
Role of Rho in Salt-Sensitive Hypertension


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Mechanisms Regulating Rho and Rac Activation
3. Renin-Angiotensin-Aldosterone System (RAAS) and Salt-Sensitive Hypertension
4. Central Nervous System
Role of Rho and Rac1 in Salt-Sensitive Hypertension
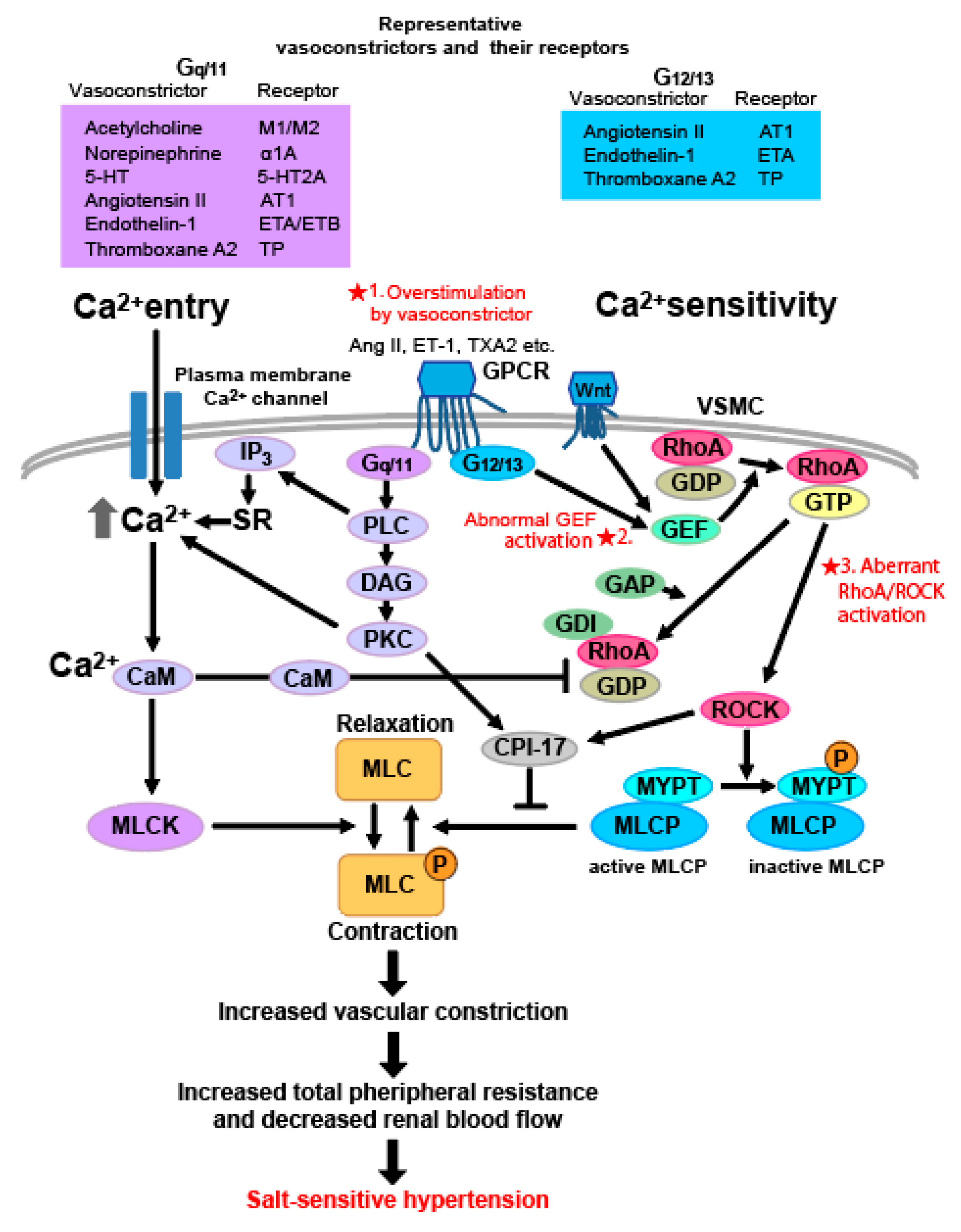
5. Vascular Smooth Muscle Cell
5.1. Role of Rho in Vascular Smooth Muscle Contraction and the Mechanism of Rho-Associated Salt-Sensitive Hypertension
5.2. Rho GEF-Related Salt-Sensitive Hypertension
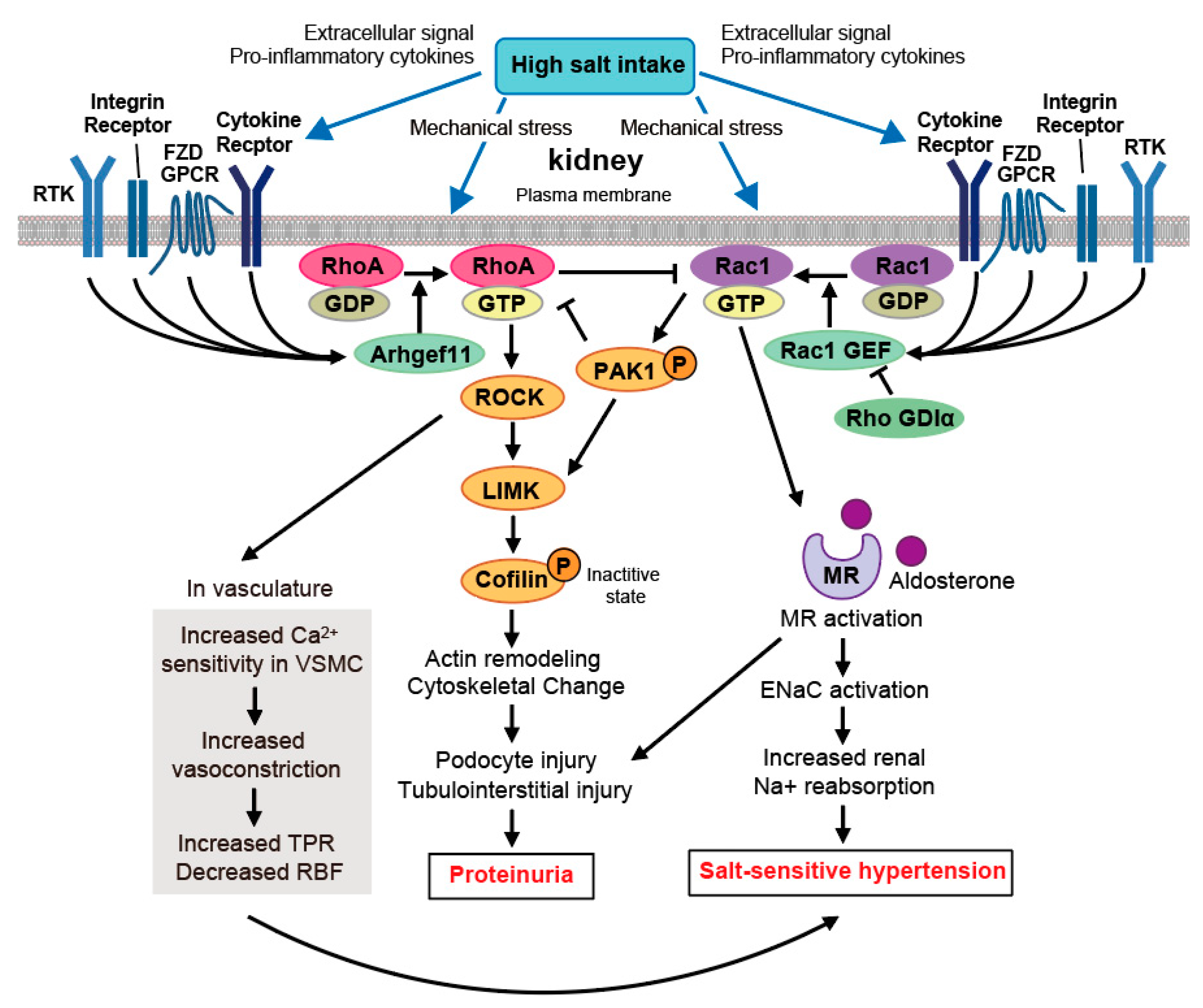
6. Kidney
Crosstalk between RhoA and Rac1 in Salt-Sensitive Hypertension
7. Aging Vasculature
7.1. Aging-Associated Hypertension via Noncanonical Wnt-RhoA/PCP Signaling
7.2. RBF Reduction via Ang II-Wnt5a-RhoA Activation in Aging-Associated Hypertension
8. Endothelium
Vascular Endothelial Dysfunction by Rho/ROCK Activation
9. Potential Role of Rho as a Therapeutic Target in Salt-Sensitive Hypertension
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement.
Informed Consent Statement.
Data Availability Statement.
Conflicts of Interest
Abbreviations
| Ang II | Angiotensin II |
| BH4 | tetrahydrobiopterin |
| BP | blood pressure |
| Cdc42 | cell division control protein 42 homolog |
| CSF | cerebrospinal fluid |
| CPI-17 | C-kinase potentiated protein Phosphatase 1 inhibitor, molecular mass 17 kDa |
| DAG | diacylglycerol |
| DOCA | deoxycorticosterone acetate |
| DS | Dahl salt-sensitive |
| ENaC | epithelial sodium channel |
| ET-1 | Endothelin-1 |
| eNOS | endothelial nitric oxide synthetase |
| FZD | Frizzled |
| GAP | GTPase-activating protein |
| GDI | GDP-dissociation inhibitor |
