1. Introduction
Under physiological conditions, bone remodelling is a continuous process that is achieved by the balanced activity of bone-forming osteoblasts and bone-resorbing osteoclasts. Disturbed bone homeostasis can be caused by aging and a reduction in oestrogen or androgen levels, chronic inflammation due to auto-immune pathologies or chronic bacterial infections, or as a consequence of the spread of cancer metastases to the bone environment. The latter is a frequent complication in multiple myeloma, a B cell lymphoma, but more typically found with solid tumours from breast, lung, prostate or thyroid cancers, renal carcinoma, as well as in melanoma, gastrointestinal tumours, and head and neck cancers [
1]. Under physiological conditions, the cytokines M-CSF and RANKL are sufficient to differentiate osteoclast precursors from the monocyte/macrophage lineage to osteoclasts. However, tumour-derived factors, such as the proinflammatory cytokines IL-1β, IL-6, TNF-α or IL-17 can enhance osteoclast formation and thus cause osteolysis, i.e., the pathological destruction of bone material [
2]. Patients suffer from severe pain as well as fractures, hypercalcaemia and spinal cord compression, all of which can reduce quality of life significantly [
3].
Cancer is now the second leading cause of death and despite the enormous progress in therapy options to treat bone cancer, there is still a need for new drugs that are able to overcome resistance of cancer cells that often arises during treatment [
4].
In an attempt to find new lead structures, plant-derived molecules, so-called phytochemicals, are studied to develop novel therapeutic tools with decreased cytotoxicity, better drug resistance and high target-specificity. Several phytochemicals show cytotoxicity against cancer cells and are already used clinically, i.e., taxol/paclitaxel from
Taxus brevifolia that kills tumour cells by inducing multipolar divisions when the cell enters mitosis [
5] or vincristine from
Cantharantus roseus that was approved as the first plant-derived cancer treatment by the FDA [
6]. Mechanistically, the effects of phytochemicals range from targeting mitochondrial activity, the induction of ROS production, the inhibition of ABC transporters that confer drug resistance, the inhibition of the proto-oncogene p53 or the induction of programmed cell death through the activation of caspases [
4].
Naphthoquinones have a naphthalene-derived structure that is for example used by Vitamin K. The plant-derived naphthoquinone plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) is a bioactive compound that was originally isolated from the roots of
Plumbago zeylanica. Plumbagin is also found in other members of the
Plumbaginaceae family (
Plumbago capensis,
Plumbago rosea, Plumbago indica), as well as in other plants such as the members of
Droseraceae and
Juglandaceae families. This naturally occurring naphthoquinone has been in medical use in eastern cultures against infectious diseases, rheumatoid arthritis, dermatological diseases and parasitic infections, respectively, because of its antimicrobial and anti-inflammatory properties. Plumbagin has gained attention in recent cancer studies and encouraging results have been reported in cases of several types of cancer including lung, prostate, breast, cervical, colorectal cancer, leukaemia and melanoma [
7]. Each of these investigations of the anti-cancer effects of plumbagin revealed an inhibitory role in multiple mitogenic signalling pathways such as NF-kB, STAT3 and AKT among others [
7]. However, to date there has been no study that addresses the detailed mechanism of its activity. Therefore, the signalling molecules known to be targets of plumbagin mostly reflect the central molecules of the respective disease model, but do not give a comprehensive overview on plumbagin-mediated effects. Like other naphthoquinones, plumbagin activates cellular signalling through redox reactions, the inhibition of cellular phosphatases and through direct protein alkylation [
8]. In cancer cells, plumbagin mainly acts through the induction of apoptosis via the upregulation of ROS [
9,
10] and cell cycle arrest through the induction of DNA strand breaks [
7,
11]. This apoptotic function is supported by other molecular mechanisms that ultimately inhibit tumour growth and metastasis. Due to these encouraging findings, it was also tested whether plumbagin affected cancer-mediated osteoclastogenesis [
12,
13]. The data suggest that plumbagin helps to improve osteolytic lesions in bone cancer models due to an inhibitory effect on RANKL signalling. The exact mechanism, however, is unclear.
