
In Translation: FcRn across the Therapeutic Spectrum



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. FcRn Structure, Binding, and Stoichiometry
2.1. pH Sensitivity of Binding
2.2. Fab Contribution to Binding
2.3. Binding Modulation by Other IgG Regions
2.4. Species Variation of Binding
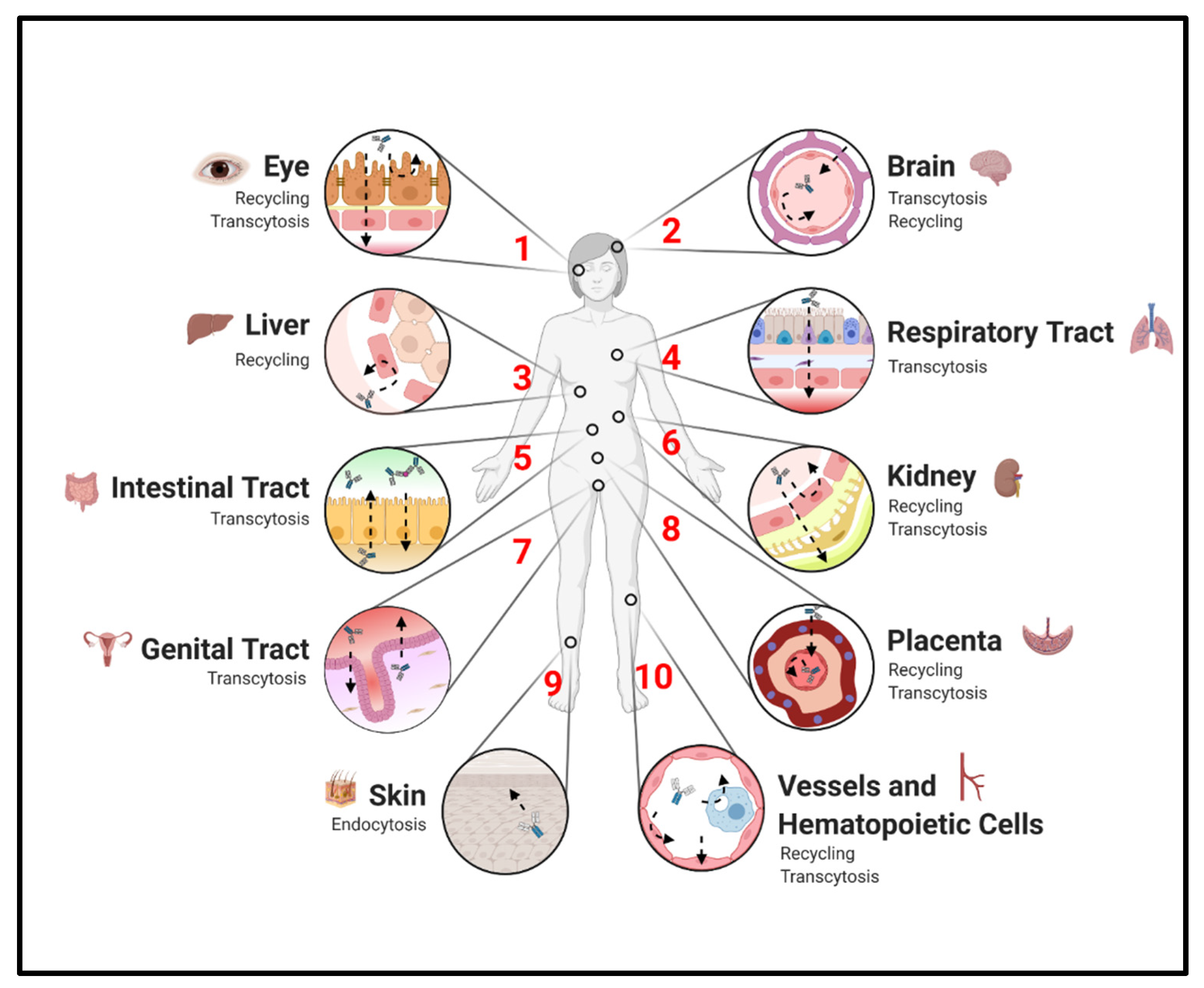
3. FcRn Expression and Its Effects on Homeostatic IgG Distribution
3.1. Endothelium and Hematopoietic Cells
3.2. Placenta
3.3. Liver and Kidneys
3.4. Other Epithelium
3.5. Brain
3.6. Tumors
3.7. Regulation of FcRn Expression
4. FcRn Effects on Immunity
4.1. Innate Immunity
4.2. Adaptive Immunity
4.3. Immune Tolerance
5. FcRn Effects on Monoclonal Antibody Pharmacokinetics
5.1. Increased Affinity for FcRn
5.2. Target-Mediated Ag Accumulation and Recycling Antibodies
5.3. Increased Antigen Endocytosis by FcRn Binding
5.4. High-Dose COVID-19 Antibodies: Is FcRn Saturated?
6. FcRn as a Modular Tool and Therapeutic Target
6.1. FcRn as a Tool
6.2. FcRn as a Target
7. FcRn in Translational Pharmacology
7.1. Preclinical Models
7.2. In Vitro Assays
7.3. In Silico Models of FcRn Recycling
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Latvala, S.; Jacobsen, B.; Otteneder, M.B.; Herrmann, A.; Kronenberg, S. Distribution of FcRn across Species and Tissues. J. Histochem. Cytochem. 2017, 65, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Montoyo, H.P.; Vaccaro, C.; Hafner, M.; Ober, R.J.; Mueller, W.; Ward, E.S. Conditional deletion of the MHC class I-related receptor FcRn reveals the sites of IgG homeostasis in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2788–2793. [Google Scholar] [CrossRef] [Green Version]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, E.S.; Devanaboyina, S.V.; Ober, R.J. Targeting FcRn for the modulation of antibody dynamics. Mol. Immunol. 2015, 67, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ober, R.J.; Martinez, C.; Vaccaro, C.; Zhou, J.; Ward, E.S. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J. Immunol. 2004, 172, 2021–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahey, J.L.; Robinson, A.G. Factors controlling serum gamma-globulin concentration. J. Exp. Med. 1963, 118, 845–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, Z.; Ram, S.; Ober, R.J.; Ward, E.S. Using multifocal plane microscopy to reveal novel trafficking processes in the recycling pathway. J. Cell Sci. 2013, 126, 1176–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ober, R.J.; Martinez, C.; Lai, X.; Zhou, J.; Ward, E.S. Exocytosis of IgG as mediated by the receptor, FcRn: An analysis at the single-molecule level. Proc. Natl. Acad. Sci. USA 2004, 101, 11076–11081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Hooghe, L.; Chalmers, A.D.; Heywood, S.; Whitley, P. Cell surface dynamics and cellular distribution of endogenous FcRn. PLoS ONE 2017, 12, e0182695. [Google Scholar] [CrossRef] [Green Version]
- Ward, E.S.; Zhou, J.; Ghetie, V.; Ober, R.J. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int. Immunol. 2003, 15, 187–195. [Google Scholar] [CrossRef]