| GEF | Guanine nucleotide exchange factors |
| GDP | Guanosine diphosphate |
| GTP | Guanosine triphosphatase |
| GPCR | G-protein-coupled receptor |
| IP3 | inositol triphosphatase |
| ICV | intracerebroventricular |
| JNK | c-Jun N-terminal kinase |
| LIMK | LIM-kinase |
| MR | mineralocorticoid receptor |
| MLC | myosin light chain |
| MLCK | myosin light chain kinase |
| MLCP | myosin light chain phosphatase |
| MYPT | myosin phosphatase target subunit |
| NO | nitric oxide |
| NOS | nitric oxide synthetase |
| NTS | nucleus of the solitary tract |
| PCP | planar-cell-polarity |
| PI3K | phosphoinositide 3-kinase |
| PLC | phospholipase C |
| PKC | protein kinase C |
| PRMT | protein arginine methyltransferases |
| PTEN | phosphatase and tensin homolog |
| PVN | paraventricular nucleus |
| RAAS | renin-angiotensin-aldosterone system |
| RAS | renin-angiotensin system |
| RBF | renal blood flow |
| RVLM | rostral ventrolateral medulla |
| ROCK | Rho-associated protein kinase |
| SDMA | symmetrical dimethylarginine |
| SFO | subfornical organ |
| SNA | sympathetic nerve activity |
| SNS | sympathetic nerve system |
| SON | supraoptic nucleus |
| TPR | total peripheral resistance |
| TXA2 | thromboxane A2 |
| VSMC | vascular smooth muscle cell |
References
- Danaei, G.; Lu, Y.; Singh, G.M.; Carnahan, E.; Stevens, G.A.; Cowan, M.J.; Farzadfar, F.; Lin, J.K.; Finucane, M.M.; Rao, M.; et al. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: A comparative risk assessment. Lancet Diabetes Endocrinol. 2014, 2, 634–647. [Google Scholar]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/hypertension (accessed on 14 March 2021).