Many investigations that addressed anti-inflammatory effects have been carried out using cancer cell lines or the macrophage cell line RAW264.7. Studies on the effect of plumbagin on M-CSF/RANKL-mediated osteoclastogenesis using primary macrophages from C57BL/6 mice, the most frequently used strain for knock-out models, have not been performed. To better understand the effect of plumbagin on RANKL-mediated differentiation of macrophages into osteoclasts, we used bone marrow-derived macrophages (BMDM) from C57BL/6 mice and stimulation with M-CSF and RANKL, co-stimulation with plumbagin or pre-stimulation with plumbagin before M-CSF/RANKL treatment. Surprisingly, in contrast to the described data, plumbagin did not inhibit the formation of multinucleated, mature osteoclasts. Particularly pre-stimulation with plumbagin showed enhanced osteoclastogenesis compared to M-CSF/RANKL alone. This was accompanied by a fast activation of NF-kB and NFATc1 signalling as well as an upregulated expression of osteoclastic markers. Our data show that plumbagin strongly increases the translational activity of the cell and that pre-incubation resulted in the enhanced surface expression of RANK. We suggest that this increased receptor expression and the general abundance of cellular proteins allow RANKL to signal more efficiently after plumbagin treatment, so that osteoclastogenesis is ultimately enhanced. However, when we performed these experiments using the Balb/c-derived murine macrophage cell line RAW264.7 or osteoclast precursors obtained from Balb/c mice we failed to corroborate the osteoclastic effect of plumbagin. Instead, plumbagin inhibited cell proliferation as well as osteoclast differentiation. The genetic background of mice is known to shape their adaptive and innate immune response [
14] and this might explain the dual effect of plumbagin. The ability of plumbagin to exert such divergent activities underlines the need for the further exploration of this molecule. At the same time, the use in an organismal context necessitates the definition of the exact cellular mechanisms involved. Our findings highlight that the use of culture conditions and the genetic background of the animals or the cell lines need to be considered in pharmacological studies as they can drastically affect the outcome and conclusions obtained from experiments.
3. Discussion
Plant-derived phytochemicals contain active substances that can be exploited pharmacologically. These chemicals are often secondary metabolites that are generated by the plant to defend itself against external challenges [
23]. Phytotherapy aims to exploit the potential of such drugs as alternative treatment options in cancer and other non-communicable diseases. Several phytochemicals are successfully being used in the clinical context, showing the relevance of this approach. The plant-derived naphthoquinone plumbagin is a bioactive compound that was originally isolated from the roots of the
Plumbago genus [
24]. Because plumbagin is an effective mediator of ROS production in a variety of different cells, the focus of research has been on its anti-cancer properties [
10]. The first report that addressed the modulatory abilities of plumbagin on macrophages was published in 1995 and suggested a potentiation of bactericidal activity [
25]. Balb/c mice have been fed with plumbagin for 6 weeks, before peritoneal macrophages were isolated and subsequently infected with
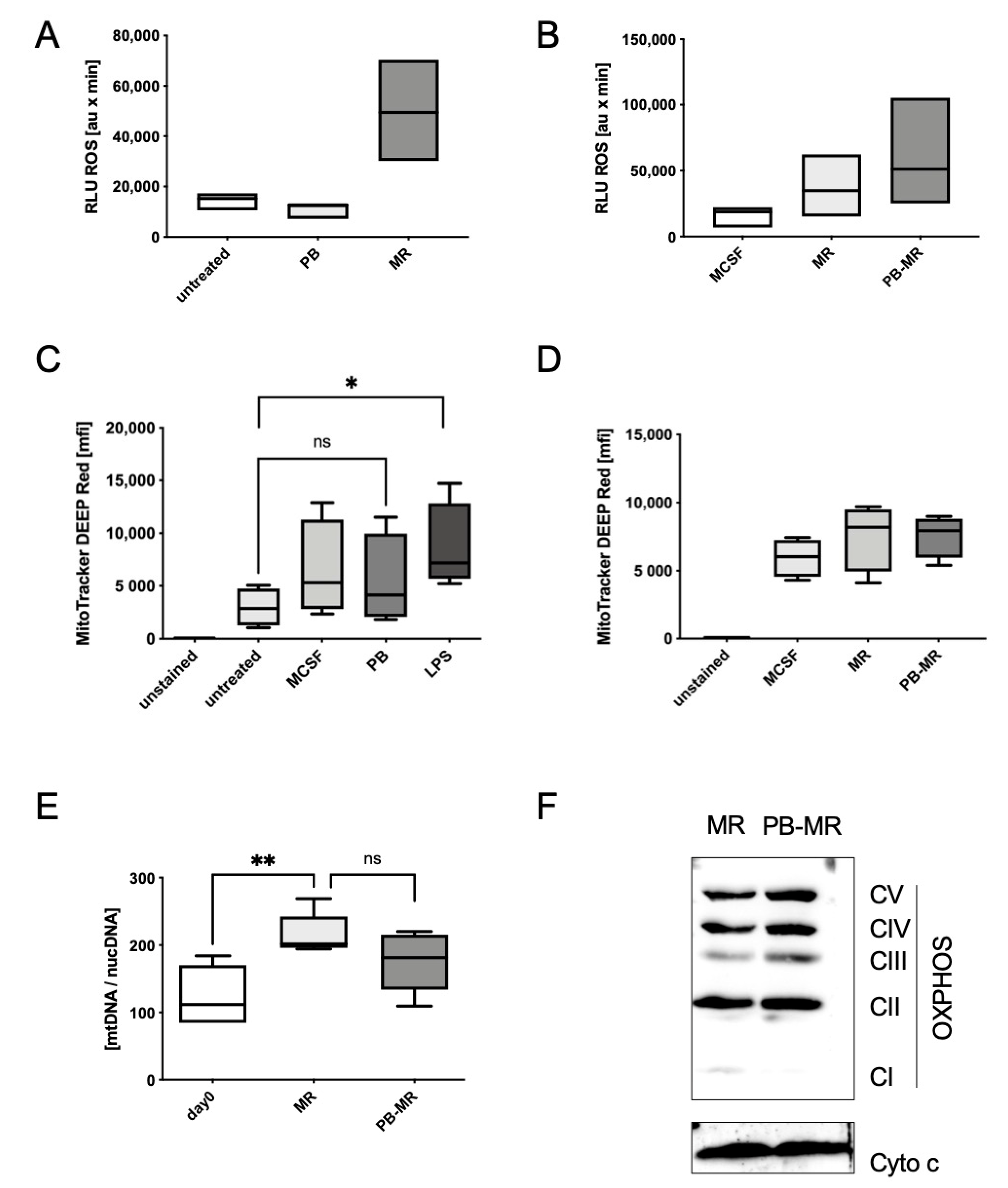
Staphylococcus aureus. The data showed a dose-dependent increase in bacterial killing in response to the plumbagin supplementation of the food. While the paper could confirm a significantly higher release of various ROS, it was not investigated if this was supported by a concomitant increase in NF-kB activity and cytokine production. Although our own data do not provide evidence for a direct effect of plumbagin on ROS production, plumbagin was still able to moderately increase ROS levels in the presence of the ROS inducer M-CSF/RANKL (
