinal Cancer—Only a Deregulation of Stem Cell Differentiation? Int. J. Mol. Med. 2006, 17, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, J.; Nasarre, P.; Gemmill, R.; Baldys, A.; Pontis, J.; Korch, C.; Guilhot, J.; Ait-Si-Ali, S.; Drabkin, H. Global Decrease of Histone H3K27 Acetylation in ZEB1-Induced Epithelial to Mesenchymal Transition in Lung Cancer Cells. Cancers 2013, 5, 334–356. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.-P.; Chua, K.; Sim, W.J.; Huang, R. Epithelial mesenchymal transition during development in fibrosis and in the progression of carcinoma. Bull. Cancer 2010, 97, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Zhou, J.; Ye, F.; Xiong, H.; Peng, L.; Zheng, Z.; Xu, F.; Cui, M.; Wei, C.; Wang, X.; et al. BRD4 inhibitor inhibits colorectal cancer growth and metastasis. Int. J. Mol. Sci. 2015, 16, 1928–1948. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [Green Version]
- James, L.I.; Barsyte-Lovejoy, D.; Zhong, N.; Krichevsky, L.; Korboukh, V.K.; Herold, J.M.; MacNevin, C.J.; Norris, J.L.; Sagum, C.A.; Tempel, W.; et al. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat. Chem. Biol. 2013, 9, 184–191. [Google Scholar] [CrossRef]
- Stuckey, J.I.; Dickson, B.M.; Cheng, N.; Liu, Y.; Norris, J.L.; Cholensky, S.H.; Tempel, W.; Qin, S.; Huber, K.G.; Sagum, C.; et al. A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat. Chem. Biol. 2016, 12, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Zeng, J.; Wang, L.; Shen, L.; Ma, X.; Li, S.; Wu, Y.; Ma, L.; Ci, X.; Guo, Q.; et al. Histone demethylase RBP2 promotes malignant progression of gastric cancer through TGF-β1-(p-Smad3)-RBP2-E-cadherin-Smad3 feedback circuit. Oncotarget 2015, 6, 17661–17674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketola, K.; Hilvo, M.; Hyötyläinen, T.; Vuoristo, A.; Ruskeepää, A.-L.; Orešič, M.; Kallioniemi, O.; Iljin, K. Salinomycin inhibits prostate cancer growth and migration via induction of oxidative stress. Br. J. Cancer 2012, 106, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y. Effects of salinomycin on cancer stem cell in human lung adenocarcinoma A549 cells. Med. Chem. 2011, 7, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Yao, H.; Liu, Q.; Chen, J.; Shi, J.; Su, F.; Song, E. Markers of tumor-initiating cells predict chemoresistance in breast cancer. PLoS ONE 2010, 5, e15630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardsley, M.R.; Horváth, V.J.; Asuzu, D.T.; Lorincz, A.; Redelman, D.; Hayashi, Y.; Popko, L.N.; Young, D.L.; Lomberk, G.A.; Urrutia, R.A.; et al. Kitlow stem cells cause resistance to Kit/platelet-derived growth factor alpha inhibitors in murine gastrointestinal stromal tumors. Gastroenterology 2010, 139, 942–952. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, D.; Daniel, V.; Sadeghi, M.; Opelz, G.; Naujokat, C. Salinomycin overcomes ABC transporter-mediated multidrug and apoptosis resistance in human leukemia stem cell-like KG-1a cells. Biochem. Biophys. Res. Commun. 2010, 394, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, T.; Pichler, M.; Neureiter, D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol. Clin. Oncol. 2013, 1, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Stintzing, S.; Kemmerling, R.; Kiesslich, T.; Alinger, B.; Ocker, M.; Neureiter, D. Myelodysplastic syndrome and histone deacetylase inhibitors: “To be or not to be acetylated”? J. Biomed. Biotechnol. 2011, 2011, 214143. [Google Scholar] [CrossRef]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [Green Version]
- Batty, N.; Malouf, G.G.; Issa, J.P.J. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett. 2009, 280, 192–200. [Google Scholar] [CrossRef]
- Di Fazio, P.; Schneider-Stock, R.; Neureiter, D.; Okamoto, K.; Wissniowski, T.; Gahr, S.; Quint, K.; Meissnitzer, M.; Alinger, B.; Montalbano, R.; et al. The pan-deacetylase inhibitor panobinostat inhibits growth of hepatocellular carcinoma models by alternative pathways of apoptosis. Cell. Oncol. 2010, 32, 285–300. [Google Scholar] [CrossRef]