- Newberry, S.J.; Chung, M.; Anderson, C.A.M.; Chen, C.; Fu, Z.; Tang, A.; Zhao, N.; Booth, M.; Marks, J.; Hollands, S.; et al. AHRQ Comparative Effectiveness Reviews. In Sodium and Potassium Intake: Effects on Chronic Disease Outcomes and Risks; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2018. [Google Scholar]
- Intersalt: An international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. Intersalt Cooperative Research Group. BMJ 1988, 297, 319–328. [CrossRef] [PubMed] [Green Version]
- Luft, F.C.; Weinberger, M.H. Heterogeneous responses to changes in dietary salt intake: The salt-sensitivity paradigm. Am. J. Clin. Nutr. 1997, 65, 612S–617S. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Delea, C.S.; Bartter, F.C.; Smith, H. The effect of high-sodium and low-sodium intakes on blood pressure and other related variables in human subjects with idiopathic hypertension. Am. J. Med. 1978, 64, 193–198. [Google Scholar] [CrossRef]
- Fujita, T.; Henry, W.L.; Bartter, F.C.; Lake, C.R.; Delea, C.S. Factors influencing blood pressure in salt-sensitive patients with hypertension. Am. J. Med. 1980, 69, 334–344. [Google Scholar] [CrossRef]
- Guyton, A.C. Kidneys and fluids in pressure regulation. Small volume but large pressure changes. Hypertension 1992, 19, I2–I8. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Ando, K.; Ogata, E. Systemic and regional hemodynamics in patients with salt-sensitive hypertension. Hypertension 1990, 16, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, T.; Noda, H.; Ando, K. Sodium susceptibility and potassium effects in young patients with borderline hypertension. Circulation 1984, 69, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Dell’Italia, L.J.; Sanders, P.W. Novel Paradigms of Salt and Hypertension. J. Am. Soc. Nephrol. 2017, 28, 1362–1369. [Google Scholar] [CrossRef] [Green Version]
- Bragulat, E.; de la Sierra, A. Salt intake, endothelial dysfunction, and salt-sensitive hypertension. J. Clin. Hypertens. 2002, 4, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Kawarazaki, W.; Mizuno, R.; Nishimoto, M.; Ayuzawa, N.; Hirohama, D.; Ueda, K.; Kawakami-Mori, F.; Oba, S.; Marumo, T.; Fujita, T. Salt causes aging-associated hypertension via vascular Wnt5a under Klotho deficiency. J. Clin. Investig. 2020, 130, 4152–4166. [Google Scholar] [CrossRef]
- Schmidlin, O.; Forman, A.; Leone, A.; Sebastian, A.; Morris, R.C., Jr. Salt sensitivity in blacks: Evidence that the initial pressor effect of NaCl involves inhibition of vasodilatation by asymmetrical dimethylarginine. Hypertension 2011, 58, 380–385. [Google Scholar] [CrossRef] [Green Version]
- Kawarazaki, W.; Nagase, M.; Yoshida, S.; Takeuchi, M.; Ishizawa, K.; Ayuzawa, N.; Ueda, K.; Fujita, T. Angiotensin II- and salt-induced kidney injury through Rac1-mediated mineralocorticoid receptor activation. J. Am. Soc. Nephrol. 2012, 23, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.; McDonough, A.A. Impact of angiotensin II-mediated stimulation of sodium transporters in the nephron assessed by computational modeling. Am. J. Physiol. Ren. Physiol. 2019, 317, F1656–F1668. [Google Scholar] [CrossRef]
- Zhao, D.; Seth, D.M.; Navar, L.G. Enhanced distal nephron sodium reabsorption in chronic angiotensin II-infused mice. Hypertension 2009, 54, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Riazi, S.; Khan, O.; Hu, X.; Ecelbarger, C.A. Aldosterone infusion with high-NaCl diet increases blood pressure in obese but not lean Zucker rats. Am. J. Physiol. Ren. Physiol. 2006, 291, F597–F605. [Google Scholar] [CrossRef] [Green Version]
- Winternitz, S.R.; Katholi, R.E.; Oparil, S. Role of the renal sympathetic nerves in the development and maintenance of hypertension in the spontaneously hypertensive rat. J. Clin. Investig. 1980, 66, 971–978. [Google Scholar] [CrossRef]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef] [Green Version]
- Campese, V.M.; Romoff, M.S.; Levitan, D.; Saglikes, Y.; Friedler, R.M.; Massry, S.G. Abnormal relationship between sodium intake and sympathetic nervous system activity in salt-sensitive patients with essential hypertension. Kidney Int. 1982, 21, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Polichnowski, A.J.; Griffin, K.A.; Long, J.; Williamson, G.A.; Bidani, A.K. Blood pressure-renal blood flow relationships in conscious angiotensin II- and phenylephrine-infused rats. Am. J. Physiol. Ren. Physiol. 2013, 305, F1074–F1084. [Google Scholar] [CrossRef] [Green Version]
- Oliver, J.A.; Cannon, P.J. The effect of altered sodium balance upon renal vascular reactivity to angiotensin II and norepinephrine in the dog. Mechanism of variation in angiotensin responses. J. Clin. Investig. 1978, 61, 610–623. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawachi, H.; Fujita, T. Podocyte as the target for aldosterone: Roles of oxidative stress and Sgk1. Hypertension 2007, 49, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Nagase, M.; Yoshida, S.; Shibata, S.; Nagase, T.; Gotoda, T.; Ando, K.; Fujita, T. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: Possible contribution of fat-derived factors. J. Am. Soc. Nephrol. 2006, 17, 3438–3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhart-Bornstein, M.; Lamounier-Zepter, V.; Schraven, A.; Langenbach, J.; Willenberg, H.S.; Barthel, A.; Hauner, H.; McCann, S.M.; Scherbaum, W.A.; Bornstein, S.R. Human adipocytes secrete mineralocorticoid-releasing factors. Proc. Natl. Acad. Sci. USA 2003, 100, 14211–14216. [Google Scholar] [CrossRef] [Green Version]
- Goodfriend, T.L.; Ball, D.L.; Egan, B.M.; Campbell, W.B.; Nithipatikom, K. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension 2004, 43, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.W.; Wang, J.; Hug, C.; Tsao, T.S.; Lodish, H.F. A family of Acrp30/adiponectin structural and functional paralogs. Proc. Natl. Acad. Sci. USA 2004, 101, 10302–10307. [Google Scholar] [CrossRef] [Green Version]