Figure 3A,B). Indeed, the ability to produce ROS can be expected to be different between cells. Cancer cells already show a predisposition to produce more ROS, which ultimately renders them more sensitive towards additional ROS [
26]. While innate immune cells are potent producers of ROS as part of their microbial killing mechanisms, differences between the cells can still be found and granulocytes or peritoneal macrophages are more likely to produce ROS than BMDMs [
27,
28].
Studies that investigated the effect of plumbagin on cytokine production describe a downregulation of NF-kB activation and a decrease in the release of pro-inflammatory cytokines [
29,
30,
31,
32,
33]. To our surprise, we found that plumbagin rapidly induces the phosphorylation of p65. In addition, our own data convincingly show that plumbagin prestimulation enhances LPS-mediated NF-kB activation and increases the production of IL-6, IL-1β and TNF-α. The increased production of cytokines fits well with our finding of an increase in p65 activation in response to plumbagin treatment that might cause an enhanced ability of the cells to react to further stimuli such as LPS or M-CSF/RANKL that both make use of the same pathway. Indeed, it would be astonishing for an inflammatory model if ROS levels increased while the production of cytokines would decrease. As mitochondrial ROS levels and the expression of pro-inflammatory cytokines are coupled [
34], it can be expected that innate immune cells that produce increased levels of ROS would also increase cytokine production. A possible explanation is that the co-stimulation of plumbagin with high doses of LPS (1 µg/mL) in combination could reduce cell viability, e.g., through an increase in ROS, and would thus cause a reduction in inflammation [
29,
31,
32]. Zhang et al. used a Balb/c macrophage model that was the closest to our own C57BL/6 model system to investigate the effect of plumbagin on immunometabolism. Again, it was claimed that plumbagin inhibits IL-1β production, however, supernatants were measured after 2 h of stimulation, which is different from the 24 h of pre-stimulation and 6 h of LPS stimulation used in this manuscript. The authors observe an increase in the oxygen consumption rate of the BDMDs, while the extracellular acidification rate, a sign of aerobic glycolysis, was reduced [
33]. Interestingly, we could observe that plumbagin has the ability to increase the expression of the electron transport chain proteins during osteoclastogenesis. It is possible that this also plays a role during inflammation. Still, our data show a pronounced early increase in mTOR-mediated anabolic pathways and protein translation as we found them during glycolysis under inflammatory conditions. mTOR also plays a central role in osteoclastogenesis under physiological and pathological conditions [
35,
36]. Our data show that protein translation is increased through plumbagin and we have evidence that RANK is among these proteins. The increase in RANK surface localisation is measurable without any other cytokine present that would sustain cell survival and thus must be quite strong. The effect is therefore most pronounced when cells are treated with plumbagin and M-CSF/RANKL. Although RANKL and M-CSF are usually described as the critical factors that determine if osteoclast differentiation takes place, the expression of the receptor RANK is also of importance. A couple of recent publications highlight the fact that the regulation of RANK expression is an effective means to change the effectiveness of osteoclast differentiation [
37,
38,
39]. Indeed, RANK expression on macrophages is quite low and it is therefore not surprising that RANK abundance can be a limiting factor in osteoclastogenesis. For example, di Ceglie et al. recently (2019) described that the endogenous DAMP and marker for sterile inflammation in rheumatoid arthritis, the alarmin S100A9, reduced M-CSF-induced RANK expression to limit osteoclastogenesis [
39].