- Di Fazio, P.; Montalbano, R.; Quint, K.; Alinger, B.; Kemmerling, R.; Kiesslich, T.; Ocker, M.; Neureiter, D. The pan-deacetylase inhibitor panobinostat modulates the expression of epithelial-mesenchymal transition markers in hepatocellular carcinoma models. Oncol. Lett. 2013, 5, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Neureiter, D.; Zopf, S.; Leu, T.; Dietze, O.; Hauser-Kronberger, C.; Hahn, E.G.; Herold, C.; Ocker, M. Apoptosis, proliferation and differentiation patterns are influenced by Zebularine and SAHA in pancreatic cancer models. Scand. J. Gastroenterol. 2007, 42, 103–116. [Google Scholar] [CrossRef]
- Takai, N.; Desmond, J.C.; Kumagai, T.; Gui, D.; Said, J.W.; Whittaker, S.; Miyakawa, I.; Koeffler, H.P. Histone Deacetylase Inhibitors Have a Profound Antigrowth Activity in Endometrial Cancer Cells. Clin. Cancer Res. 2004, 10, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.-T.; Mock, K.; Ruh, M.; Schüler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Zhang, J.; Wu, M.; Zhang, Y. A Dynamic 3D Tumor Spheroid Chip Enables More Accurate Nanomedicine Uptake Evaluation. Adv. Sci. 2019, 6, 1901462. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Cho, S.K. Targeting miRNAs by histone deacetylase inhibitors (HDACi): Rationalizing epigenetics-based therapies for breast cancer. Pharmacol. Ther. 2020, 206, 107437. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Rudek, M.A.; Piekarz, R. Entinostat: A promising treatment option for patients with advanced breast cancer. Future Oncol. 2017, 13, 1137–1148. [Google Scholar] [CrossRef]
- Yeruva, S.L.H.; Zhao, F.; Miller, K.D.; Tevaarwerk, A.J.; Wagner, L.I.; Gray, R.J.; Sparano, J.A.; Connolly, R.M. E2112: Randomized phase III trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. NPJ Breast Cancer 2018, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-C.M.; Dowhan, D.H.; Muscat, G.E.O. Epigenetic arginine methylation in breast cancer: Emerging therapeutic strategies. J. Mol. Endocrinol. 2019, 62, R223–R237. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Davies, C.C. Linking PRMT5 to breast cancer stem cells: New therapeutic opportunities? Mol. Cell. Oncol. 2018, 5, e1441628. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Zhong, J.; Liu, J.; Liu, K.; Zhao, J.; Xu, T.; Zeng, T.; Li, Z.; Chen, Y.; Ding, W.; et al. Protein arginine N-methyltransferase 2 reverses tamoxifen resistance in breast cancer cells through suppression of ER-α36. Oncol. Rep. 2018, 39, 2604–2612. [Google Scholar] [CrossRef]
- Hiken, J.F.; McDonald, J.I.; Decker, K.F.; Sanchez, C.; Hoog, J.; VanderKraats, N.D.; Jung, K.L.; Akinhanmi, M.; Rois, L.E.; Ellis, M.J.; et al. Epigenetic activation of the prostaglandin receptor EP4 promotes resistance to endocrine therapy for breast cancer. Oncogene 2017, 36, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Q.; Wu, K.-J. Epigenetic regulation of epithelial-mesenchymal transition by hypoxia in cancer: Targets and therapy. Curr. Pharm. Des. 2015, 21, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Sidoli, S.; Kulej, K.; Ross, K.; Wu, C.H.; Garcia, B.A. Coordination between TGF-β cellular signaling and epigenetic regulation during epithelial to mesenchymal transition. Epigenet. Chromatin 2019, 12, 11. [Google Scholar] [CrossRef]
- Open-Label, Multicenter, Phase 1/2 Study of Tazemetostat (EZH2 Histone Methyl Transferase [HMT] Inhibitor) as a Single Agent in Subjects with Adv. Solid Tumors or with B-cell Lymphomas and Tazemetostat in Combination with Prednisolone in Subjects with DLBCL. Available online: https://clinicaltrials.gov/ct2/show/NCT01897571 (accessed on 21 January 2021).
- A Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics and Clinical Activity of GSK2816126 in Subjects with Relapsed/Refractory Diffuse Large B Cell Lymphoma, Transformed Follicular Lymphoma, Other Non-Hodgkin’s Lymphomas, Solid Tumors and Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT02082977 (accessed on 21 January 2021).