- Huby, A.C.; Antonova, G.; Groenendyk, J.; Gomez-Sanchez, C.E.; Bollag, W.B.; Filosa, J.A.; Belin de Chantemèle, E.J. Adipocyte-Derived Hormone Leptin Is a Direct Regulator of Aldosterone Secretion, Which Promotes Endothelial Dysfunction and Cardiac Fibrosis. Circulation 2015, 132, 2134–2145. [Google Scholar] [CrossRef]
- Fujita, M.; Ando, K.; Nagae, A.; Fujita, T. Sympathoexcitation by Oxidative Stress in the Brain Mediates Arterial Pressure Elevation in Salt-Sensitive Hypertension. Hypertension 2007, 50, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawarazaki, W.; Kurihara, H.; Tanaka, H.; Miyoshi, J.; Takai, Y.; Fujita, T. Modification of mineralocorticoid receptor function by Rac1 GTPase: Implication in proteinuric kidney disease. Nat. Med. 2008, 14, 1370–1376. [Google Scholar] [CrossRef]
- Shibata, S.; Mu, S.; Kawarazaki, H.; Muraoka, K.; Ishizawa, K.; Yoshida, S.; Kawarazaki, W.; Takeuchi, M.; Ayuzawa, N.; Miyoshi, J.; et al. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J. Clin. Investig. 2011, 121, 3233–3243. [Google Scholar] [CrossRef]
- Han, X.; Hu, Z.; Chen, J.; Huang, J.; Huang, C.; Liu, F.; Gu, C.; Yang, X.; Hixson, J.E.; Lu, X.; et al. Associations Between Genetic Variants of NADPH Oxidase-Related Genes and Blood Pressure Responses to Dietary Sodium Intervention: The GenSalt Study. Am. J. Hypertens. 2017, 30, 427–434. [Google Scholar] [CrossRef]
- Su, Q.; Huo, C.J.; Li, H.B.; Liu, K.L.; Li, X.; Yang, Q.; Song, X.A.; Chen, W.S.; Cui, W.; Zhu, G.Q.; et al. Renin-angiotensin system acting on reactive oxygen species in paraventricular nucleus induces sympathetic activation via AT1R/PKCγ/Rac1 pathway in salt-induced hypertension. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Kawarazaki, W.; Fujita, T. Aberrant Rac1-mineralocorticoid receptor pathways in salt-sensitive hypertension. Clin. Exp. Pharmacol. Physiol. 2013, 40, 929–936. [Google Scholar] [CrossRef]
- Nagase, M. Role of Rac1 GTPase in salt-sensitive hypertension. Curr. Opin. Nephrol. Hypertens. 2013, 22, 148–155. [Google Scholar] [CrossRef]
- Kawarazaki, W.; Fujita, T. Kidney and epigenetic mechanisms of salt-sensitive hypertension. Nat. Rev. Nephrol. 2021, 1–14. [Google Scholar] [CrossRef]
- Kawakami-Mori, F.; Nishimoto, M.; Reheman, L.; Kawarazaki, W.; Ayuzawa, N.; Ueda, K.; Hirohama, D.; Kohno, D.; Oba, S.; Shimosawa, T.; et al. Aberrant DNA methylation of hypothalamic angiotensin receptor in prenatal programmed hypertension. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Hall, J.E.; Do Carmo, J.M.; Da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity, kidney dysfunction and hypertension: Mechanistic links. Nat. Rev. Nephrol. 2019, 15, 367–385. [Google Scholar] [CrossRef]
- Elijovich, F.; Weinberger, M.H.; Anderson, C.A.; Appel, L.J.; Bursztyn, M.; Cook, N.R.; Dart, R.A.; Newton-Cheh, C.H.; Sacks, F.M.; Laffer, C.L. Salt Sensitivity of Blood Pressure: A Scientific Statement From the American Heart Association. Hypertension 2016, 68, e7–e46. [Google Scholar] [CrossRef] [Green Version]
- Wolf-Maier, K.; Cooper, R.S.; Banegas, J.R.; Giampaoli, S.; Hense, H.W.; Joffres, M.; Kastarinen, M.; Poulter, N.; Primatesta, P.; Rodríguez-Artalejo, F.; et al. Hypertension prevalence and blood pressure levels in 6 European countries, Canada, and the United States. JAMA 2003, 289, 2363–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilluy, C.; Brégeon, J.; Toumaniantz, G.; Rolli-Derkinderen, M.; Retailleau, K.; Loufrani, L.; Henrion, D.; Scalbert, E.; Bril, A.; Torres, R.M.; et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 2010, 16, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.L.; Brégeon, J.; Devos, N.; Chadeuf, G.; Blanchard, A.; Azizi, M.; Pacaud, P.; Jeunemaître, X.; Loirand, G. Angiotensin II activates the RhoA exchange factor Arhgef1 in humans. Hypertension 2015, 65, 1273–1278. [Google Scholar] [CrossRef] [Green Version]
- Satoh, S.; Ueda, Y.; Koyanagi, M.; Kadokami, T.; Sugano, M.; Yoshikawa, Y.; Makino, N. Chronic inhibition of Rho kinase blunts the process of left ventricular hypertrophy leading to cardiac contractile dysfunction in hypertension-induced heart failure. J. Mol. Cell. Cardiol. 2003, 35, 59–70. [Google Scholar] [CrossRef]
- Cao, Y.; Fang, Y.; Mu, J.; Liu, X. High salt medium activates RhoA/ROCK and downregulates eNOS expression via the upregulation of ADMA. Mol. Med. Rep. 2016, 14, 606–612. [Google Scholar] [CrossRef]
- Ito, K.; Hirooka, Y.; Sunagawa, K. Acquisition of brain Na sensitivity contributes to salt-induced sympathoexcitation and cardiac dysfunction in mice with pressure overload. Circ. Res. 2009, 104, 1004–1011. [Google Scholar] [CrossRef] [Green Version]
- Behuliak, M.; Bencze, M.; Vaněčková, I.; Kuneš, J.; Zicha, J. Basal and Activated Calcium Sensitization Mediated by RhoA/Rho Kinase Pathway in Rats with Genetic and Salt Hypertension. BioMed Res. Inte. 2017, 2017, 8029728. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.C.; Wu, W.; Attipoe, E.M.; Sasser, J.M.; Taylor, E.B.; Showmaker, K.C.; Kyle, P.B.; Lindsey, M.L.; Garrett, M.R. Loss of Arhgef11 in the Dahl Salt-Sensitive Rat Protects Against Hypertension-Induced Renal Injury. Hypertension 2020, 75, 1012–1024. [Google Scholar] [CrossRef]
- Seccia, T.M.; Rigato, M.; Ravarotto, V.; Calò, L.A. ROCK (RhoA/Rho Kinase) in Cardiovascular-Renal Pathophysiology: A Review of New Advancements. J. Clin. Med. 2020, 9, 1328. [Google Scholar] [CrossRef]
- Calò, L.A.; Pessina, A.C. RhoA/Rho-kinase pathway: Much more than just a modulation of vascular tone. Evidence from studies in humans. J. Hypertens. 2007, 25, 259–264. [Google Scholar] [CrossRef]
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The Ras protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012, 196, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Park, H.O.; Bi, E. Central roles of small GTPases in the development of cell polarity in yeast and beyond. Microbiol. Mol. Biol. Rev. 2007, 71, 48–96. [Google Scholar] [CrossRef] [Green Version]