At this point, we do not know how signalling pathways are activated by plumbagin and what the central regulator of plumbagin-transduced signals is, as we specifically looked for signalling molecules that are important during osteoclastogenesis. Using the swiss target prediction tool, we verified some of the already published targets [
40], but more work is needed to understand how plumbagin activates specific signalling events in these cells. While there are numerous experimental set-ups that looked at signalling molecules that might be relevant for their respective experimental system, a comprehensive study that identifies the mode of activation is missing. Predictive software suggests a number of quite different molecules; however, the predictions are based on a similarity search and ultimately rely on the experimental data that have been obtained for plumbagin or similar naphthoquinones. Klotz et al. suggest a variety of possible scenarios regarding naphthoquinone-mediated signalling: (a) the activation of RTK and subsequent downstream signalling; (b) the inhibition of phosphatases and the resulting increase in protein phosphorylation and the subsequent activation of downstream molecules; (c) the direct redox-mediated activity change of molecules; as well as (d) the alkylation of proteins [
8]. Our data suggest that the activation of signalling pathways occurs rapidly, i.e., within minutes and that a strong increase in protein tyrosine phosphorylation goes along with it (data not shown). This suggests that it is possible that specific kinases are activated or phosphatases inhibited. PTP1B, for example, is an important protein tyrosine phosphatase in haematopoietic cells and macrophages and CD45-deficient mice have a defect in osteoclast formation due to reduced cellular motility. Of note, the inhibition of phosphatases is frequently used in osteoporosis therapy with alendronate [