- Simister, N.E.; Mostov, K.E. An Fc receptor structurally related to MHC class I antigens. Nature 1989, 337, 184–187. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Huber, A.H.; Bjorkman, P.J. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 1994, 372, 379–383. [Google Scholar] [CrossRef]
- West, A.P.; Bjorkman, P.J. Crystal structure and IgG binding properties of the human MHC-related Fc receptor. Biochemistry 2000, 39, 9698–9708. [Google Scholar] [CrossRef]
- Martin, W.L.; West, A.P., Jr.; Gan, L.; Bjorkman, P.J. Crystal structure at 2.8 Å of an FcRn/heterodimeric Fc complex: Mechanism of pH-dependent binding. Mol. Cell 2001, 7, 867–877. [Google Scholar] [CrossRef]
- Idusogie, E.E.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Wong, P.Y.; Ultsch, M.; Meng, Y.G.; Mulkerrin, M.G. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J. Immunol. 2000, 164, 4178–4184. [Google Scholar] [CrossRef] [Green Version]
- Vaughn, D.E.; Bjorkman, P.J. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure 1998, 6, 63–73. [Google Scholar] [CrossRef]
- Abdiche, Y.N.; Yeung, Y.A.; Chaparro-Riggers, J.; Barman, I.; Strop, P.; Chin, S.M.; Pham, A.; Bolton, G.; McDonough, D.; Lindquist, K.; et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. mAbs 2015, 7, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, T.A.; Terry, W.D. Familial hypercatabolic hypoproteinemia. A disorder of endogenous catabolism of albumin and immunoglobulin. J. Clin. Investig. 1990, 86, 2093–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, M.A.; Haynes, L.D.; Kim, J.; Bronson, C.L.; Chaudhury, C.; Mohanty, S.; Waldmann, T.A.; Robinson, J.M.; Anderson, C.L. Familial hypercatabolic hypoproteinemia caused by deficiency of the neonatal Fc receptor, FcRn, due to a mutant beta2-microglobulin gene. Proc. Natl. Acad. Sci. USA 2006, 103, 5084–5089. [Google Scholar] [CrossRef] [Green Version]
- Oganesyan, V.; Damschroder, M.M.; Cook, K.E.; Li, Q.; Gao, C.; Wu, H.; Dall’Acqua, W.F. Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 2014, 289, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Bern, M.; Sand, K.M.; Nilsen, J.; Sandlie, I.; Andersen, J.T. The role of albumin receptors in regulation of albumin homeostasis: Implications for drug delivery. J. Control. Release 2015, 211, 144–162. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Andersen, J.T.; Daba, M.B.; Berntzen, G.; Michaelsen, T.E.; Sandlie, I. Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J. Biol. Chem. 2010, 285, 4826–4836. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, N.M.; Andersen, J.T.; Stemerding, A.M.; Bjarnarson, S.P.; Verheul, R.C.; Gerritsen, J.; Zhao, Y.; Kleijer, M.; Sandlie, I.; de Haas, M.; et al. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat. Commun. 2011, 2, 599. [Google Scholar] [CrossRef] [Green Version]
