
A New Anti-Estrogen Discovery Platform Identifies FDA-Approved Imidazole Anti-Fungal Drugs as Bioactive Compounds against ERα Expressing Breast Cancer Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Ability of FDA-Approved Drugs to Bind ERα and Affect ERα Signaling and Cell Proliferation in Cell Lines Modelling Primary and MBC
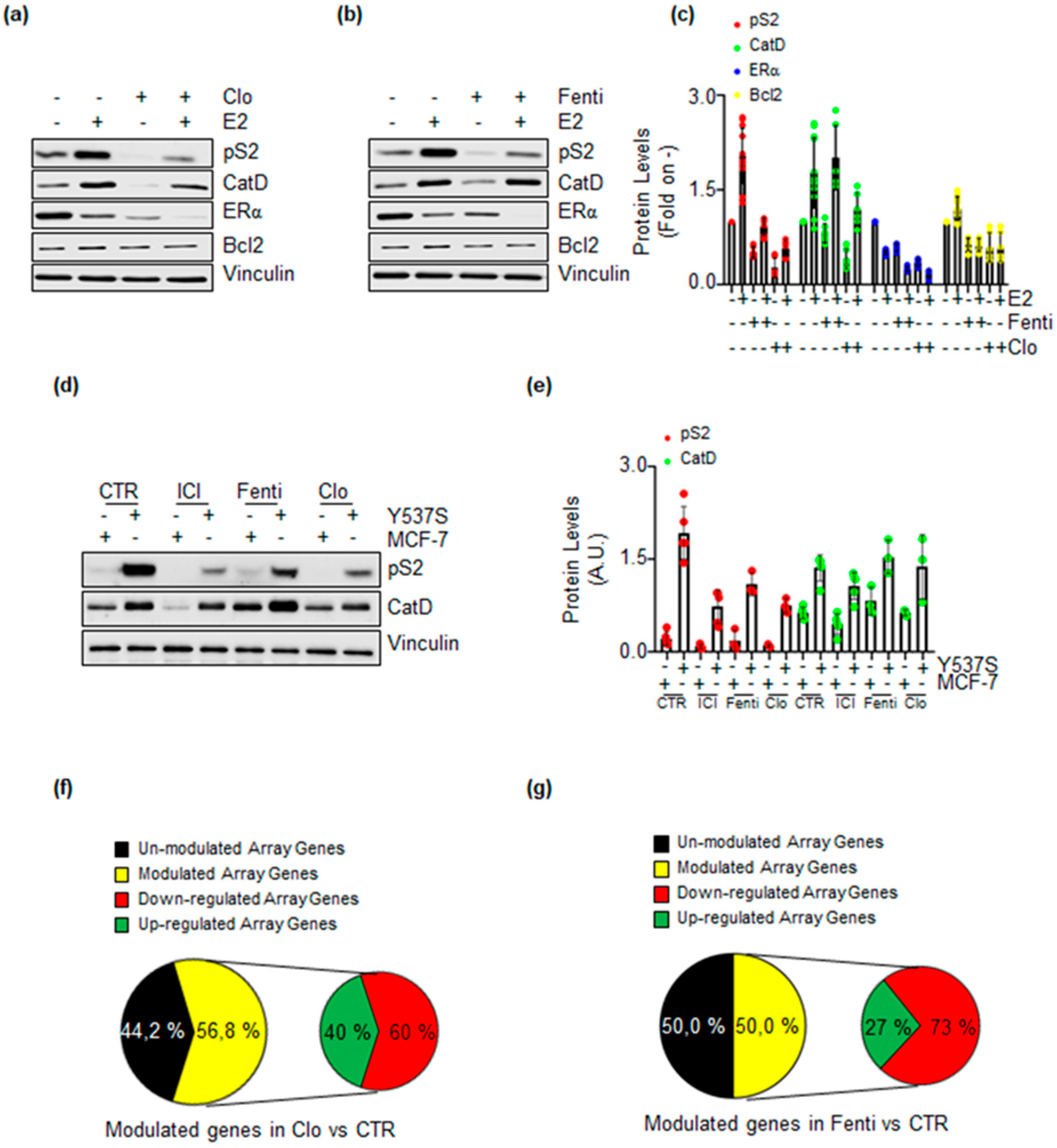
2.2. Validation of Clotrimazole and Fenticonazole to Affect ERα Signaling and Cell Proliferation
2.3. Clotrimazole- and Fenticonazole-Dependent Mechanism for the Control of ERα Intracellular Concentration
2.4. Clotrimazole and Fenticonazole Administration Impairs ERα Transcriptional Activity in MCF-7 and Y537S Cells
2.5. Clotrimazole and Fenticonazole Administration Prevents ERα Target Gene Expression
2.6. Clotrimazole and Fenticonazole Administration Prevents E2-Induced Cell Proliferation
2.7. Pre-Clinical Evaluation of Clotrimazole and Fenticonazole as Novel Drugs for Treatment of MBC
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. In Vitro ERα Binding Assay
4.3. In-Cell Western Blotting
4.4. In-Cell Propidium Iodide (PI) Staining
4.5. Real-Time Measurement of ERα Transcriptional Activity
4.6. Cell Manipulation for Western Blotting Analyses
4.7. Cell Proliferation and Cell Cycle Assays
4.8. RNA Isolation and qPCR Analysis
4.9. Bromodeoxyuridine Incorporation Assay
4.10. Tumor Spheroid Formation
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nassa, G.; Salvati, A.; Tarallo, R.; Gigantino, V.; Alexandrova, E.; Memoli, D.; Sellitto, A.; Rizzo, F.; Malanga, D.; Mirante, T.; et al. Inhibition of histone methyltransferase DOT1L silences ERalpha gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci. Adv. 2019, 5, eaav5590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-dependent estrogen receptor alpha membrane localization: Regulation by 17 beta-estradiol. Mol. Biol. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Razandi, M.; Pedram, A.; Greene, G.L.; Levin, E.R. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: Studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol. Endocrinol. 1999, 13, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Laios, I.; Journe, F.; Nonclercq, D.; Vidal, D.S.; Toillon, R.A.; Laurent, G.; Leclercq, G. Role of the proteasome in the regulation of estrogen receptor alpha turnover and function in MCF-7 breast carcinoma cells. J. Steroid Biochem. Mol. Biol. 2005, 94, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Busonero, C.; Leone, S.; Bartoloni, S.; Acconcia, F. Strategies to degrade estrogen receptor alpha in primary and ESR1 mutant-expressing metastatic breast cancer. Mol. Cell. Endocrinol. 2019, 480, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; Gigantino, V.; Nassa, G.; Cappa, V.M.; Ventola, G.M.; Cracas, D.G.C.; Mastrocinque, R.; Rizzo, F.; Tarallo, R.; Weisz, A.; et al. Global View of Candidate Therapeutic Target Genes in Hormone-Responsive Breast Cancer. Int. J. Mol. Sci. 2020, 21, 4068. [Google Scholar] [CrossRef] [PubMed]