41]. Clearly, further studies are needed for plumbagin to assess its suggested usefulness for therapeutic purposes.
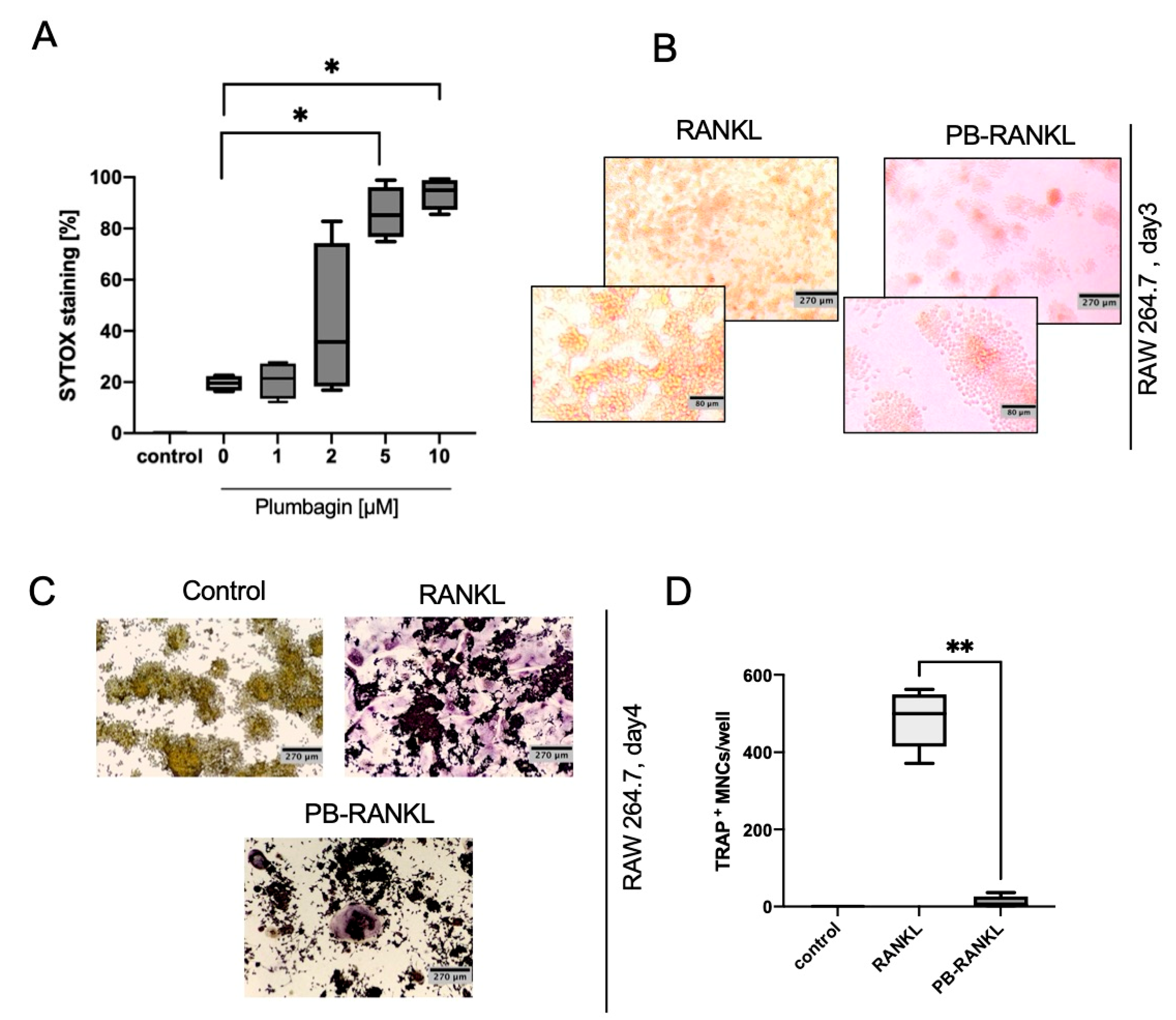
Currently, there are two other reports on the effect of plumbagin on osteoclast formation in the context of breast cancer [
12,
13] that both suggest an inhibitory effect of plumbagin on osteoclastogenesis. We can corroborate this negative impact on osteoclast formation found by Sung et al. who used a RAW264.6 model [
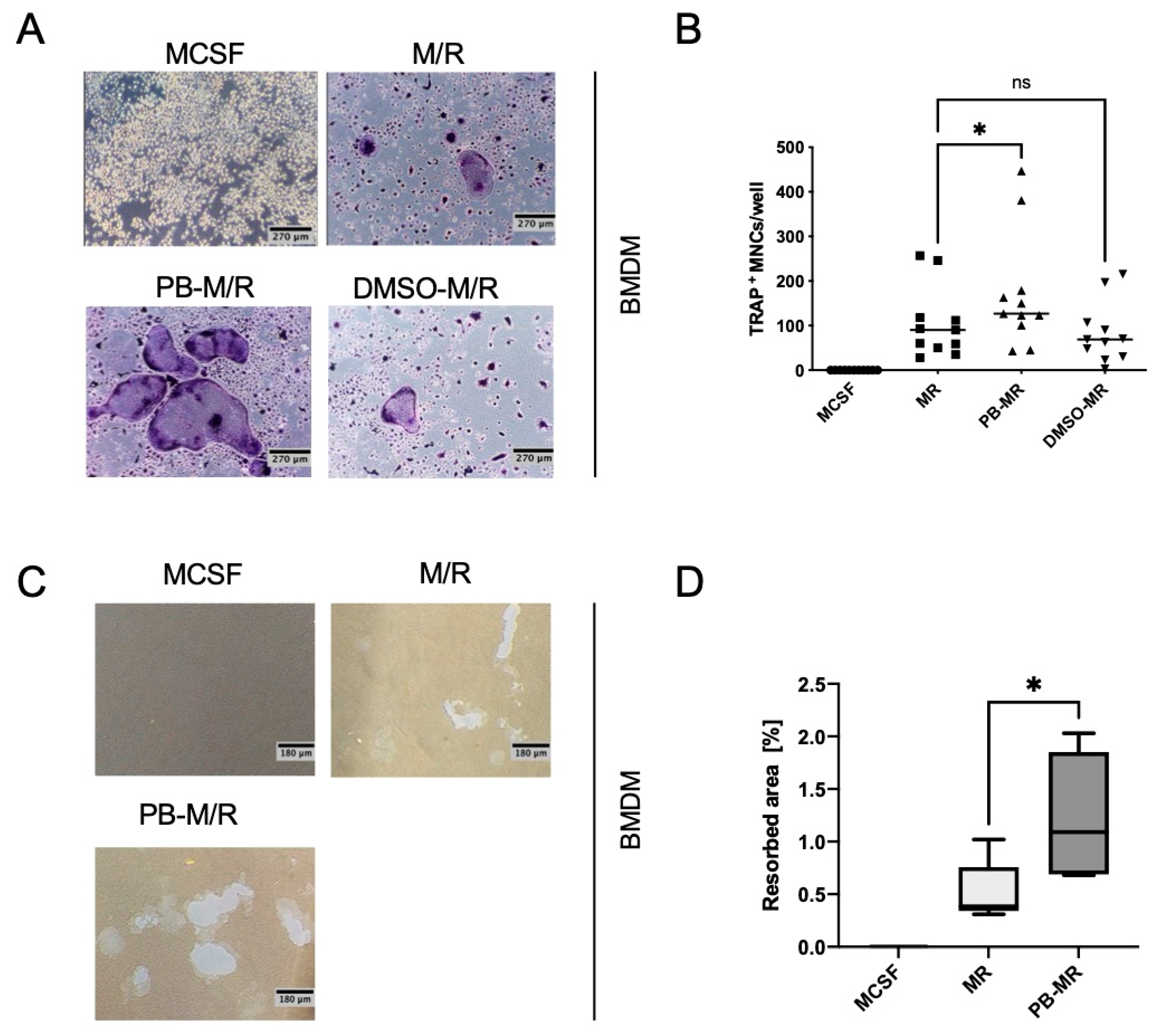
13]. Li et al. directly cultured primary bone marrow cells from C57BL/6 mice with plumbagin that, compared to bone marrow-derived macrophages, might show a different tolerance towards the effects of plumbagin. In contrast, when we studied the effect of plumbagin on C57BL/6 BMDM, we did not see any difference in the ability to differentiate into osteoclasts between M-CSF/RANKL and plumbagin/M-CSF/RANKL co-cultured cells (
Figure S1A,B). Instead, pre-treatment even enhanced the differentiation process. Because RAW264.7 macrophages are derived from Balb/c mice, we hypothesize that the observed differential effects of plumbagin could be dependent on the genetic background of the mice. It has been known for a long time that these mice act differently in the context of an infection. In mice infected with Leishmania, it was found that Balb/c mice had a strong Th2 response while C57BL/6 mice primarily exerted a Th1 phenotype response [
14,
42]. Our data clearly indicate that the differences between experimental systems and the reproducibility of data may need to be discussed in the context of differential immune activation, especially in the context of pharmacological treatment.
4. Materials and Methods
4.1. Mice