- Greiner, D.; Bonaldi, T.; Eskeland, R.; Roemer, E.; Imhof, A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat. Chem. Biol. 2005, 1, 143–145. [Google Scholar] [CrossRef]
- Lakshmikuttyamma, A.; Scott, S.A.; DeCoteau, J.F.; Geyer, C.R. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene 2010, 29, 576–588. [Google Scholar] [CrossRef] [Green Version]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Zhang, X.; Horton, J.R.; Upadhyay, A.K.; Spannhoff, A.; Liu, J.; Snyder, J.P.; Bedford, M.T.; Cheng, X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 2009, 16, 312–317. [Google Scholar] [CrossRef]
- Pan, M.-R.; Hsu, M.-C.; Chen, L.-T.; Hung, W.-C. G9a orchestrates PCL3 and KDM7A to promote histone H3K27 methylation. Sci. Rep. 2015, 5, 18709. [Google Scholar] [CrossRef] [Green Version]
- Ferrari-Amorotti, G.; Fragliasso, V.; Esteki, R.; Prudente, Z.; Soliera, A.R.; Cattelani, S.; Manzotti, G.; Grisendi, G.; Dominici, M.; Pieraccioli, M.; et al. Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer Res. 2013, 73, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.-I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Lv, T.; Yuan, D.; Miao, X.; Lv, Y.; Zhan, P.; Shen, X.; Song, Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE 2012, 7, e35065. [Google Scholar] [CrossRef] [Green Version]
- Investigation of GSK2879552 in Subjects with Relapsed/Refractory Small Cell Lung Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02034123 (accessed on 21 January 2021).
- Maes, T.; Tirapu, I.; Mascaró, C.; Ortega, A.; Estiarte, A.; Valls, N.; Castro-Palomino, J.; Buesa Arjol, C.; Kurz, G. Preclinical characterization of a potent and selective inhibitor of the histone demethylase KDM1A for MLL leukemia. J. Clin. Oncol. 2013, 31, e13543. [Google Scholar] [CrossRef]
- Ryningen, A.; Stapnes, C.; Bruserud, Ø. Clonogenic acute myelogenous leukemia cells are heterogeneous with regard to regulation of differentiation and effect of epigenetic pharmacological targeting. Leuk. Res. 2007, 31, 1303–1313. [Google Scholar] [CrossRef]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Jones, P.A.; Taylor, S.M.; Wilson, V. DNA modification, differentiation, and transformation. J. Exp. Zool. 1983, 228, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.-J.; Kahata, K.; Idås, O.; Thuault, S.; Heldin, C.-H.; Moustakas, A. The high mobility group A2 protein epigenetically silences the Cdh1 gene during epithelial-to-mesenchymal transition. Nucleic Acids Res. 2015, 43, 162–178. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.-S.; Ino, Y.; Kanai, Y.; Sakamoto, M.; Hirohashi, S. 5-aza-2′-deoxycytidine restores the E-cadherin system in E-cadherin-silenced cancer cells and reduces cancer metastasis. Clin. Exp. Metastasis 2004, 21, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, T.; Zouridis, H.; Wu, Y.; Cheng, L.L.; Tan, I.B.; Gopalakrishnan, V.; Ooi, C.H.; Lee, J.; Qin, L.; Wu, J.; et al. Integrated epigenomics identifies BMP4 as a modulator of cisplatin sensitivity in gastric cancer. Gut 2013, 62, 22–33. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Wu, K.-J. Epigenetic regulation of epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β signaling. J. Biomed. Sci. 2020, 27, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, M.; Mattheolabakis, G.; Suresh, M.; Amiji, M. Reversing epigenetic mechanisms of drug resistance in solid tumors using targeted microRNA delivery. Expert Opin. Drug Deliv. 2016, 13, 987–998. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markouli, M.; Strepkos, D.; Basdra, E.K.; Papavassiliou, A.G.; Piperi, C. Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis. Int. J. Mol. Sci. 2021, 22, 2778. https://doi.org/10.3390/ijms22052778
Markouli M, Strepkos D, Basdra EK, Papavassiliou AG, Piperi C. Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis. International Journal of Molecular Sciences. 2021; 22(5):2778. https://doi.org/10.3390/ijms22052778
Chicago/Turabian StyleMarkouli, Mariam, Dimitrios Strepkos, Efthimia K. Basdra, Athanasios G. Papavassiliou, and Christina Piperi. 2021. "Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis" International Journal of Molecular Sciences 22, no. 5: 2778. https://doi.org/10.3390/ijms22052778
APA StyleMarkouli, M., Strepkos, D., Basdra, E. K., Papavassiliou, A. G., & Piperi, C. (2021). Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis. International Journal of Molecular Sciences, 22(5), 2778. https://doi.org/10.3390/ijms22052778