- Metivier, R.; Penot, G.; Hubner, M.R.; Reid, G.; Brand, H.; Kos, M.; Gannon, F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 2003, 115, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Reid, G.; Hubner, M.R.; Metivier, R.; Brand, H.; Denger, S.; Manu, D.; Beaudouin, J.; Ellenberg, J.; Gannon, F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 2003, 11, 695–707. [Google Scholar] [CrossRef]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; de Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumachi, F.; Luisetto, G.; Basso, S.M.; Basso, U.; Brunello, A.; Camozzi, V. Endocrine therapy of breast cancer. Curr. Med. Chem. 2011, 18, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Gimple, R.C.; Zhou, N.; Zhao, L.; Gustafsson, J.A.; Zhou, S. Targeting Nuclear Receptors for Cancer Therapy: Premises, Promises, and Challenges. Trends Cancer 2020. [Google Scholar] [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5, e12792. [Google Scholar] [CrossRef]
- Harrod, A.; Fulton, J.; Nguyen, V.T.M.; Periyasamy, M.; Ramos-Garcia, L.; Lai, C.F.; Metodieva, G.; de Giorgio, A.; Williams, R.L.; Santos, D.B.; et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017, 36, 2286–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Levine, K.M.; Bahreini, A.; Wang, P.; Chu, D.; Park, B.H.; Oesterreich, S.; Lee, A.V. Upregulation of IRS1 enhances IGF1 response in Y537S and D538G ESR1 mutant breast cancer cells. Endocrinology 2018, 159, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Rumble, R.B.; Macrae, E.; Barton, D.L.; Connolly, H.K.; Dickler, M.N.; Fallowfield, L.; Fowble, B.; Ingle, J.N.; Jahanzeb, M.; et al. Endocrine Therapy for Hormone Receptor-Positive Metastatic Breast Cancer: American Society of Clinical Oncology Guideline. J. Clin. Oncol. 2016, 34, 3069–3103. [Google Scholar] [CrossRef] [PubMed]
- Weir, H.M.; Bradbury, R.H.; Lawson, M.; Rabow, A.A.; Buttar, D.; Callis, R.J.; Curwen, J.O.; de Almeida, C.; Ballard, P.; Hulse, M.; et al. AZD9496: An Oral Estrogen Receptor Inhibitor That Blocks the Growth of ER-Positive and ESR1-Mutant Breast Tumors in Preclinical Models. Cancer Res. 2016, 76, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, J.D.; Darimont, B.; Zhou, W.; Arrazate, A.; Young, A.; Ingalla, E.; Walter, K.; Blake, R.A.; Nonomiya, J.; Guan, Z.; et al. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. eLife 2016, 5, e15828. [Google Scholar] [CrossRef] [PubMed]
- Cipolletti, M.; Leone, S.; Bartoloni, S.; Busonero, C.; Acconcia, F. Real-time measurement of E2:E:ERalpha transcriptional activity in living cells. J. Cell. Physiol. 2020, 235, 6697–6710. [Google Scholar] [CrossRef] [PubMed]
