
The Time Has Come to Explore Plasma Biomarkers in Genetic Cardiomyopathies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Genes Implicated in Cardiomyopathies
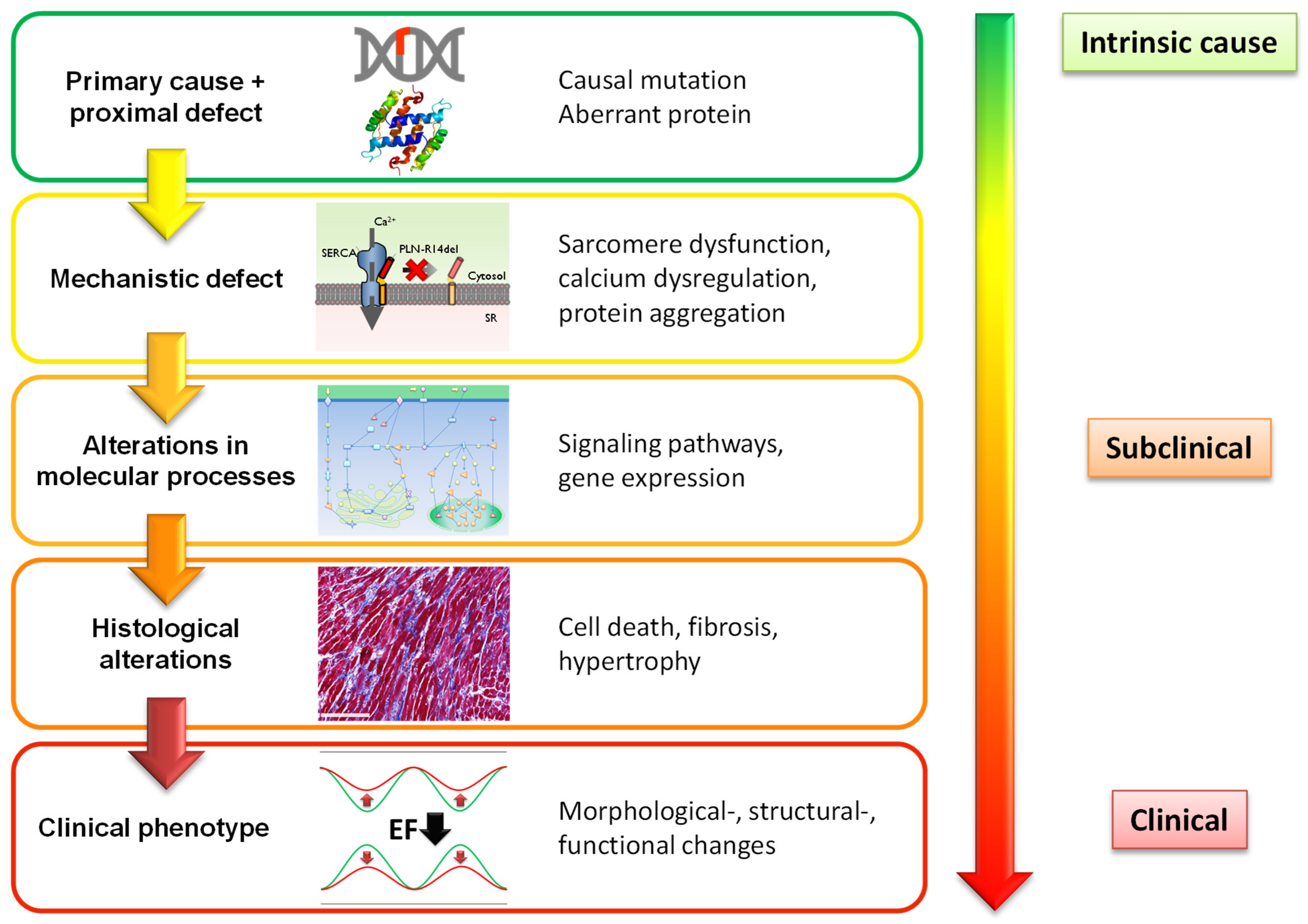
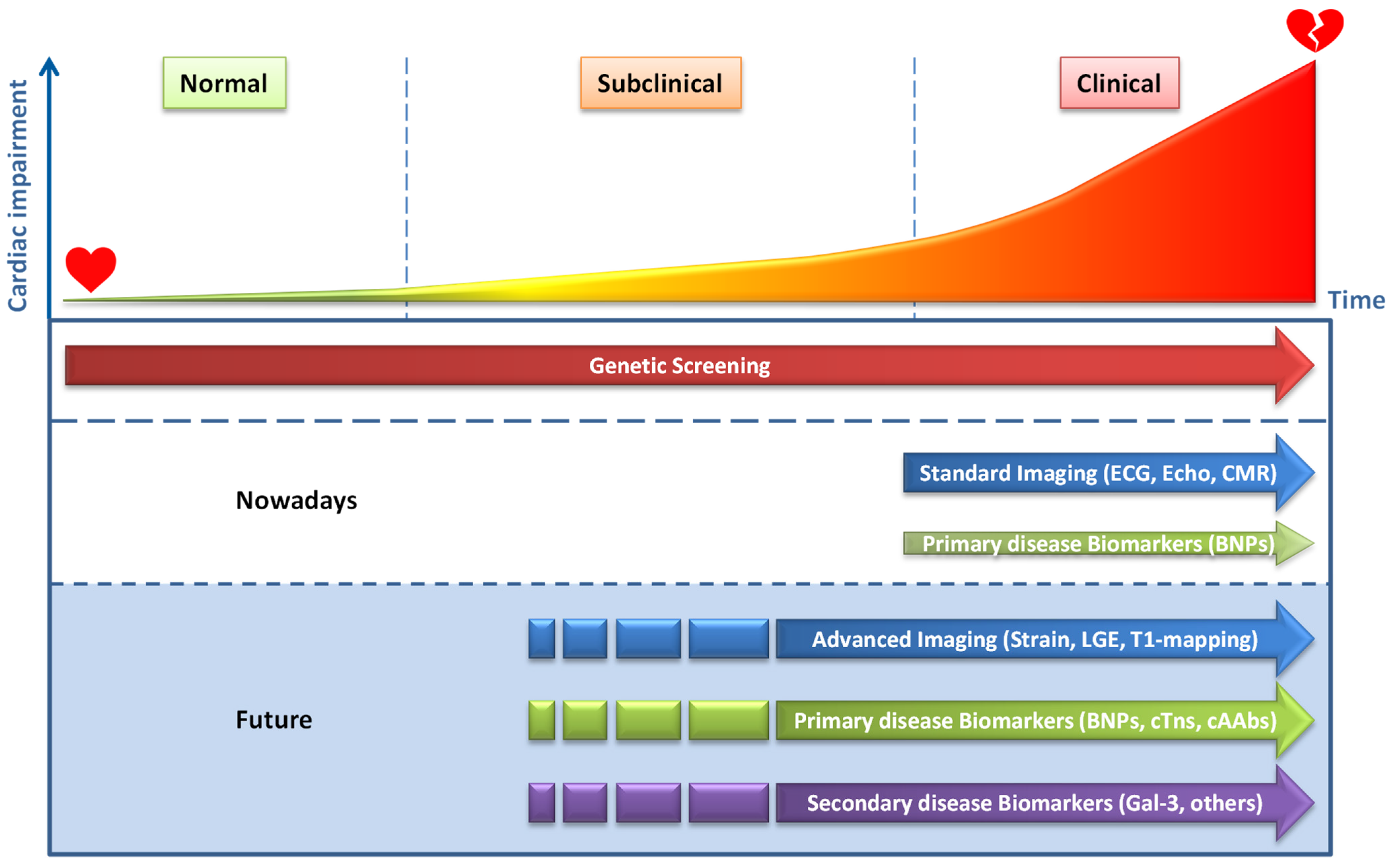
3. Development of Cardiac Dysfunction
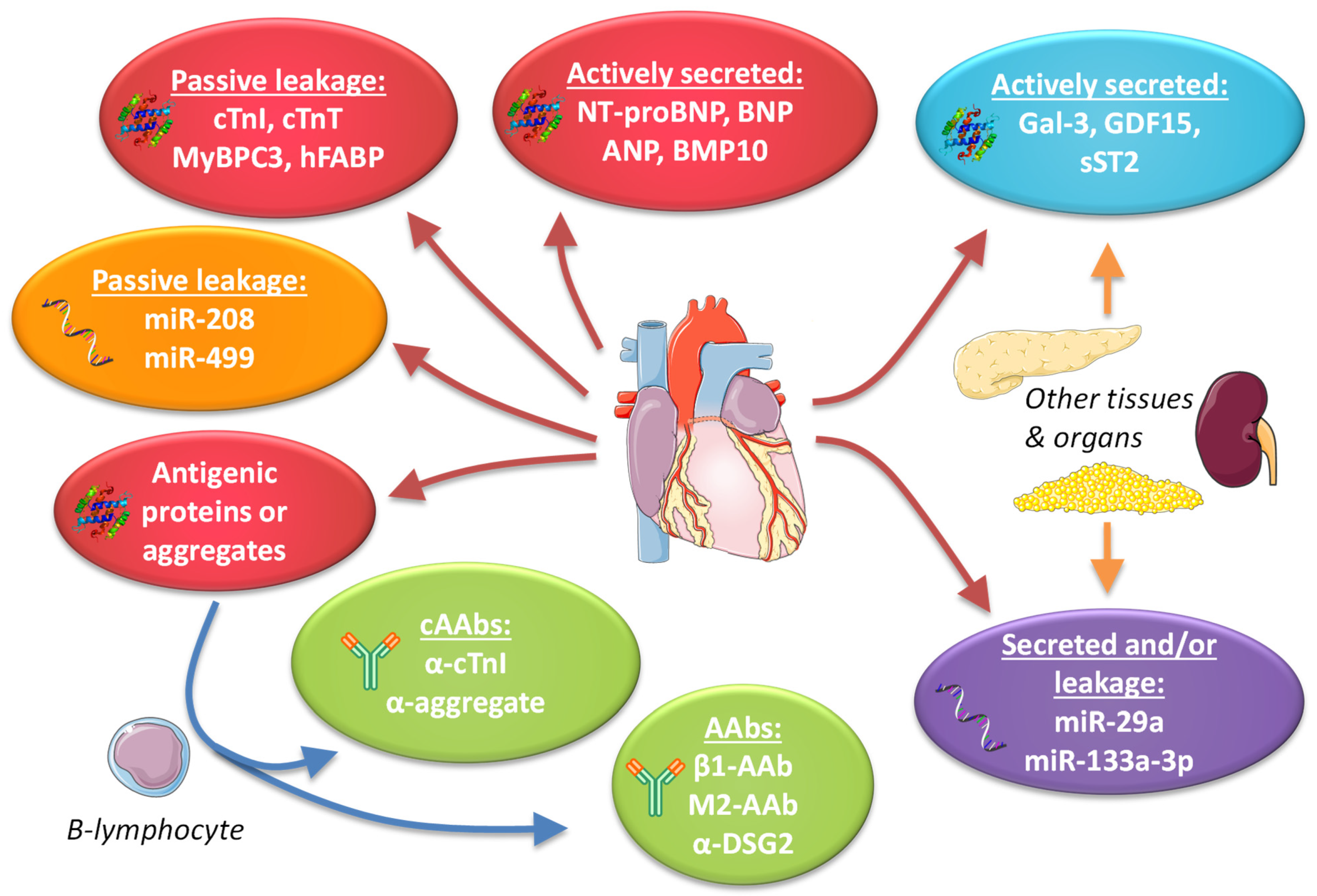
4. Cardiac-Specific Plasma Protein Biomarkers
4.1. NPs and Troponins in HCM, DCM and ACM
4.2. BNPs and Troponins in Subclinical Disease
5. Non-Cardiac-Specific Plasma Protein Biomarkers
5.1. Non-Cardiac-Specific Plasma Biomarkers in HCM, DCM and ACM
5.2. Non-Cardiac-Specific Plasma Biomarkers in Subclinical Heart Disease
6. Noncoding RNA Biomarkers
6.1. ncRNA Biomarkers in HCM, DCM and ACM
6.2. ncRNA Biomarkers in Subclinical Heart Disease
7. Autoantibodies
7.1. Autoantibody Biomarkers in HCM, DCM and ACM
7.2. Autoantibody Biomarkers in Subclinical Heart Disease
8. Future Directions and Challenges
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Geisterfer-Lowrance, A.A.T.; Kass, S.; Tanigawa, G.; Vosberg, H.-P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A β cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.; McKenna, R.W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestroni, L.; Rocco, C.; Gregori, D.; Sinagra, G.; Di Lenarda, A.; Miocic, S.; Vatta, M.; Pinamonti, B.; Muntoni, F.; Caforio, A.L.; et al. Familial dilated cardiomyopathy: Evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J. Am. Coll. Cardiol. 1999, 34, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right Ventricular Cardiomyopathy and Sudden Death in Young People. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Angelini, A.; Melacini, P.; Barbero, F.; Thiene, G. Evolutionary Persistence of Spongy Myocardium in Humans. Circulation 1999, 99, 2475. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531. [Google Scholar] [CrossRef]
- Elmaghawry, M.; Alhashemi, M.; Zorzi, A.; Yacoub, M.H. A global perspective of arrhythmogenic right ventricular cardiomyopathy. Glob. Cardiol. Sci. Pract. 2012, 2012, 26. [Google Scholar] [CrossRef]
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive Cardiomyopathy. Circ. Res. 2017, 121, 819–837. [Google Scholar] [CrossRef]
- Towbin, J.A.; Jefferies, J.L. Cardiomyopathies Due to Left Ventricular Noncompaction, Mitochondrial and Storage Diseases, and Inborn Errors of Metabolism. Circ. Res. 2017, 121, 838–854. [Google Scholar] [CrossRef] [Green Version]
- Čelutkienė, J.; Plymen, C.M.; Flachskampf, F.A.; de Boer, R.A.; Grapsa, J.; Manka, R.; Anderson, L.; Garbi, M.; Barberis, V.; Filardi, P.P.; et al. Innovative imaging methods in heart failure: A shifting paradigm in cardiac assessment. Position statement on behalf of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2018, 20, 1615–1633. [Google Scholar] [CrossRef] [Green Version]
- Zamorano, J.L.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. J. Am. Coll. Cardiol. 2018, 72, e91–e220. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Bloma, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the Europea. Eur. Heart J. 2015, 36, 2793–2867l. [Google Scholar] [CrossRef] [Green Version]
- Mueller, C.; McDonald, K.; de Boer, R.A.; Maisel, A.; Cleland, J.G.F.; Kozhuharov, N.; Coats, A.J.S.; Metra, M.; Mebazaa, A.; Ruschitzka, F.; et al. Heart Failure Association of the European Society of Cardiology practical guidance on the use of natriuretic peptide concentrations. Eur. J. Heart Fail. 2019, 21, 715–731. [Google Scholar] [CrossRef] [Green Version]
- Ravassa, S.; Delles, C.; Currie, G.; Díez, J. Biomarkers of Cardiovascular Disease. In Textbook of Vascular Medicine; Touyz, R.M., Delles, C., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 319–330. ISBN 978-3-030-16481-2. [Google Scholar]
- McCauley, M.D.; Wehrens, X.H.T. Animal models of arrhythmogenic cardiomyopathy. Dis. Model. Mech. 2009, 2, 563–570. [Google Scholar] [CrossRef] [Green Version]
- van der Zwaag, P.A.; van Rijsingen, I.A.W.; Asimaki, A.; Jongbloed, J.D.H.; van Veldhuisen, D.J.; Wiesfeld, A.C.P.; Cox, M.G.P.J.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.W.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Greaves, S.C.; Roche, A.H.G.; Neutze, J.M.; Whitlock, R.M.L.; Veale, A.M.O. Inheritance of hypertrophic cardiomyopathy: A cross sectional and M mode echocardiographic study of 50 families. Heart 1987, 58, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic Cardiomyopathy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef]
- Erdmann, J.; Daehmlow, S.; Wischke, S.; Senyuva, M.; Werner, U.; Raible, J.; Tanis, N.; Dyachenko, S.; Hummel, M.; Hetzer, R.; et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin. Genet. 2003, 64, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.P.; Syrris, P.; Esteban, M.T.T.; Jenkins, S.; Pantazis, A.; Deanfield, J.E.; McKenna, W.J.; Elliott, P.M. Prevalence of Sarcomere Protein Gene Mutations in Preadolescent Children With Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Dauphin, C.; Simon Jouk, P.; Da Costa, A.; Prieur, F.; Bresson, J.-L.; Faivre, L.; Eicher, J.-C.; et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur. J. Med. Genet. 2010, 53, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, L.; Watkins, H.; MacRae, C.; Lamas, R.; McKenna, W.; Vosberg, H.-P.; Seldman, J.G.; Seidman, C.E. α-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994, 77, 701–712. [Google Scholar] [CrossRef]