- Vaughn, D.E.; Milburn, C.M.; Penny, D.M.; Martin, W.L.; Johnson, J.L.; Bjorkman, P.J. Identification of critical IgG binding epitopes on the neonatal Fc receptor. J. Mol. Biol. 1997, 274, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Alt, N.; Zhang, T.Y.; Motchnik, P.; Taticek, R.; Quarmby, V.; Schlothauer, T.; Beck, H.; Emrich, T.; Harris, R.J. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biologicals 2016, 44, 291–305. [Google Scholar] [CrossRef]
- Tsuchida, D.; Yamazaki, K.; Akashi, S. Comprehensive Characterization of Relationship between Higher-Order Structure and FcRn Binding Affinity of Stress-Exposed Monoclonal Antibodies. Pharm. Res. 2016, 33, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti-Ciarlet, A.; Wang, W.; Lownes, R.; Pristatsky, P.; Fang, Y.; McKelvey, T.; Li, Y.; Li, Y.; Drummond, J.; Prueksaritanont, T.; et al. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc gamma receptors. Mol. Immunol. 2009, 46, 1878–1882. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Chen, K.; Chu, L.; Kinderman, F.; Apostol, I.; Huang, G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009, 18, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Spiekermann, G.M.; Finn, P.W.; Ward, E.S.; Dumont, J.; Dickinson, B.L.; Blumberg, R.S.; Lencer, W.I. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: Functional expression of FcRn in the mammalian lung. J. Exp. Med. 2002, 196, 303–310. [Google Scholar] [CrossRef]
- Liu, L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell 2018, 9, 15–32. [Google Scholar] [CrossRef]
- Wang, W.; Lu, P.; Fang, Y.; Hamuro, L.; Pittman, T.; Carr, B.; Hochman, J.; Prueksaritanont, T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug. Metab. Dispos. 2011, 39, 1469–1477. [Google Scholar] [CrossRef] [Green Version]
- Schoch, A.; Kettenberger, H.; Mundigl, O.; Winter, G.; Engert, J.; Heinrich, J.; Emrich, T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc. Natl. Acad. Sci. USA 2015, 112, 5997–6002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piche-Nicholas, N.M.; Avery, L.B.; King, A.C.; Kavosi, M.; Wang, M.; O’Hara, D.M.; Tchistiakova, L.; Katragadda, M. Changes in complementarity-determining regions significantly alter IgG binding to the neonatal Fc receptor (FcRn) and pharmacokinetics. mAbs 2018, 10, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlothauer, T.; Rueger, P.; Stracke, J.O.; Hertenberger, H.; Fingas, F.; Kling, L.; Emrich, T.; Drabner, G.; Seeber, S.; Auer, J.; et al. Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. mAbs 2013, 5, 576–586. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.F.; Larraillet, V.; Schlothauer, T.; Kettenberger, H.; Hilger, M.; Rand, K.D. Investigating the interaction between the neonatal Fc receptor and monoclonal antibody variants by hydrogen/deuterium exchange mass spectrometry. Mol. Cell Proteomics 2015, 14, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Rossini, S.; Noé, R.; Daventure, V.; Lecerf, M.; Justesen, S.; Dimitrov, J.D. V Region of IgG Controls the Molecular Properties of the Binding Site for Neonatal Fc Receptor. J. Immunol. 2020, 205, 2850–2860. [Google Scholar] [CrossRef] [PubMed]