C57BL/6 wild-type mice were purchased from Janvier Labs (Le Genest St. Isle, France). Mice were maintained under specific pathogen-free (SPF) conditions in compliance with the German policies on animal welfare (approval number: T-61/18, 10/2018).
4.2. Reagents
Tissue culture reagents were purchased from Anprotec (Bruckberg, Germany), Biochrom GmbH (Berlin, Germany), PAN biotech (Aidenbach, Germany), Thermo Scientific (Langenselbold, Germany), Merck and Sigma. Antibodies against phosphorylated NF-kB, AMPKα (Thr172), 4E-BP1 (Thr37/46), p70 S6 Kinase (Thr 389), Tyr-100, IKKα/β and total NFATc1 as well as RelB were purchased from Cell Signaling Technology (Leiden, The Netherlands). Antibodies against GAPDH, β-actin and cytochrome C were obtained from Proteintech (Manchester, UK). OXPHOS antibody cocktail was purchased from abcam (Cambridge, UK). Secondary horseradish peroxidase (HRP)-linked antibodies were obtained from Cell Signaling Technology (anti-rabbit IgG, anti-Mouse IgG). PE-conjugated anti-F4/80, PE-conjugated anti-CD86, PE-conjugated CD-115, CD265 (RANK), and PE- conjugated IgG2a were provided by BioLegend (San Diego, CA, USA). PE-conjugated anti-CD80 was purchased from BD Biosciences (Heidelberg, Germany). SYTOX Green was procured from Thermo Scientific (Langenselbold, Germany), and MitoTracker DEEP Red FM stain from Cell Signaling Technology (Leiden, The Netherlands). PCR primers were purchased from Apara (Denzlingen, Germany) or Biomers (Ulm, Germany).
4.3. Differentiation of BMDMs
Bone marrow (BM) cells were isolated from the femur, tibia and humerus 12-20-week-old C57BL/6 female mice. To generate BMDMs, BMC were treated with L929-cell conditioned medium (LCCM) as described previously [
15]. On day 1, BM cells were resuspended in 20 mL of DMEM medium. On day 4, the cells were restimulated with 30% LCCM and incubated for an additional 3 days.