- Kimura, A.; Harada, H.; Park, J.-E.; Nishi, H.; Satoh, M.; Takahashi, M.; Hiroi, S.; Sasaoka, T.; Ohbuchi, N.; Nakamura, T.; et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat. Genet. 1997, 16, 379–382. [Google Scholar] [CrossRef]
- Geier, C.; Gehmlich, K.; Ehler, E.; Hassfeld, S.; Perrot, A.; Hayess, K.; Cardim, N.; Wenzel, K.; Erdmann, B.; Krackhardt, F.; et al. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum. Mol. Genet. 2008, 17, 2753–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poetter, K.; Jiang, H.; Hassanzadeh, S.; Master, S.R.; Chang, A.; Dalakas, M.C.; Rayment, I.; Sellers, J.R.; Fananapazir, L.; Epstein, N.D. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat. Genet. 1996, 13, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; Klausen, I.C.; Pedersen, A.K.; Egeblad, H.; Bross, P.; Kruse, T.A.; Gregersen, N.; Hansen, P.S.; Baandrup, U.; Børglum, A.D. α-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J. Clin. Investig. 1999, 103, R39–R43. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic Variation in Titin in Arrhythmogenic Right Ventricular Cardiomyopathy–Overlap Syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [Green Version]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Grünig, E.; Tasman, J.A.; Kücherer, H.; Franz, W.; Kübler, W.; Katus, H.A. Frequency and Phenotypes of Familial Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 1998, 31, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Mestroni, L.; Maisch, B.; McKenna, W.J.; Schwartz, K.; Charron, P.; Rocco, C.; Tesson, F.; Richter, A.; Wilke, A.; Komajda, M. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur. Heart J. 1999, 2, 93–102. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.G.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of Titin Causing Dilated Cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Nat. Rev. Cardiol. 2020, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Dellefave, L.; McNally, E.M. The genetics of dilated cardiomyopathy. Curr. Opin. Cardiol. 2010, 25, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, L.; Brun, F.; Spezzacatene, A.; Sinagra, G.; Taylor, M.R.G. Genetic causes of dilated cardiomyopathy. Prog. Pediatr. Cardiol. 2014, 37, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, N.; Hayashi, T.; Arimura, T.; Koga, Y.; Takahashi, M.; Shibata, H.; Teraoka, K.; Chikamori, T.; Yamashina, A.; Kimura, A. αB-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 342, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Brunckhorst, C.; Duru, F.; Saguner, A.M. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythmia Electrophysiol. Rev. 2016, 5, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoorntje, E.T.; te Rijdt, W.P.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef]
- Ohno, S. The genetic background of arrhythmogenic right ventricular cardiomyopathy. J. Arrhythmia 2016, 32, 398–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacoby, D.; McKenna, W.J. Genetics of inherited cardiomyopathy. Eur. Heart J. 2012, 33, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Sawant, A.C.; te Riele, A.S.J.M.; Tichnell, C.; Murray, B.; Bhonsale, A.; Tandri, H.; Judge, D.P.; Calkins, H.; James, C.A. Safety of American Heart Association-recommended minimum exercise for desmosomal mutation carriers. Heart Rhythm 2016, 13, 199–207. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise Increases Age-Related Penetrance and Arrhythmic Risk in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy–Associated Desmosomal Mutation Carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A. Inherited Cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [Green Version]
- Dirkx, E.; da Costa Martins, P.A.; De Windt, L.J. Regulation of fetal gene expression in heart failure. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2414–2424. [Google Scholar] [CrossRef] [Green Version]
- Eijgenraam, T.R.; Silljé, H.H.W.; de Boer, R.A. Current understanding of fibrosis in genetic cardiomyopathies. Trends Cardiovasc. Med. 2020, 30, 353–361. [Google Scholar] [CrossRef]
- Vigneault, D.M.; Yang, E.; Jensen, P.J.; Tee, M.W.; Farhad, H.; Chu, L.; Noble, J.A.; Day, S.M.; Colan, S.D.; Russell, M.W.; et al. Left Ventricular Strain Is Abnormal in Preclinical and Overt Hypertrophic Cardiomyopathy: Cardiac MR Feature Tracking. Radiology 2019, 290, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.K.; Misurka, J.; Ho, C.Y.; Chan, W.-X.; Agmon, Y.; Seidman, C.; Rakowski, H.; Carasso, S. Multilayer Myocardial Mechanics in Genotype-Positive Left Ventricular Hypertrophy-Negative Patients With Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2018, 122, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Taha, K.; Te Rijdt, W.P.; Verstraelen, T.E.; Cramer, M.J.; de Boer, R.A.; de Bruin-Bon, R.H.A.C.M.; Bouma, B.J.; Asselbergs, F.W.; Wilde, A.A.M.; van den Berg, M.P.; et al. Early Mechanical Alterations in Phospholamban Mutation Carriers. JACC Cardiovasc. Imaging 2020. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Piek, A.; Schouten, E.M.; van de Kolk, C.W.A.; Mueller, C.; Mebazaa, A.; Voors, A.A.; de Boer, R.A.; Silljé, H.H.W. Plasma levels of heart failure biomarkers are primarily a reflection of extracardiac production. Theranostics 2018, 8, 4155–4169. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of Amer. J. Am. Coll. Cardiol. 2017, 70, 776–803. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA Guideline for the Management of Heart Failure. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef] [Green Version]