- Leone, S.; Busonero, C.; Acconcia, F. A high throughput method to study the physiology of E2:E:ERalpha signaling in breast cancer cells. J. Cell. Physiol 2018, 233, 3713–3722. [Google Scholar] [CrossRef]
- Cipolletti, M.; Pescatori, S.; Acconcia, F. Real-time challenging of ERα Y537S mutant transcriptional activity in living cells. Endocrines 2021, 2, 6. [Google Scholar] [CrossRef]
- Filgueira, C.S.; Benod, C.; Lou, X.; Gunamalai, P.S.; Villagomez, R.A.; Strom, A.; Gustafsson, J.A.; Berkenstam, A.L.; Webb, P. A screening cascade to identify ERbeta ligands. Nucl. Recept. Signal. 2014, 12, e003. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D. Illustration of SSMD, z score, SSMD*, z* score, and t statistic for hit selection in RNAi high-throughput screens. J. Biomol. Screen. 2011, 16, 775–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Metzger, D.; Bornert, J.M.; Chambon, P. Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J. 1993, 12, 1153–1160. [Google Scholar] [CrossRef]
- La Rosa, P.; Pesiri, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation Regulates 17beta-Estradiol-Induced Estrogen Receptor-alpha Degradation and Transcriptional Activity. Mol. Endocrinol. 2012, 26, 762–774. [Google Scholar] [CrossRef] [Green Version]
- Acconcia, F.; Totta, P.; Ogawa, S.; Cardillo, I.; Inoue, S.; Leone, S.; Trentalance, A.; Muramatsu, M.; Marino, M. Survival versus apoptotic 17beta-estradiol effect: Role of ER alpha and ER beta activated non-genomic signaling. J. Cell. Physiol 2005, 203, 193–201. [Google Scholar] [CrossRef]
- Bartoloni, S.; Leone, S.; Acconcia, F. Unexpected Impact of a Hepatitis C Virus Inhibitor on 17beta-Estradiol Signaling in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 3418. [Google Scholar] [CrossRef]
- Busonero, C.; Leone, S.; Bianchi, F.; Maspero, E.; Fiocchetti, M.; Palumbo, O.; Cipolletti, M.; Bartoloni, S.; Acconcia, F. Ouabain and Digoxin Activate the Proteasome and the Degradation of the ERα in Cells Modeling Primary and Metastatic Breast Cancer. Cancers 2020, 12, 3840. [Google Scholar] [CrossRef]
- Campbell, J.; Ryan, C.J.; Brough, R.; Bajrami, I.; Pemberton, H.N.; Chong, I.Y.; Costa-Cabral, S.; Frankum, J.; Gulati, A.; Holme, H.; et al. Large-Scale Profiling of Kinase Dependencies in Cancer Cell Lines. Cell Rep. 2016, 14, 2490–2501. [Google Scholar] [CrossRef] [Green Version]
- Overk, C.R.; Yao, P.; Chen, S.; Deng, S.; Imai, A.; Main, M.; Schinkovitz, A.; Farnsworth, N.R.; Pauli, G.F.; Bolton, J.L. High-content screening and mechanism-based evaluation of estrogenic botanical extracts. Comb. Chem. High Throughput Screen. 2008, 11, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Giamas, G.; Filipovic, A.; Jacob, J.; Messier, W.; Zhang, H.; Yang, D.; Zhang, W.; Shifa, B.A.; Photiou, A.; Tralau-Stewart, C.; et al. Kinome screening for regulators of the estrogen receptor identifies LMTK3 as a new therapeutic target in breast cancer. Nat. Med. 2011, 17, 715–719. [Google Scholar] [CrossRef]