4.4. Stimulation
Cells were stimulated with 2 µM plumbagin (Cayman Chemical, Ann Arbor, USA, purity ≥98%) or 50 ng/mL rec. mouse sRANKL, 25 ng/mL rec. mouse M-CSF (all from Biotechne, Abington, UK). Plumbagin was used as 53 mM stock dissolved in DMSO and diluted with medium to a final concentration of 1–10 µM. LPS was from Salmonella enterica serotype abortus equi (L5886 Sigma, Taufkirchen, Germany) and used at 100 ng/mL.
4.5. TRAP Staining
A total of 5 × 104 cells were seeded in 200 µL of medium in 96-well plates and treated as described. Cells were then fixed and stained using Acid Phosphatase, Leukocyte (TRAP) Kit (Sigma-Aldrich, Taufkirchen, Germany). TRAP-positive cells with three or more nuclei were counted as osteoclasts.
4.6. Bone Resorption Assay
A total of 2 × 106 cells were seeded in 1 mL of medium in Osteo Assay Surface 24-well plates (Corning, NY, USA) for 10 days and stimulated as indicated. On day 10, cells were washed with PBS and incubated in 250 µL sodium–hypochlorite (5–7%) for 5 min at RT. Wells were washed twice with H2O and left to dry overnight. The resorbed area was measured with ImageJ software.
4.7. Cell Viability Assay
As indicated, 5 × 104 cells per well were seeded in a translucent 96 well microplate and stimulated with plumbagin. To quantify ATP levels, the supernatant was removed and the CellTiter-Glo Reagent (Promega, Mannheim, Germany) was added according to the manufacturer’s protocol. Cell lysates were transferred to white 96-well assay plates and measured with a LUMIstar Optima (BMG, Offenburg, Germany) luminometer.
4.8. Quantitative Real-Time PCR
BMDM cells (2 × 10
5 per well) were seeded and the cells were stimulated in a 24-well plate as indicated. RNA isolation was carried out by using RNeasy Plus Micro Kit (QIAGEN), according to the manufacturer’s protocol. cDNA was prepared by using the Biozym cDNA Synthesis Kit (Biozym Scientific GmbH, Hessisch Oldendorf, Germany). Quantitative RT-PCR was performed using qPCRBIO Syber Green Mix Hi-ROX (PCR Biosystems, London, UK) and run with the StepOne Real-Time PCR System (Applied Biosystems, Darmstadt, Germany). An initial denaturation step was 2 min at 95 °C, followed by 40 cycles of denaturation at 95 °C for 5 s and amplification at 60 °C for 20 s. All primer pairs used were recently described in [
43].
4.9. Western Blot Analysis
A total of 2 × 106 cells were stimulated in 2 mL of complete medium in a 6-well plate with plumbagin. Cells were washed with ice-cold PBS. Cell lysis was carried out in 200 μL of 1× RIPA buffer, freshly supplemented with a Phosphatase and Protease-Inhibitor Cocktail (Roche, Mannheim, Germany). Lysates were collected and run on an SDS-PAGE 4–20% gradient polyacrylamide gel (Anamed, Gross-Bieberau, Germany). Proteins were transferred to nitrocellulose membrane via semi-dry Western blot, blocked with 1× Blue Block locking buffer (SERVA, Heidelberg, Germany) for 30 min at RT. Membranes were incubated overnight at 4°C with the primary antibody diluted as suggested by the manufacturer’s protocol. After 1 h incubation with the secondary antibody (HRP-coupled), protein bands were detected by enhanced chemiluminescence (Intas Science Imaging, Göttingen, Germany).
4.10. Nuclear Translocation of NFATc1
Bone marrow derived macrophages (BMDMs) from Balb/c mice were seeded onto glass coverslips and incubated in complete media containing M-CSF (30 ng/mL), RANKL (40 ng/mL), with or without plumbagin for 48 h. After incubation, the cells were washed with PBS and fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS for 5 min. The cells were blocked with 5% bovine serum albumin (BSA) for 1 h. The cells were incubated with the mouse anti-NFATc1 antibody (BD Biosciences, San Jose, CA, USA), followed by the anti-mouse FITC secondary antibody (Invitrogen, Frankfurt, Germany) for 45 min. Cells were washed and mounted using antifade mounting medium containing DAPI (Abcam, Bristol, UK) and the localisation of NFATc1 was observed under A1R HD25 Nikon confocal microscope under 60× magnification.