- Hollenberg, S.M.; Warner Stevenson, L.; Ahmad, T.; Amin, V.J.; Bozkurt, B.; Butler, J.; Davis, L.L.; Drazner, M.H.; Kirkpatrick, J.N.; Peterson, P.N.; et al. 2019 ACC Expert Consensus Decision Pathway on Risk Assessment, Management, and Clinical Trajectory of Patients Hospitalized With Heart Failure. J. Am. Coll. Cardiol. 2019, 74, 1966–2011. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.A.; Barry, J.; Roberts, P.R.; Morgan, J.M. Brain natriuretic peptide for the prediction of sudden cardiac death and ventricular arrhythmias: A meta-analysis. Eur. J. Heart Fail. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Levine, Y.C.; Rosenberg, M.A.; Mittleman, M.; Samuel, M.; Methachittiphan, N.; Link, M.; Josephson, M.E.; Buxton, A.E. B-type natriuretic peptide is a major predictor of ventricular tachyarrhythmias. Heart Rhythm 2014, 11, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Piek, A.; Suthahar, N.; Voors, A.A.; Boer, R.A.; Silljé, H.H.W. A combined bioinformatics, experimental and clinical approach to identify novel cardiac-specific heart failure biomarkers: Is Dickkopf -3 (DKK3) a possible candidate? Eur. J. Heart Fail. 2020, 22, 2065–2074. [Google Scholar] [CrossRef]
- Reyat, J.S.; Chua, W.; Cardoso, V.R.; Witten, A.; Kastner, P.M.; Kabir, S.N.; Sinner, M.F.; Wesselink, R.; Holmes, A.P.; Pavlovic, D.; et al. Reduced left atrial cardiomyocyte PITX2 and elevated circulating BMP10 predict atrial fibrillation after ablation. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.-P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2020. [Google Scholar] [CrossRef]
- Amsterdam, E.A.; Wenger, N.K.; Brindis, R.G.; Casey, D.E.; Ganiats, T.G.; Holmes, D.R.; Jaffe, A.S.; Jneid, H.; Kelly, R.F.; Kontos, M.C.; et al. 2014 AHA/ACC Guideline for the Management of Patients With Non–ST-Elevation Acute Coronary Syndromes. Circulation 2014, 130. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D.; Thygesen, K.; Alpert, J.S.; White, H.D.; Jaffe, A.S.; et al. Third universal definition of myocardial infarction. Eur. Heart J. 2012, 33, 2551–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twerenbold, R.; Jaffe, A.; Reichlin, T.; Reiter, M.; Mueller, C. High-sensitive troponin T measurements: What do we gain and what are the challenges? Eur. Heart J. 2012, 33, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Rezar, R.; Jirak, P.; Gschwandtner, M.; Derler, R.; Felder, T.K.; Haslinger, M.; Kopp, K.; Seelmaier, C.; Granitz, C.; Hoppe, U.C.; et al. Heart-Type Fatty Acid-Binding Protein (H-FABP) and Its Role as a Biomarker in Heart Failure: What Do We Know So Far? J. Clin. Med. 2020, 9, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaier, T.E.; Alaour, B.; Marber, M. Cardiac Myosin-Binding Protein C—From Bench to Improved Diagnosis of Acute Myocardial Infarction. Cardiovasc. Drugs Ther. 2019, 33, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, C.; Barwari, T.; Joshi, A.; Theofilatos, K.; Zampetaki, A.; Barallobre-Barreiro, J.; Singh, B.; Sörensen, N.A.; Neumann, J.T.; Zeller, T.; et al. Comparative Analysis of Circulating Noncoding RNAs Versus Protein Biomarkers in the Detection of Myocardial Injury. Circ. Res. 2019, 125, 328–340. [Google Scholar] [CrossRef]
- Ho, C.Y.; López, B.; Coelho-Filho, O.R.; Lakdawala, N.K.; Cirino, A.L.; Jarolim, P.; Kwong, R.; González, A.; Colan, S.D.; Seidman, J.G.; et al. Myocardial Fibrosis as an Early Manifestation of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2010, 363, 552–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.E.; Shi, L.; Day, S.M.; Colan, S.D.; Russell, M.W.; Towbin, J.A.; Sherrid, M.V.; Canter, C.E.; Jefferies, J.L.; Murphy, A.; et al. Biomarkers of cardiovascular stress and fibrosis in preclinical hypertrophic cardiomyopathy. Open Heart 2017, 4, e000615. [Google Scholar] [CrossRef]
- Cramer, G.E.; Gommans, D.H.F.; Dieker, H.-J.; Michels, M.; Verheugt, F.; de Boer, M.-J.; Bakker, J.; Fouraux, M.A.; Timmermans, J.; Kofflard, M.; et al. Exercise and myocardial injury in hypertrophic cardiomyopathy. Heart 2020, 106, 1169–1175. [Google Scholar] [CrossRef]
- Nakamura, T.; Sakamoto, K.; Yamano, T.; Kikkawa, M.; Zen, K.; Hikosaka, T.; Kubota, T.; Azuma, A.; Nishimura, T. Increased plasma brain natriuretic peptide level as a guide for silent myocardial ischemia in patients with non-obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2002. [Google Scholar] [CrossRef] [Green Version]
- Hinton, J.; Gabara, L.; Curzen, N. Is the true clinical value of high-sensitivity troponins as a biomarker of risk? The concept that detection of high-sensitivity troponin ‘never means nothing’. Expert Rev. Cardiovasc. Ther. 2020, 18, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Gommans, D.H.F.; Cramer, G.E.; Fouraux, M.A.; Bakker, J.; Michels, M.; Dieker, H.-J.; Timmermans, J.; Marcelis, C.L.M.; Verheugt, F.W.A.; de Boer, M.-J.; et al. Prediction of Extensive Myocardial Fibrosis in Nonhigh Risk Patients With Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2018, 122, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Lu, M.; Hou, C.; Chen, X.; Wang, J.; Yin, G.; Chu, J.; Zhang, S.; Prasad, S.K.; Pu, J.; et al. Relation Between N-Terminal Pro-Brain Natriuretic Peptide and Cardiac Remodeling and Function Assessed by Cardiovascular Magnetic Resonance Imaging in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2015, 115, 341–347. [Google Scholar] [CrossRef]