- Mulloy, J.C.; Cancelas, J.A.; Filippi, M.D.; Kalfa, T.A.; Guo, F.; Zheng, Y. Rho GTPases in hematopoiesis and hemopathies. Blood 2010, 115, 936–947. [Google Scholar] [CrossRef]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. Rho family GTPases. Biochem. Soc. Trans. 2012, 40, 1378–1382. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.K.; Kholodenko, B.N.; von Kriegsheim, A. Rac1 and RhoA: Networks, loops and bistability. Small GTPases 2018, 9, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Ueda, T.; Kikuchi, A.; Ohga, N.; Yamamoto, J.; Takai, Y. Purification and characterization from bovine brain cytosol of a novel regulatory protein inhibiting the dissociation of GDP from and the subsequent binding of GTP to rhoB p20, a ras p21-like GTP-binding protein. J. Biol. Chem. 1990, 265, 9373–9380. [Google Scholar] [CrossRef]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The ‘invisible hand’: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef] [Green Version]
- Maguire, J.J.; Davenport, A.P. Regulation of vascular reactivity by established and emerging GPCRs. Trends Pharmacol. Sci. 2005, 26, 448–454. [Google Scholar] [CrossRef]
- Gohla, A.; Schultz, G.; Offermanns, S. Role for G(12)/G(13) in agonist-induced vascular smooth muscle cell contraction. Circ. Res. 2000, 87, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuhara, S.; Chikumi, H.; Gutkind, J.S. RGS-containing RhoGEFs: The missing link between transforming G proteins and Rho? Oncogene 2001, 20, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazawa, T.; Eto, M.; Woodsome, T.P.; Brautigan, D.L. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J. Biol. Chem. 2000, 275, 9897–9900. [Google Scholar] [CrossRef] [Green Version]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef] [PubMed]
- MIALL, W.E.; OLDHAM, P.D. The hereditary factor in arterial blood-pressure. Br. Med. J. 1963, 1, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Majid, D.S.; Prieto, M.C.; Navar, L.G. Salt-Sensitive Hypertension: Perspectives on Intrarenal Mechanisms. Curr. Hypertens. Rev. 2015, 11, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Armando, I.; Villar, V.A.; Jose, P.A. Genomics and Pharmacogenomics of Salt-sensitive Hypertension. Curr. Hypertens. Rev. 2015, 11, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyton, A.C. The surprising kidney-fluid mechanism for pressure control--its infinite gain! Hypertension 1990, 16, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Kobori, H.; Nishiyama, A.; Abe, Y.; Navar, L.G. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension 2003, 41, 592–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, A.; Benyó, Z.; Lukasova, M.; Leutgeb, B.; Wettschureck, N.; Gorbey, S.; Orsy, P.; Horváth, B.; Maser-Gluth, C.; Greiner, E.; et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008, 14, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Hirooka, Y.; Kishi, T.; Ito, K.; Sunagawa, K. Potential clinical application of recently discovered brain mechanisms involved in hypertension. Hypertension 2013, 62, 995–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malpas, S.C. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol. Rev. 2010, 90, 513–557. [Google Scholar] [CrossRef] [PubMed]
- Guyenet, P.G. The sympathetic control of blood pressure. Nat. Rev. Neurosci. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Ito, K.; Hirooka, Y.; Sakai, K.; Kishi, T.; Kaibuchi, K.; Shimokawa, H.; Takeshita, A. Rho/Rho-kinase pathway in brain stem contributes to blood pressure regulation via sympathetic nervous system: Possible involvement in neural mechanisms of hypertension. Circ. Res. 2003, 92, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Nozoe, M.; Hirooka, Y.; Koga, Y.; Sagara, Y.; Kishi, T.; Engelhardt, J.F.; Sunagawa, K. Inhibition of Rac1-derived reactive oxygen species in nucleus tractus solitarius decreases blood pressure and heart rate in stroke-prone spontaneously hypertensive rats. Hypertension 2007, 50, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, M.; Ohtsuka, K.; Nanbu, A.; Takahashi, H.; Yoshimura, M. Benzamil blockade of brain Na+ channels averts Na(+)-induced hypertension in rats. Am. J. Physiol. 1998, 274, R635–R644. [Google Scholar] [CrossRef]
- Huang, B.S.; Amin, M.S.; Leenen, F.H. The central role of the brain in salt-sensitive hypertension. Curr. Opin. Cardiol. 2006, 21, 295–304. [Google Scholar] [CrossRef]
- Huang, B.S.; Leenen, F.H. Blockade of brain mineralocorticoid receptors or Na+ channels prevents sympathetic hyperactivity and improves cardiac function in rats post-MI. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2491–H2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drenjančević-Perić, I.; Jelaković, B.; Lombard, J.H.; Kunert, M.P.; Kibel, A.; Gros, M. High-salt diet and hypertension: Focus on the renin-angiotensin system. Kidney Blood Press. Res. 2011, 34, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaustein, M.P.; Leenen, F.H.; Chen, L.; Golovina, V.A.; Hamlyn, J.M.; Pallone, T.L.; Van Huysse, J.W.; Zhang, J.; Wier, W.G. How NaCl raises blood pressure: A new paradigm for the pathogenesis of salt-dependent hypertension. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1031–H1049. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.S.; White, R.A.; Bi, L.; Leenen, F.H. Central infusion of aliskiren prevents sympathetic hyperactivity and hypertension in Dahl salt-sensitive rats on high salt intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R825–R832. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.S.; Van Vliet, B.N.; Leenen, F.H. Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1160–H1166. [Google Scholar] [CrossRef] [PubMed]
- Simchon, S.; Manger, W.; Golanov, E.; Kamen, J.; Sommer, G.; Marshall, C.H. Handling 22NaCl by the blood-brain barrier and kidney: Its relevance to salt-induced hypertension in dahl rats. Hypertension 1999, 33, 517–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.M.; Veerasingham, S.J.; Tan, J.; Leenen, F.H. Effects of high salt intake on brain AT1 receptor densities in Dahl rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1949–H1955. [Google Scholar] [CrossRef] [Green Version]