4.11. ELISA
A total of 2 × 106 cells were stimulated in 2 mL of medium in a cell culture dish with plumbagin and LPS. Supernatants were collected and ELISA measurements were performed in a 96-well plate with BD OptEIA Elisa Kits (BD Biosciences, Heidelberg, Germany) according to the manufacturer’s protocol.
4.12. Mitochondrial Extract Preparation
A total of 2 × 107 cells were seeded in 10 mL of complete medium in a 10-cm dish and stimulated for 3 days. For mitochondria extraction, a Mitochondria Isolation Kit (Thermo Scientific, Frankfurt, Germany) with Dounce Homogenization was used following the manufacturer’s protocol.
4.13. Mitochondrial Copy Number
A total of 7 × 104 cells were seeded in 1 mL of complete medium in 48 well-plate. On day 4, total genomic DNA was isolated with the innuPREP DNA Mini Kit (Analytik Jena, Jena, Germany). Subsequently, qPCR was run with 2 ng/μL of DNA to amplify and relatively quantify the amount of nuclear and mitochondrial DNA. The mitochondrial copy number in variously stimulated cells was determined by comparing the nuclear DNA amount with the amount of mitochondrial DNA in the cells.
4.14. Translation Assay
A total of 1 × 106 cells were seeded in 1 mL of medium in a 24-well plate for 24 h before additional treatment. Cells were stimulated for 6 h. After 6 h of stimulation, the medium was changed to l-methionine free Medium (Gibco DMEM supplemented with 200 µM l-cystine, 2 mM l-glutamine, 1 mM Sodium Pyruvate, 10 mM HEPES) with homopropargylglycine (HPG) according to the Click-iT™ HPG Alexa Fluor™ 488 Protein Synthesis Assay Kit (Thermo Scientific, Darmstadt, Germany) protocol. Cells were incubated for 30 min at 37 °C, 5% CO2. Then, the cells were washed with DPBS and fixed with a 4% PFA fixation buffer (BioLegend, London, OK) for 10 min at RT. Cells were washed twice with 3% BSA in PBS and incubated with 200 µL permeabilization buffer (0.5% Saponin) for 10 min at RT. The washing step with DPBS was repeated twice. Cells were then incubated for 30 min protected from light in 250 µL Click-iT reaction cocktail containing Alexa Fluor 488 azide (recipe as prescribed on the kit protocol). The final washing step was carried out with the rinse buffer. The cells were scraped in 400 µL DPBS. FITC fluorescence levels were measured by FACS. Translation was inhibited by adding 10 µM cycloheximide (Santa Cruz Biotechnologies Inc., Dallas, TX, USA).
4.15. ROS Measurement
A total of 1 × 105 cells were seeded in 100 µL of medium in white 96-well plates for 48 h. On day 2, the medium was removed. Fifty microlitres (50 μL) Hank’s buffered salt solution (HBSS) for 30 min was added to each well before luminescence measurement. Luminol buffer (10 mg/mL Luminol (Sigma-Aldrich), and HRP 100 U/mL (Sigma-Aldrich) in HBSS) were prepared with and without 60 ng/mL of phorbol 12-myristate 13-acetate (PMA) (Cayman Chemical, Ann Arbor, MI, USA). Fifty microlitres (50 μL) luminol buffer was added to the cells and the luminescence measurement was started as soon as possible upon the addition because of the immediate reaction. The bioluminescence was measured over a time period of around 2 h. To quantify ROS production, the area under the curve was calculated with Graph Pad Prism software (San Diego, CA, USA).
4.16. FACS Analysis
For FACS analysis, 1 × 106 cells were used per sample. Cells were blocked for 15 min in PBS, 2% BSA on ice in a total volume of 100 μL. The staining step was carried out on ice for one hour with the appropriate antibody or incubated in the corresponding isotype control diluted as suggested by the manufacturer’s protocol. The surface expression of RANK (R12-31) IgG2a (RTK2758), CD11b (M1/70), CD80 (16-10A1), CD86 (GL-1), F4/80 (BM8) was quantified by flow cytometry by using FACS Canto cytometer (BD Biosciences, Heidelberg, Germany) and BD FACS Diva Software. For the live/dead staining, cells were washed once with DPBS and were stained with SYTOX dye. The cells could be analysed within the FITC channel directly, without further incubation or washing. For measuring mitochondrial activity, the cells were stained with 100 nM MitoTracker (Cell Signaling Technology, Leiden, The Netherlands). After 30 min incubation, the cells were washed with PBS and analysed within the APC-Cy7 channel.
4.17. Statistical Analyses
Data are presented as the means ± SD on Graph Pad Prism (San Diego, CA, USA). Comparison between 2 groups was performed as indicated in the figure legend for each experiment.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