- Matsuo, K.; Nishikimi, T.; Yutani, C.; Kurita, T.; Shimizu, W.; Taguchi, A.; Suyama, K.; Aihara, N.; Kamakura, S.; Kangawa, K.; et al. Diagnostic Value of Plasma Levels of Brain Natriuretic Peptide in Arrhythmogenic Right Ventricular Dysplasia. Circulation 1998, 98, 2433–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, K.; Fujiwara, H.; Doyama, K.; Miyamae, M.; Fujiwara, T.; Suga, S.; Mukoyama, M.; Nakao, K.; Imura, H.; Sasayama, S. Ventricular expression of brain natriuretic peptide in hypertrophic cardiomyopathy. Circulation 1993, 88, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Coats, C.J.; Gallagher, M.J.; Foley, M.; O’Mahony, C.; Critoph, C.; Gimeno, J.; Dawnay, A.; McKenna, W.J.; Elliott, P.M. Relation between serum N-terminal pro-brain natriuretic peptide and prognosis in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2013, 34, 2529–2537. [Google Scholar] [CrossRef] [Green Version]
- Geske, J.B.; McKie, P.M.; Ommen, S.R.; Sorajja, P. B-Type Natriuretic Peptide and Survival in Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2013, 61, 2456–2460. [Google Scholar] [CrossRef] [Green Version]
- Stadiotti, I.; Pompilio, G.; Maione, A.S.; Pilato, C.A.; D’Alessandra, Y.; Sommariva, E. Arrhythmogenic cardiomyopathy: What blood can reveal? Heart Rhythm 2019, 16, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Ochi, Y.; Baba, Y.; Sugiura, K.; Takahashi, A.; Hirota, T.; Yamanaka, S.; Yamasaki, N.; Doi, Y.L.; Kitaoka, H. Elevation of high-sensitivity cardiac troponin T and left ventricular remodelling in hypertrophic cardiomyopathy. ESC Heart Fail. 2020, 7, 3593–3600. [Google Scholar] [CrossRef] [PubMed]
- Daniels, L.B.; Maisel, A.S. Natriuretic Peptides. J. Am. Coll. Cardiol. 2007, 50, 2357–2368. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.; Larson, M.G.; Levy, D.; Benjamin, E.J.; Leip, E.P.; Omland, T.; Wolf, P.A.; Vasan, R.S. Plasma Natriuretic Peptide Levels and the Risk of Cardiovascular Events and Death. N. Engl. J. Med. 2004, 350, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J. Assessing the Role of Circulating, Genetic, and Imaging Biomarkers in Cardiovascular Risk Prediction. Circulation 2011, 123, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Patton, K.K.; Ellinor, P.T.; Heckbert, S.R.; Christenson, R.H.; DeFilippi, C.; Gottdiener, J.S.; Kronmal, R.A. N-Terminal Pro-B-Type Natriuretic Peptide Is a Major Predictor of the Development of Atrial Fibrillation. Circulation 2009, 120, 1768–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, M.H.; Hansen, T.W.; Christensen, M.K.; Gustafsson, F.; Rasmussen, S.; Wachtell, K.; Ibsen, H.; Torp-Pedersen, C.; Hildebrandt, P.R. N-terminal pro-brain natriuretic peptide, but not high sensitivity C-reactive protein, improves cardiovascular risk prediction in the general population. Eur. Heart J. 2007, 28, 1374–1381. [Google Scholar] [CrossRef] [Green Version]
- Suthahar, N.; Lau, E.S.; Blaha, M.J.; Paniagua, S.M.; Larson, M.G.; Psaty, B.M.; Benjamin, E.J.; Allison, M.A.; Bartz, T.M.; Januzzi, J.L.; et al. Sex-Specific Associations of Cardiovascular Risk Factors and Biomarkers With Incident Heart Failure. J. Am. Coll. Cardiol. 2020, 76, 1455–1465. [Google Scholar] [CrossRef]
- Wang, T.J.; Wollert, K.C.; Larson, M.G.; Coglianese, E.; McCabe, E.L.; Cheng, S.; Ho, J.E.; Fradley, M.G.; Ghorbani, A.; Xanthakis, V.; et al. Prognostic Utility of Novel Biomarkers of Cardiovascular Stress. Circulation 2012, 126, 1596–1604. [Google Scholar] [CrossRef] [Green Version]
- Suthahar, N.; Meems, L.M.G.; van Veldhuisen, D.J.; Walter, J.E.; Gansevoort, R.T.; Heymans, S.; Schroen, B.; van der Harst, P.; Kootstra-Ros, J.E.; van Empel, V.; et al. High-Sensitivity Troponin-T and Cardiovascular Outcomes in the Community: Differences Between Women and Men. Mayo Clin. Proc. 2020, 95, 1158–1168. [Google Scholar] [CrossRef]
- Kaura, A.; Panoulas, V.; Glampson, B.; Davies, J.; Mulla, A.; Woods, K.; Omigie, J.; Shah, A.D.; Channon, K.M.; Weber, J.N.; et al. Association of troponin level and age with mortality in 250,000 patients: Cohort study across five UK acute care centres. BMJ 2019, l6055. [Google Scholar] [CrossRef]
- Korngold, E.C.; Januzzi, J.L.; Lou Gantzer, M.; Moorthy, M.V.; Cook, N.R.; Albert, C.M. Amino-Terminal Pro-B-Type Natriuretic Peptide and High-Sensitivity C-Reactive Protein as Predictors of Sudden Cardiac Death Among Women. Circulation 2009, 119, 2868–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton, K.K.; Sotoodehnia, N.; DeFilippi, C.; Siscovick, D.S.; Gottdiener, J.S.; Kronmal, R.A. N-terminal pro-B-type natriuretic peptide is associated with sudden cardiac death risk: The Cardiovascular Health Study. Heart Rhythm 2011, 8, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, P.; Michalak, E.; Kowalik, I.; Franaszczyk, M.; Sobieszczanska-Malek, M.; Truszkowska, G.; Stepien-Wojno, M.; Biernacka, E.K.; Foss-Nieradko, B.; Lewandowski, M.; et al. Can Circulating Cardiac Biomarkers Be Helpful in the Assessment of LMNA Mutation Carriers? J. Clin. Med. 2020, 9, 1443. [Google Scholar] [CrossRef] [PubMed]
- Tidholm, A.; Häggström, J.; Borgarelli, M.; Tarducci, A. Canine Idiopathic Dilated Cardiomyopathy. Part I: Aetiology, Clinical Characteristics, Epidemiology and Pathology. Vet. J. 2001, 162, 92–107. [Google Scholar] [CrossRef]