- Pick, E. Role of the Rho GTPase Rac in the activation of the phagocyte NADPH oxidase: Outsourcing a key task. Small GTPases 2014, 5, e27952. [Google Scholar] [CrossRef] [Green Version]
- Garrido, A.M.; Griendling, K.K. NADPH oxidases and angiotensin II receptor signaling. Mol. Cell. Endocrinol. 2009, 302, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakumar, P.; Jagadeesh, G. A century old renin-angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell. Signal. 2014, 26, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; Zheng, H.; Tan, J.; Patel, K.P.; Leenen, F.H. Regulation of hypothalamic renin-angiotensin system and oxidative stress by aldosterone. Exp. Physiol. 2011, 96, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Misárková, E.; Behuliak, M.; Bencze, M.; Zicha, J. Excitation-contraction coupling and excitation-transcription coupling in blood vessels: Their possible interactions in hypertensive vascular remodeling. Physiol. Res. 2016, 65, 173–191. [Google Scholar] [CrossRef]
- Goulopoulou, S.; Webb, R.C. Symphony of vascular contraction: How smooth muscle cells lose harmony to signal increased vascular resistance in hypertension. Hypertension 2014, 63, e33–e39. [Google Scholar] [CrossRef] [Green Version]
- Walsh, M.P. Vascular smooth muscle myosin light chain diphosphorylation: Mechanism, function, and pathological implications. IUBMB Life 2011, 63, 987–1000. [Google Scholar] [CrossRef]
- Eto, M.; Kitazawa, T. Diversity and plasticity in signaling pathways that regulate smooth muscle responsiveness: Paradigms and paradoxes for the myosin phosphatase, the master regulator of smooth muscle contraction. J. Smooth Muscle Res. 2017, 53, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Shirai, H.; Autieri, M.; Eguchi, S. Small GTP-binding proteins and mitogen-activated protein kinases as promising therapeutic targets of vascular remodeling. Curr. Opin. Nephrol. Hypertens. 2007, 16, 111–115. [Google Scholar] [CrossRef]
- Loirand, G.; Pacaud, P. The role of Rho protein signaling in hypertension. Nat. Rev. Cardiol. 2010, 7, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Crestani, S.; Webb, R.C.; da Silva-Santos, J.E. High-Salt Intake Augments the Activity of the RhoA/ROCK Pathway and Reduces Intracellular Calcium in Arteries From Rats. Am. J. Hypertens. 2017, 30, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komers, R.; Rogers, S.; Oyama, T.T.; Xu, B.; Yang, C.L.; McCormick, J.; Ellison, D.H. Enhanced phosphorylation of Na(+)-Cl- co-transporter in experimental metabolic syndrome: Role of insulin. Clin. Sci. 2012, 123, 635–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, D.; Gordon, K.L.; Polinsky, P.; Suga, S.; Schwartz, S.M.; Johnson, R.J. Salt-sensitive hypertension develops after short-term exposure to Angiotensin II. Hypertension 1999, 33, 1013–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Nagase, M.; Shibata, S.; Yoshida, S.; Nagase, T.; Gotoda, T.; Fujita, T. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension 2006, 47, 1084–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, M.R.; Joe, B.; Yerga-Woolwine, S. Genetic linkage of urinary albumin excretion in Dahl salt-sensitive rats: Influence of dietary salt and confirmation using congenic strains. Physiol. Genom. 2006, 25, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.M.; Johnson, A.C.; Stelloh, C.; Dreisbach, A.W.; Franceschini, N.; Regner, K.R.; Townsend, R.R.; Roman, R.J.; Garrett, M.R. Genetic variants in Arhgef11 are associated with kidney injury in the Dahl salt-sensitive rat. Hypertension 2012, 60, 1157–1168. [Google Scholar] [CrossRef] [Green Version]
- Garrett, M.R.; Dene, H.; Rapp, J.P. Time-course genetic analysis of albuminuria in Dahl salt-sensitive rats on low-salt diet. J. Am. Soc. Nephrol. 2003, 14, 1175–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, H.Y.; Saisawat, P.; Ashraf, S.; Hurd, T.W.; Vega-Warner, V.; Fang, H.; Beck, B.B.; Gribouval, O.; Zhou, W.; Diaz, K.A.; et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J. Clin. Investig. 2013, 123, 3243–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, I.R.; Baldwin, C.; Auguste, D.; Ha, K.C.; El Andalousi, J.; Fahiminiya, S.; Bitzan, M.; Bernard, C.; Akbari, M.R.; Narod, S.A.; et al. ARHGDIA: A novel gene implicated in nephrotic syndrome. J. Med. Genet. 2013, 50, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumiyama, K.; Osanai, T.; Sagara, S.; Yamamoto, Y.; Itoh, T.; Sukekawa, T.; Nishizaki, F.; Magota, K.; Okumura, K. Estrogen attenuates coupling factor 6-induced salt-sensitive hypertension and cardiac systolic dysfunction in mice. Hypertens. Res. 2012, 35, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawarazaki, H.; Ando, K.; Shibata, S.; Muraoka, K.; Fujita, M.; Kawarasaki, C.; Fujita, T. Mineralocorticoid receptor--Rac1 activation and oxidative stress play major roles in salt-induced hypertension and kidney injury in prepubertal rats. J. Hypertens. 2012, 30, 1977–1985. [Google Scholar] [CrossRef]
- Symons, M.; Segall, J.E. Rac and Rho driving tumor invasion: Who’s at the wheel? Genome Biol. 2009, 10, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, B.N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006, 7, 165–176. [Google Scholar] [CrossRef]
- Byrne, K.M.; Monsefi, N.; Dawson, J.C.; Degasperi, A.; Bukowski-Wills, J.C.; Volinsky, N.; Dobrzyński, M.; Birtwistle, M.R.; Tsyganov, M.A.; Kiyatkin, A.; et al. Bistability in the Rac1, PAK, and RhoA Signaling Network Drives Actin Cytoskeleton Dynamics and Cell Motility Switches. Cell Syst. 2016, 2, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef]
- Hirohama, D.; Ayuzawa, N.; Ueda, K.; Nishimoto, M.; Kawarazaki, W.; Watanabe, A.; Shimosawa, T.; Marumo, T.; Shibata, S.; Fujita, T. Aldosterone Is Essential for Angiotensin II-Induced Upregulation of Pendrin. J. Am. Soc. Nephrol. 2018, 29, 57–68. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Babwah, A.V.; Ferguson, S.S. Small GTP-binding protein-coupled receptors. Biochem. Soc. Trans. 2004, 32, 1040–1044. [Google Scholar] [CrossRef]