- Singh, K.; Munuganti, R.S.; Leblanc, E.; Lin, Y.L.; Leung, E.; Lallous, N.; Butler, M.; Cherkasov, A.; Rennie, P.S. In silico discovery and validation of potent small-molecule inhibitors targeting the activation function 2 site of human oestrogen receptor alpha. Breast Cancer Res. 2015, 17, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, S.S.; Chang, Y.C.; Lin, W.C.; Lee, W.S.; Hsueh, P.R.; Hsu, C.W. Epidemiology, Treatment, and Prevention of Nosocomial Bacterial Pneumonia. J. Clin. Med. 2020, 9, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Muramatsu, T.; Kanai, Y.; Ojima, H.; Sukeda, A.; Hiraoka, N.; Arai, E.; Sugiyama, Y.; Matsuzaki, J.; Uchida, R.; et al. Establishment of Patient-Derived Organoids and Drug Screening for Biliary Tract Carcinoma. Cell Rep. 2019, 27, 1265–1276.e1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadavakollu, S.; Stailey, C.; Kunapareddy, C.S.; White, S. Clotrimazole as a Cancer Drug: A Short Review. Med. Chem. 2014, 4, 722–724. [Google Scholar] [CrossRef] [Green Version]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Juan, G. Analysis of DNA content and BrdU incorporation. Curr. Protoc. Cytom. 2001, 2, 7.7.1–7.7.9. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cipolletti, M.; Bartoloni, S.; Busonero, C.; Parente, M.; Leone, S.; Acconcia, F. A New Anti-Estrogen Discovery Platform Identifies FDA-Approved Imidazole Anti-Fungal Drugs as Bioactive Compounds against ERα Expressing Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 2915. https://doi.org/10.3390/ijms22062915
Cipolletti M, Bartoloni S, Busonero C, Parente M, Leone S, Acconcia F. A New Anti-Estrogen Discovery Platform Identifies FDA-Approved Imidazole Anti-Fungal Drugs as Bioactive Compounds against ERα Expressing Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(6):2915. https://doi.org/10.3390/ijms22062915
Chicago/Turabian StyleCipolletti, Manuela, Stefania Bartoloni, Claudia Busonero, Martina Parente, Stefano Leone, and Filippo Acconcia. 2021. "A New Anti-Estrogen Discovery Platform Identifies FDA-Approved Imidazole Anti-Fungal Drugs as Bioactive Compounds against ERα Expressing Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 6: 2915. https://doi.org/10.3390/ijms22062915
APA StyleCipolletti, M., Bartoloni, S., Busonero, C., Parente, M., Leone, S., & Acconcia, F. (2021). A New Anti-Estrogen Discovery Platform Identifies FDA-Approved Imidazole Anti-Fungal Drugs as Bioactive Compounds against ERα Expressing Breast Cancer Cells. International Journal of Molecular Sciences, 22(6), 2915. https://doi.org/10.3390/ijms22062915