- Wess, G.; Domenech, O.; Dukes-McEwan, J.; Häggström, J.; Gordon, S. European Society of Veterinary Cardiology screening guidelines for dilated cardiomyopathy in Doberman Pinschers. J. Vet. Cardiol. 2017, 19, 405–415. [Google Scholar] [CrossRef]
- Wess, G.; Butz, V.; Mahling, M.; Hartmann, K. Evaluation of N-terminal pro-B-type natriuretic peptide as a diagnostic marker of various stages of cardiomyopathy in Doberman Pinschers. Am. J. Vet. Res. 2011, 72, 642–649. [Google Scholar] [CrossRef]
- Klüser, L.; Maier, E.T.; Wess, G. Evaluation of a high-sensitivity cardiac troponin I assay compared to a first-generation cardiac troponin I assay in Doberman Pinschers with and without dilated cardiomyopathy. J. Vet. Intern. Med. 2019, 33, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Gehlken, C.; Suthahar, N.; Meijers, W.C.; de Boer, R.A. Galectin-3 in Heart Failure. Heart Fail. Clin. 2018, 14, 75–92. [Google Scholar] [CrossRef]
- van der Velde, A.R.; Gullestad, L.; Ueland, T.; Aukrust, P.; Guo, Y.; Adourian, A.; Muntendam, P.; van Veldhuisen, D.J.; de Boer, R.A. Prognostic Value of Changes in Galectin-3 Levels Over Time in Patients With Heart Failure. Circ. Heart Fail. 2013, 6, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Stevens, S.R.; Lucas, J.; Fiuzat, M.; Adams, K.F.; Whellan, D.J.; Donahue, M.P.; Kitzman, D.W.; Piña, I.L.; Zannad, F.; et al. Utility of Growth Differentiation Factor-15, A Marker of Oxidative Stress and Inflammation, in Chronic Heart Failure. JACC Heart Fail. 2017, 5, 724–734. [Google Scholar] [CrossRef]
- Wollert, K.C.; Kempf, T. Growth Differentiation Factor 15 in Heart Failure: An Update. Curr. Heart Fail. Rep. 2012, 9, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, R.; Lee, R.T. The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat. Rev. Drug Discov. 2008, 7, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Lotierzo, M.; Dupuy, A.M.; Kalmanovich, E.; Roubille, F.; Cristol, J.P. sST2 as a value-added biomarker in heart failure. Clin. Chim. Acta 2020, 501, 120–130. [Google Scholar] [CrossRef]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; Ho, J.E.; Liu, F.-T.; de Boer, R.A. Galectin-3 Activation and Inhibition in Heart Failure and Cardiovascular Disease: An Update. Theranostics 2018, 8, 593–609. [Google Scholar] [CrossRef] [PubMed]
- Piek, A.; Du, W.; de Boer, R.A.; Silljé, H.H.W. Novel heart failure biomarkers: Why do we fail to exploit their potential? Crit. Rev. Clin. Lab. Sci. 2018, 55, 246–263. [Google Scholar] [CrossRef]
- Gawor, M.; Śpiewak, M.; Janas, J.; Kożuch, K.; Wróbel, A.; Mazurkiewicz, Ł.; Baranowski, R.; Marczak, M.; Grzybowski, J. The usefulness of sST2 and galectin-3 as novel biomarkers for better risk stratification in hypertrophic cardiomyopathy. Kardiol. Pol. 2017, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Yakar Tuluce, S.; Tuluce, K.; Cil, Z.; Volkan Emren, S.; İlke Akyildiz, Z.; Ergene, O. Galectin-3 levels in patients with hypertrophic cardiomyopathy and its relationship with left ventricular mass index and function. Anatol. J. Cardiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.-J.; Xu, J.; Du, W.; Zhang, J.-X.; Zhong, M.; Zhou, Y.-N. Cardiac magnetic resonance and galectin-3 level as predictors of prognostic outcomes for non-ischemic cardiomyopathy patients. Int. J. Cardiovasc. Imaging 2016, 32, 1725–1733. [Google Scholar] [CrossRef]
- Montoro-García, S.; Hernández-Romero, D.; Jover, E.; García-Honrubia, A.; Vilchez, J.A.; Casas, T.; Martínez, P.; Climent, V.; Caballero, L.; Valdés, M.; et al. Growth differentiation factor-15, a novel biomarker related with disease severity in patients with hypertrophic cardiomyopathy. Eur. J. Intern. Med. 2012, 23, 169–174. [Google Scholar] [CrossRef]
- Lichtenauer, M.; Jirak, P.; Wernly, B.; Paar, V.; Rohm, I.; Jung, C.; Schernthaner, C.; Kraus, J.; Motloch, L.J.; Yilmaz, A.; et al. A comparative analysis of novel cardiovascular biomarkers in patients with chronic heart failure. Eur. J. Intern. Med. 2017, 44, 31–38. [Google Scholar] [CrossRef]
- Vergaro, G.; Del Franco, A.; Giannoni, A.; Prontera, C.; Ripoli, A.; Barison, A.; Masci, P.G.; Aquaro, G.D.; Cohen Solal, A.; Padeletti, L.; et al. Galectin-3 and myocardial fibrosis in nonischemic dilated cardiomyopathy. Int. J. Cardiol. 2015, 184, 96–100. [Google Scholar] [CrossRef]
- Stojkovic, S.; Kaider, A.; Koller, L.; Brekalo, M.; Wojta, J.; Diedrich, A.; Demyanets, S.; Pezawas, T. GDF-15 is a better complimentary marker for risk stratification of arrhythmic death in non-ischaemic, dilated cardiomyopathy than soluble ST 2. J. Cell. Mol. Med. 2018, 22, 2422–2429. [Google Scholar] [CrossRef] [Green Version]
- Lok, S.I.; Winkens, B.; Goldschmeding, R.; van Geffen, A.J.P.; Nous, F.M.A.; van Kuik, J.; van der Weide, P.; Klöpping, C.; Kirkels, J.H.; Lahpor, J.R.; et al. Circulating growth differentiation factor-15 correlates with myocardial fibrosis in patients with non-ischaemic dilated cardiomyopathy and decreases rapidly after left ventricular assist device support. Eur. J. Heart Fail. 2012, 14, 1249–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binas, D.; Daniel, H.; Richter, A.; Ruppert, V.; Schlüter, K.-D.; Schieffer, B.; Pankuweit, S. The prognostic value of sST2 and galectin-3 considering different aetiologies in non-ischaemic heart failure. Open Heart 2018, 5, e000750. [Google Scholar] [CrossRef] [PubMed]