- Schiller, M.R. Coupling receptor tyrosine kinases to Rho GTPases--GEFs what’s the link. Cell. Signal. 2006, 18, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Faruqi, T.R.; Gomez, D.; Bustelo, X.R.; Bar-Sagi, D.; Reich, N.C. Rac1 mediates STAT3 activation by autocrine IL-6. Proc. Natl. Acad. Sci. USA 2001, 98, 9014–9019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotchin, N.A.; Hall, A. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J. Cell Biol. 1995, 131, 1857–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redmond, L.; Ghosh, A. The role of Notch and Rho GTPase signaling in the control of dendritic development. Curr. Opin. Neurobiol. 2001, 11, 111–117. [Google Scholar] [CrossRef]
- Park, S.Y.; Kang, M.J.; Han, J.S. Interleukin-1 beta promotes neuronal differentiation through the Wnt5a/RhoA/JNK pathway in cortical neural precursor cells. Mol. Brain 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Maldonado, M.D.M.; Medina, J.I.; Velazquez, L.; Dharmawardhane, S. Targeting Rac and Cdc42 GEFs in Metastatic Cancer. Front. Cell Dev. Biol. 2020, 8, 201. [Google Scholar] [CrossRef]
- Yadav, S.; Barton, M.; Nguyen, N.T. Stretching Induces Overexpression of RhoA and Rac1 GTPases in Breast Cancer Cells. Adv. Biosyst. 2020, 4, e1900222. [Google Scholar] [CrossRef]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mahler, A.; Balogh, A.; Marko, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Zheng, Q.; Wang, W.; Xin, N.; Song, X.; Zhao, C. Biological functions of macrophage-derived Wnt5a, and its roles in human diseases. Oncotarget 2016, 7, 67674–67684. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Yan, T.; Hao, L.; Zhu, Y. Wnt5a induces ROR1 and ROR2 to activate RhoA in esophageal squamous cell carcinoma cells. Cancer Manag. Res. 2019, 11, 2803–2815. [Google Scholar] [CrossRef] [Green Version]
- Kühl, M.; Sheldahl, L.C.; Park, M.; Miller, J.R.; Moon, R.T. The Wnt/Ca2+ pathway: A new vertebrate Wnt signaling pathway takes shape. Trends Genet. TIG 2000, 16, 279–283. [Google Scholar] [CrossRef]
- Dejana, E. The role of wnt signaling in physiological and pathological angiogenesis. Circ. Res. 2010, 107, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharmacol. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef]
- Marinou, K.; Christodoulides, C.; Antoniades, C.; Koutsilieris, M. Wnt signaling in cardiovascular physiology. Trends Endocrinol. Metab. TEM 2012, 23, 628–636. [Google Scholar] [CrossRef]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Sun, Z. Molecular basis of Klotho: From gene to function in aging. Endocr. Rev. 2015, 36, 174–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, D.; Sato, Y.; Aizawa, M.; Maki, T.; Kurosawa, M.; Kuro-o, M.; Furukawa, Y. Soluble alphaKlotho as a candidate for the biomarker of aging. Biochem. Biophys. Res. Commun. 2015, 467, 1019–1025. [Google Scholar] [CrossRef]
- Xiao, N.M.; Zhang, Y.M.; Zheng, Q.; Gu, J. Klotho is a serum factor related to human aging. Chin. Med. J. 2004, 117, 742–747. [Google Scholar]
- Citterio, L.; Delli Carpini, S.; Lupoli, S.; Brioni, E.; Simonini, M.; Fontana, S.; Zagato, L.; Messaggio, E.; Barlassina, C.; Cusi, D.; et al. Klotho Gene in Human Salt-Sensitive Hypertension. Clin. J. Am. Soc. Nephrol. 2020, 15, 375–383. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Habas, R.; Dawid, I.B. Dishevelled and Wnt signaling: Is the nucleus the final frontier? J. Biol. 2005, 4, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y. Wnt/Planar cell polarity signaling: A new paradigm for cancer therapy. Mol. Cancer Ther. 2009, 8, 2103–2109. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Brusco, L.; Gu, G.; Selever, J.; Beyer, A.; Covington, K.R.; Tsimelzon, A.; Wang, T.; Hilsenbeck, S.G.; Chamness, G.C.; et al. Loss of Rho GDIα and resistance to tamoxifen via effects on estrogen receptor α. J. Natl. Cancer Inst. 2011, 103, 538–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuster, J.J.; Zuriaga, M.A.; Ngo, D.T.-M.; Farb, M.G.; Aprahamian, T.; Yamaguchi, T.P.; Gokce, N.; Walsh, K. Noncanonical Wnt Signaling Promotes Obesity-Induced Adipose Tissue Inflammation and Metabolic Dysfunction Independent of Adipose Tissue Expansion. Diabetes 2015, 64, 1235–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.; Lateef, A.M.; Engels, K.; Samsell, L.; Baylis, C. Basal and stimulated nitric oxide in control of kidney function in the aging rat. Am. J. Physiol. 1997, 272, R1747–R1753. [Google Scholar] [CrossRef]
- De Nicola, L.; Blantz, R.C.; Gabbai, F.B. Nitric oxide and angiotensin II. Glomerular and tubular interaction in the rat. J. Clin. Investig. 1992, 89, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.J.; Vaziri, N.D.; Zhang, J.; Wang, H.W.; Wang, X.Q. Association of renal injury with nitric oxide deficiency in aged SHR: Prevention by hypertension control with AT1 blockade. Kidney Int. 2002, 62, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Redgrave, J.; Rabinowe, S.; Hollenberg, N.K.; Williams, G.H. Correction of abnormal renal blood flow response to angiotensin II by converting enzyme inhibition in essential hypertensives. J. Clin. Investig. 1985, 75, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Campese, V.M.; Parise, M.; Karubian, F.; Bigazzi, R. Abnormal renal hemodynamics in black salt-sensitive patients with hypertension. Hypertension 1991, 18, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Shimamoto, H.; Shimamoto, Y. Time course of hemodynamic responses to sodium in elderly hypertensive patients. Hypertension 1990, 16, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhar, M.; Najumuddin; Gul, M.; Rashid, S. Antagonistic role of Klotho-derived peptides dynamics in the pancreatic cancer treatment through obstructing WNT-1 and Frizzled binding. Biophys. Chem. 2018, 240, 107–117. [Google Scholar] [CrossRef]
- Miyoshi, A.; Suzuki, H.; Fujiwara, M.; Masai, M.; Iwasaki, T. Impairment of endothelial function in salt-sensitive hypertension in humans. Am. J. Hypertens. 1997, 10, 1083–1090. [Google Scholar] [CrossRef]