- Oz, F.; Onur, I.; Elitok, A.; Ademoglu, E.; Altun, I.; Bilge, A.K.; Adalet, K. Galectin-3 correlates with arrhythmogenic right ventricular cardiomyopathy and predicts the risk of ventricular arrhythmias in patients with implantable defibrillators. Acta Cardiol. 2017, 72, 453–459. [Google Scholar] [CrossRef]
- Akdis, D.; Chen, L.; Saguner, A.; Zhang, N.; Gawinecka, J.; Saleh, L.; Von Eckardstein, A.; Ren, J.; Matter, C.; Hu, Z.; et al. Novel plasma biomarkers in arrhythmogenic cardiomyopathy: The role of ST2 and GDF-15 in predicting biventricular involvement. Eur. Heart J. 2020, 41. [Google Scholar] [CrossRef]
- Broch, K.; Leren, I.S.; Saberniak, J.; Ueland, T.; Edvardsen, T.; Gullestad, L.; Haugaa, K.H. Soluble ST2 is associated with disease severity in arrhythmogenic right ventricular cardiomyopathy. Biomarkers 2017, 22, 367–371. [Google Scholar] [CrossRef] [Green Version]
- de Boer, R.A.; Nayor, M.; DeFilippi, C.R.; Enserro, D.; Bhambhani, V.; Kizer, J.R.; Blaha, M.J.; Brouwers, F.P.; Cushman, M.; Lima, J.A.C.; et al. Association of Cardiovascular Biomarkers With Incident Heart Failure With Preserved and Reduced Ejection Fraction. JAMA Cardiol. 2018, 3, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Velde, A.R.; Meijers, W.C.; van den Heuvel, E.R.; Bakker, S.J.; van Gilst, W.H.; van der Harst, P.; Hillege, H.; de Boer, R.A. Determinants of temporal changes in galectin-3 level in the general population: Data of PREVEND. Int. J. Cardiol. 2016, 222, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, A.; Bhambhani, V.; Christenson, R.H.; Meijers, W.C.; de Boer, R.A.; Levy, D.; Larson, M.G.; Ho, J.E. Longitudinal Change in Galectin-3 and Incident Cardiovascular Outcomes. J. Am. Coll. Cardiol. 2018, 72, 3246–3254. [Google Scholar] [CrossRef]
- Captur, G.; Heywood, W.E.; Coats, C.; Rosmini, S.; Patel, V.; Lopes, L.R.; Collis, R.; Patel, N.; Syrris, P.; Bassett, P.; et al. Identification of a Multiplex Biomarker Panel for Hypertrophic Cardiomyopathy Using Quantitative Proteomics and Machine Learning. Mol. Cell. Proteom. 2020, 19, 114–127. [Google Scholar] [CrossRef]
- Das, S.; Shah, R.; Dimmeler, S.; Freedman, J.E.; Holley, C.; Lee, J.-M.; Moore, K.; Musunuru, K.; Wang, D.-Z.; Xiao, J.; et al. Noncoding RNAs in Cardiovascular Disease: Current Knowledge, Tools and Technologies for Investigation, and Future Directions: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2020, 13. [Google Scholar] [CrossRef]
- Viereck, J.; Thum, T. Circulating Noncoding RNAs as Biomarkers of Cardiovascular Disease and Injury. Circ. Res. 2017, 120, 381–399. [Google Scholar] [CrossRef]
- Schulte, C.; Barwari, T.; Joshi, A.; Zeller, T.; Mayr, M. Noncoding RNAs versus Protein Biomarkers in Cardiovascular Disease. Trends Mol. Med. 2020, 26, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Stępień, E.; Costa, M.C.; Kurc, S.; Drożdż, A.; Cortez-Dias, N.; Enguita, F.J. The circulating non-coding RNA landscape for biomarker research: Lessons and prospects from cardiovascular diseases. Acta Pharmacol. Sin. 2018, 39, 1085–1099. [Google Scholar] [CrossRef]
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2016; pp. 3–17. [Google Scholar]
- Luo, W.; Dai, Y.; Chen, Z.; Yue, X.; Andrade-Powell, K.C.; Chang, J. Spatial and temporal tracking of cardiac exosomes in mouse using a nano-luciferase-CD63 fusion protein. Commun. Biol. 2020, 3, 114. [Google Scholar] [CrossRef]
- Bellin, G.; Gardin, C.; Ferroni, L.; Chachques, J.; Rogante, M.; Mitrečić, D.; Ferrari, R.; Zavan, B. Exosome in Cardiovascular Diseases: A Complex World Full of Hope. Cells 2019, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, A.; Tandon, M.; Alevizos, I.; Illei, G.G. The Majority of MicroRNAs Detectable in Serum and Saliva Is Concentrated in Exosomes. PLoS ONE 2012, 7, e30679. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [Green Version]
- Jansen, F.; Yang, X.; Proebsting, S.; Hoelscher, M.; Przybilla, D.; Baumann, K.; Schmitz, T.; Dolf, A.; Endl, E.; Franklin, B.S.; et al. MicroRNA Expression in Circulating Microvesicles Predicts Cardiovascular Events in Patients With Coronary Artery Disease. J. Am. Heart Assoc. 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Gidlöf, O.; Evander, M.; Rezeli, M.; Marko-Varga, G.; Laurell, T.; Erlinge, D. Proteomic profiling of extracellular vesicles reveals additional diagnostic biomarkers for myocardial infarction compared to plasma alone. Sci. Rep. 2019, 9, 8991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.-K.; Zhu, J.-Q.; Zhang, J.-T.; Li, Q.; Li, Y.; He, J.; Qin, Y.-W.; Jing, Q. Circulating microRNA: A novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J. 2010, 31, 659–666. [Google Scholar] [CrossRef]
- Olivieri, F.; Antonicelli, R.; Lorenzi, M.; D’Alessandra, Y.; Lazzarini, R.; Santini, G.; Spazzafumo, L.; Lisa, R.; La Sala, L.; Galeazzi, R.; et al. Diagnostic potential of circulating miR-499-5p in elderly patients with acute non ST-elevation myocardial infarction. Int. J. Cardiol. 2013, 167, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, R.; Viviani Anselmi, C.; Losi, M.A.; Papa, L.; Cavarretta, E.; Da Costa Martins, P.; Contaldi, C.; Saccani Jotti, G.; Franzone, A.; Galastri, L.; et al. Circulating miR-29a, Among Other Up-Regulated MicroRNAs, Is the Only Biomarker for Both Hypertrophy and Fibrosis in Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Ellims, A.H.; Moore, X.; White, D.A.; Taylor, A.J.; Chin-Dusting, J.; Dart, A.M. Circulating microRNAs as biomarkers for diffuse myocardial fibrosis in patients with hypertrophic cardiomyopathy. J. Transl. Med. 2015, 13, 314. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntelios, D.; Meditskou, S.; Efthimiadis, G.; Pitsis, A.; Nikolakaki, E.; Girtovitis, F.; Parcharidou, D.; Zegkos, T.; Kouidou, S.; Karvounis, H.; et al. Elevated plasma levels of miR-29a are associated with hemolysis in patients with hypertrophic cardiomyopathy. Clin. Chim. Acta 2017, 471, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, K.; Wilczek, A.L.; de Gonzalo-Calvo, D.; Pfanne, A.; Derda, A.A.; Zwadlo, C.; Bavendiek, U.; Bauersachs, J.; Fiedler, J.; Thum, T. Serum circular RNAs act as blood-based biomarkers for hypertrophic obstructive cardiomyopathy. Sci. Rep. 2019, 9, 20350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S.D.; Karimpour-Fard, A.; Peterson, V.; Auerbach, S.R.; Stenmark, K.R.; Stauffer, B.L.; Sucharov, C.C. Circulating microRNA as a biomarker for recovery in pediatric dilated cardiomyopathy. J. Heart Lung Transplant. 2015, 34, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.-L.; Zhang, H.-F.; Shen, J.; Zhang, Q.; Li, X.-L. Circulating microRNAs levels in Chinese heart failure patients caused by dilated cardiomyopathy. Indian Heart J. 2013, 65, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Bueno Marinas, M.; Celeghin, R.; Cason, M.; Bariani, R.; Frigo, A.C.; Jager, J.; Syrris, P.; Elliott, P.M.; Bauce, B.; Thiene, G.; et al. A microRNA Expression Profile as Non-Invasive Biomarker in a Large Arrhythmogenic Cardiomyopathy Cohort. Int. J. Mol. Sci. 2020, 21, 1536. [Google Scholar] [CrossRef] [Green Version]
- Bye, A.; Røsjø, H.; Nauman, J.; Silva, G.J.J.; Follestad, T.; Omland, T.; Wisløff, U. Circulating microRNAs predict future fatal myocardial infarction in healthy individuals—The HUNT study. J. Mol. Cell. Cardiol. 2016, 97, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Zampetaki, A.; Willeit, P.; Tilling, L.; Drozdov, I.; Prokopi, M.; Renard, J.-M.; Mayr, A.; Weger, S.; Schett, G.; Shah, A.; et al. Prospective Study on Circulating MicroRNAs and Risk of Myocardial Infarction. J. Am. Coll. Cardiol. 2012, 60, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Maisch, B. Cardio-Immunology of Myocarditis: Focus on Immune Mechanisms and Treatment Options. Front. Cardiovasc. Med. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Kaya, Z.; Schwab, J.; Westenfeld, R.; Ehlermann, P.; Bachelier, K.; Oettl, R.; von Kaisenberg, C.S.; Katus, H.A.; Bauersachs, J.; et al. Evidence of autoantibodies against cardiac troponin I and sarcomeric myosin in peripartum cardiomyopathy. Basic Res. Cardiol. 2015, 110, 60. [Google Scholar] [CrossRef]
- Peukert, S.; Fu, M.L.X.; Eftekhari, P.; Poepping, I.; Voss, A.; Thalhammer, C.; Hempel, A.; Menz, M.; Dietz, R.; Osterziel, K.J. The Frequency of Occurrence of Anti-cardiac Receptor Autoantibodies and their Correlation with Clinical Manifestation in Patients with Hypertrophic Cardiomyopathy. Autoimmunity 1999, 29, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.L.-X.; Hoebeke, J.; Matsui, S.; Matoba, M.; Magnusson, Y.; Hedner, T.; Herlitz, H.; Hjalmarson, Å. Autoantibodies against Cardiac G-Protein-Coupled Receptors Define Different Populations with Cardiomyopathies but Not with Hypertension. Clin. Immunol. Immunopathol. 1994, 72, 15–20. [Google Scholar] [CrossRef]
- Boudonas, G.; Boura, P.; Lefkos, N.; Zacharioydaki, E.; Efthymiadis, A. A Possible Role for Autoantibodies in Left Ventricular Hypertrophy. Cardiology 1994, 84, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Liu, R.; Luo, X.; Gao, X.; Hu, F.; Guo, C.; Wang, J.; Hu, X.; Chun, Y.; Yuan, J.; et al. The relationship between β 1 -adrenergic and M 2 -muscarinic receptor autoantibodies and hypertrophic cardiomyopathy. Exp. Physiol. 2020, 105, 522–530. [Google Scholar] [CrossRef]
- Sánchez, D.; Gregor, P.; Čurila, K.; Hoffmanová, I.; Hábová, V.; Tučková, L.; Tlaskalová-Hogenová, H. Anti-calreticulin antibodies and calreticulin in sera of patients diagnosed with dilated or hypertrophic cardiomyopathy. Autoimmunity 2016, 49, 554–562. [Google Scholar] [CrossRef]
- Boehm, J.; Orth, T.; Van Nguyen, P.; Söling, H.-D. Systemic lupus erythematosus is associated with increased auto-antibody titers against calreticulin and Grp94, but calreticulin is not the Ro/SS-A antigen. Eur. J. Clin. Investig. 1994, 24, 248–257. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, J.; Liu, Z.; Fu, H.; Huo, Q.; Gu, Y.; Liu, Y. Association of calreticulin expression with disease activity and organ damage in systemic lupus erythematosus patients. Exp. Ther. Med. 2017, 13, 2577–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caforio, A.L.; Keeling, P.; McKenna, W.; Mann, J.; Caforio, A.L.; Bottazzo, G.; Zachara, E.; Mestroni, L.; Camerini, F. Evidence from family studies for autoimmunity in dilated cardiomyopathy. Lancet 1994, 344, 773–777. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Vinci, A.; Iliceto, S. Anti-heart autoantibodies in familial dilated cardiomyopathy. Autoimmunity 2008, 41, 462–469. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Mahon, N.G.; Baig, M.K.; Tona, F.; Murphy, R.T.; Elliott, P.M.; McKenna, W.J. Prospective Familial Assessment in Dilated Cardiomyopathy. Circulation 2007, 115, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Vilela, E.M.; Bettencourt-Silva, R.; da Costa, J.T.; Barbosa, A.R.; Silva, M.P.; Teixeira, M.; Primo, J.; Gama Ribeiro, V.; Nunes, J.P.L. Anti-cardiac troponin antibodies in clinical human disease: A systematic review. Ann. Transl. Med. 2017, 5, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, D.; Pieroni, M.; Fatah, M.; Charpentier, F.; Cunningham, K.S.; Spears, D.A.; Chatterjee, D.; Suna, G.; Bos, J.M.; Ackerman, M.J.; et al. An autoantibody profile detects Brugada syndrome and identifies abnormally expressed myocardial proteins. Eur. Heart J. 2020, 41, 2878–2890. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef] [Green Version]