- Bragulat, E.; de la Sierra, A.; Antonio, M.T.; Coca, A. Endothelial dysfunction in salt-sensitive essential hypertension. Hypertension 2001, 37, 444–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Félétou, M.; Vanhoutte, P.M. Endothelial dysfunction: A multifaceted disorder (The Wiggers Award Lecture). Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H985–H1002. [Google Scholar] [CrossRef]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Laufs, U.; Liao, J.K. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J. Biol. Chem. 1998, 273, 24266–24271. [Google Scholar] [CrossRef] [Green Version]
- Wolfrum, S.; Dendorfer, A.; Rikitake, Y.; Stalker, T.J.; Gong, Y.; Scalia, R.; Dominiak, P.; Liao, J.K. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1842–1847. [Google Scholar] [CrossRef] [Green Version]
- Ming, X.F.; Viswambharan, H.; Barandier, C.; Ruffieux, J.; Kaibuchi, K.; Rusconi, S.; Yang, Z. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol. Cell Biol. 2002, 22, 8467–8477. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Dong, X.; Wang, Z.; Liu, W.; Deng, N.; Ding, Y.; Tang, L.; Hla, T.; Zeng, R.; Li, L.; et al. Regulation of PTEN by Rho small GTPases. Nat. Cell Biol. 2005, 7, 399–404. [Google Scholar] [CrossRef]
- Romero, M.J.; Platt, D.H.; Tawfik, H.E.; Labazi, M.; El-Remessy, A.B.; Bartoli, M.; Caldwell, R.B.; Caldwell, R.W. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ. Res. 2008, 102, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Arakawa, K. Salt-induced hemodynamic regulation mediated by nitric oxide. J. Hypertens. 2011, 29, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, L.R.; Wojciak-Stothard, B. The DDAH/ADMA pathway in the control of endothelial cell migration and angiogenesis. Biochem. Soc. Trans. 2009, 37, 1243–1247. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Mu, J.J.; Fang, Y.; Yuan, Z.Y.; Liu, F.Q. Impact of high salt independent of blood pressure on PRMT/ADMA/DDAH pathway in the aorta of Dahl salt-sensitive rats. Int. J. Mol. Sci. 2013, 14, 8062–8072. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, L. The DDAH/ADMA pathway is a critical regulator of NO signalling in vascular homeostasis. Cell Adhes. Migr. 2008, 2, 149–150. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, M.; Suzuki, Y.; Sugita, K.; Saito, I.; Sasaki, T.; Takakura, K.; Nagata, I.; Kikuchi, H.; Takemae, T.; Hidaka, H.; et al. Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Results of a prospective placebo-controlled double-blind trial. J. Neurosurg. 1992, 76, 571–577. [Google Scholar] [CrossRef]
- Serle, J.B.; Katz, L.J.; McLaurin, E.; Heah, T.; Ramirez-Davis, N.; Usner, D.W.; Novack, G.D.; Kopczynski, C.C. Two Phase 3 Clinical Trials Comparing the Safety and Efficacy of Netarsudil to Timolol in Patients With Elevated Intraocular Pressure: Rho Kinase Elevated IOP Treatment Trial 1 and 2 (ROCKET-1 and ROCKET-2). Am. J. Ophthalmol. 2018, 186, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanihara, H.; Inoue, T.; Yamamoto, T.; Kuwayama, Y.; Abe, H.; Suganami, H.; Araie, M. Intra-ocular pressure-lowering effects of a Rho kinase inhibitor, ripasudil (K-115), over 24 hours in primary open-angle glaucoma and ocular hypertension: A randomized, open-label, crossover study. Acta Ophthalmol. 2015, 93, e254–e260. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Fukumoto, Y.; Saji, K.; Sugimura, K.; Demachi, J.; Nawata, J.; Shimokawa, H. Acute vasodilator effects of inhaled fasudil, a specific Rho-kinase inhibitor, in patients with pulmonary arterial hypertension. Heart Vessels 2010, 25, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, Y.; Yamada, N.; Matsubara, H.; Mizoguchi, M.; Uchino, K.; Yao, A.; Kihara, Y.; Kawano, M.; Watanabe, H.; Takeda, Y.; et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ. J. 2013, 77, 2619–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defert, O.; Boland, S. Rho kinase inhibitors: A patent review (2014–2016). Expert Opin. Ther. Pat. 2017, 27, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Berrino, E.; Supuran, C.T. Rho-kinase inhibitors in the management of glaucoma. Expert Opin. Ther. Pat. 2019, 29, 817–827. [Google Scholar] [CrossRef]
- McLeod, R.; Kumar, R.; Papadatos-Pastos, D.; Mateo, J.; Brown, J.S.; Garces, A.H.I.; Ruddle, R.; Decordova, S.; Jueliger, S.; Ferraldeschi, R.; et al. First-in-Human Study of AT13148, a Dual ROCK-AKT Inhibitor in Patients with Solid Tumors. Clin. Cancer Res. 2020, 26, 4777–4784. [Google Scholar] [CrossRef]
- Mudigonda, P.; Mudigonda, T.; Feneran, A.N.; Alamdari, H.S.; Sandoval, L.; Feldman, S.R. Interleukin-23 and interleukin-17: Importance in pathogenesis and therapy of psoriasis. Dermatol. Online J. 2012, 18, 1. [Google Scholar] [PubMed]
- Biswas, P.S.; Gupta, S.; Chang, E.; Song, L.; Stirzaker, R.A.; Liao, J.K.; Bhagat, G.; Pernis, A.B. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Investig. 2010, 120, 3280–3295. [Google Scholar] [CrossRef] [Green Version]
- Zanin-Zhorov, A.; Weiss, J.M.; Trzeciak, A.; Chen, W.; Zhang, J.; Nyuydzefe, M.S.; Arencibia, C.; Polimera, S.; Schueller, O.; Fuentes-Duculan, J.; et al. Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10. J. Immunol. 2017, 198, 3809–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masumoto, A.; Hirooka, Y.; Shimokawa, H.; Hironaga, K.; Setoguchi, S.; Takeshita, A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 2001, 38, 1307–1310. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawarazaki, W.; Fujita, T. Role of Rho in Salt-Sensitive Hypertension. Int. J. Mol. Sci. 2021, 22, 2958. https://doi.org/10.3390/ijms22062958
Kawarazaki W, Fujita T. Role of Rho in Salt-Sensitive Hypertension. International Journal of Molecular Sciences. 2021; 22(6):2958. https://doi.org/10.3390/ijms22062958
Chicago/Turabian StyleKawarazaki, Wakako, and Toshiro Fujita. 2021. "Role of Rho in Salt-Sensitive Hypertension" International Journal of Molecular Sciences 22, no. 6: 2958. https://doi.org/10.3390/ijms22062958
APA StyleKawarazaki, W., & Fujita, T. (2021). Role of Rho in Salt-Sensitive Hypertension. International Journal of Molecular Sciences, 22(6), 2958. https://doi.org/10.3390/ijms22062958