- Calkins, H. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy: Is this too good to be true? Eur. Heart J. 2018, 39, 3945–3946. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.-Y.; et al. Evidence From Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Lodder, E.M. A highly specific biomarker for Brugada syndrome. Also too good to be true? Eur. Heart J. 2020, 41, 2891–2893. [Google Scholar] [CrossRef] [PubMed]
- Stiles, M.K.; Wilde, A.A.M.; Abrams, D.J.; Ackerman, M.J.; Albert, C.M.; Behr, E.R.; Chugh, S.S.; Cornel, M.C.; Gardner, K.; Ingles, J.; et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm 2021, 18, e1–e50. [Google Scholar] [CrossRef]
- Wang, X.; Osinska, H.; Klevitsky, R.; Gerdes, A.M.; Nieman, M.; Lorenz, J.; Hewett, T.; Robbins, J. Expression of R120G–αB-Crystallin Causes Aberrant Desmin and αB-Crystallin Aggregation and Cardiomyopathy in Mice. Circ. Res. 2001, 89, 84–91. [Google Scholar] [CrossRef] [Green Version]
- McLendon, P.M.; Robbins, J. Desmin-related cardiomyopathy: An unfolding story. Am. J. Physiol. Circ. Physiol. 2011, 301, H1220–H1228. [Google Scholar] [CrossRef] [Green Version]
- te Rijdt, W.P.; van Tintelen, J.P.; Vink, A.; van der Wal, A.C.; de Boer, R.A.; van den Berg, M.P.; Suurmeijer, A.J.H. Phospholamban p.Arg14del cardiomyopathy is characterized by phospholamban aggregates, aggresomes, and autophagic degradation. Histopathology 2016, 69, 542–550. [Google Scholar] [CrossRef]
- Padrón-Barthe, L.; Villalba-Orero, M.; Gómez-Salinero, J.M.; Domínguez, F.; Román, M.; Larrasa-Alonso, J.; Ortiz-Sánchez, P.; Martínez, F.; López-Olañeta, M.; Bonzón-Kulichenko, E.; et al. Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3β. Circulation 2019, 140, 1188–1204. [Google Scholar] [CrossRef]
- Wong, L.L.; Zou, R.; Zhou, L.; Lim, J.Y.; Phua, D.C.Y.; Liu, C.; Chong, J.P.C.; Ng, J.Y.X.; Liew, O.W.; Chan, S.P.; et al. Combining Circulating MicroRNA and NT-proBNP to Detect and Categorize Heart Failure Subtypes. J. Am. Coll. Cardiol. 2019, 73, 1300–1313. [Google Scholar] [CrossRef] [PubMed]
- Meijers, W.C.; van der Velde, A.R.; Muller Kobold, A.C.; Dijck-Brouwer, J.; Wu, A.H.; Jaffe, A.; de Boer, R.A. Variability of biomarkers in patients with chronic heart failure and healthy controls. Eur. J. Heart Fail. 2017, 19, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Brann, A.; Chang, K.-W.; Maisel, A.S. The Confounding Effects of Non-cardiac Pathologies on the Interpretation of Cardiac Biomarkers. Curr. Heart Fail. Rep. 2018, 15, 239–249. [Google Scholar] [CrossRef] [PubMed]
- The Role of Biomarkers in Inherited Cardiac Conditions. Available online: https://clinicaltrials.gov/ct2/show/NCT04312230 (accessed on 14 January 2021).
- Biomarkers of Inherited Cardiovascular Conditions. Available online: https://clinicaltrials.gov/ct2/show/NCT02804256 (accessed on 14 January 2021).
- Molecular and Imaging Studies of Cardiovascular Health and Disease (Biobank). Available online: https://clinicaltrials.gov/ct2/show/NCT02804269 (accessed on 1 February 2021).
- Defining the Genetics, Biomarkers and Outcomes for Dilated Cardiomyopathy (Go-DCM). Available online: https://clinicaltrials.gov/ct2/show/NCT03843255 (accessed on 1 February 2021).
- An Integrative-“Omics” Study of Cardiomyopathy Patients for Diagnosis and Prognosis in China (AOCC). Available online: https://clinicaltrials.gov/ct2/show/NCT03076580 (accessed on 1 February 2021).
- HCMR—Novel Markers of Prognosis in Hypertrophic Cardiomyopathy (HCMR). Available online: https://clinicaltrials.gov/ct2/show/NCT01915615 (accessed on 1 February 2021).
- Jansen, M.; Christiaans, I.; van der Crabben, S.N.; Michels, M.; Huurman, R.; Hoedemaekers, Y.M.; Dooijes, D.; Jongbloed, J.D.H.; Boven, L.G.; Lekanne Deprez, R.H.; et al. BIO FOr CARE: Biomarkers of hypertrophic cardiomyopathy development and progression in carriers of Dutch founder truncating MYBPC3 variants—design and status. Netherlands Heart J. 2021. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stege, N.M.; de Boer, R.A.; van den Berg, M.P.; Silljé, H.H.W. The Time Has Come to Explore Plasma Biomarkers in Genetic Cardiomyopathies. Int. J. Mol. Sci. 2021, 22, 2955. https://doi.org/10.3390/ijms22062955
Stege NM, de Boer RA, van den Berg MP, Silljé HHW. The Time Has Come to Explore Plasma Biomarkers in Genetic Cardiomyopathies. International Journal of Molecular Sciences. 2021; 22(6):2955. https://doi.org/10.3390/ijms22062955
Chicago/Turabian StyleStege, Nienke M., Rudolf A. de Boer, Maarten P. van den Berg, and Herman H. W. Silljé. 2021. "The Time Has Come to Explore Plasma Biomarkers in Genetic Cardiomyopathies" International Journal of Molecular Sciences 22, no. 6: 2955. https://doi.org/10.3390/ijms22062955
APA StyleStege, N. M., de Boer, R. A., van den Berg, M. P., & Silljé, H. H. W. (2021). The Time Has Come to Explore Plasma Biomarkers in Genetic Cardiomyopathies. International Journal of Molecular Sciences, 22(6), 2955. https://doi.org/10.3390/ijms22062955