- Gurbaxani, B.; Dela Cruz, L.L.; Chintalacharuvu, K.; Morrison, S.L. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol. Immunol. 2006, 43, 1462–1473. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Estevez, A.; Schlothauer, T.; Wecksler, A.T. Antigen physiochemical properties allosterically effect the IgG Fc-region and Fc neonatal receptor affinity. mAbs 2020, 12, 1802135. [Google Scholar] [CrossRef]
- Ternant, D.; Arnoult, C.; Pugnière, M.; Dhommée, C.; Drocourt, D.; Perouzel, E.; Passot, C.; Baroukh, N.; Mulleman, D.; Tiraby, G.; et al. IgG1 Allotypes Influence the Pharmacokinetics of Therapeutic Monoclonal Antibodies through FcRn Binding. J. Immunol. 2016, 196, 607–613. [Google Scholar] [CrossRef]
- Monnet, C.; Jorieux, S.; Urbain, R.; Fournier, N.; Bouayadi, K.; De Romeuf, C.; Behrens, C.K.; Fontayne, A.; Mondon, P. Selection of IgG Variants with Increased FcRn Binding Using Random and Directed Mutagenesis: Impact on Effector Functions. Front. Immunol. 2015, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Mateos, F.; Ober, R.J.; Ward, E.S. Conferring the binding properties of the mouse MHC class I-related receptor, FcRn, onto the human ortholog by sequential rounds of site-directed mutagenesis. J. Mol. Biol. 2005, 345, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, W.F.; Woods, R.M.; Ward, E.S.; Palaszynski, S.R.; Patel, N.K.; Brewah, Y.A.; Wu, H.; Kiener, P.A.; Langermann, S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: Biological consequences. J. Immunol. 2002, 169, 5171–5180. [Google Scholar] [PubMed] [Green Version]
- Zhou, J.; Johnson, J.E.; Ghetie, V.; Ober, R.J.; Ward, E.S. Generation of mutated variants of the human form of the MHC class I-related receptor, FcRn, with increased affinity for mouse immunoglobulin G. J. Mol. Biol. 2003, 332, 901–913. [Google Scholar] [CrossRef]
- Vaccaro, C.; Bawdon, R.; Wanjie, S.; Ober, R.J.; Ward, E.S. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 18709–18714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ober, R.J.; Radu, C.G.; Ghetie, V.; Ward, E.S. Differences in promiscuity for antibody-FcRn interactions across species: Implications for therapeutic antibodies. Int. Immunol. 2001, 13, 1551–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambell, F.W. The transmission of immunity from mother to young and the catabolism of immunoglobulins. Lancet 1966, 2, 1087–1093. [Google Scholar] [CrossRef]
- Kim, H.; Robinson, S.B.; Csaky, K.G. FcRn receptor-mediated pharmacokinetics of therapeutic IgG in the eye. Mol. Vis. 2009, 15, 2803–2812. [Google Scholar] [PubMed]
- Haymann, J.P.; Levraud, J.P.; Bouet, S.; Kappes, V.; Hagège, J.; Nguyen, G.; Xu, Y.; Rondeau, E.; Sraer, J.D. Characterization and localization of the neonatal Fc receptor in adult human kidney. J. Am. Soc. Nephrol. 2000, 11, 632–639. [Google Scholar] [PubMed]
- Blumberg, R.S.; Koss, T.; Story, C.M.; Barisani, D.; Polischuk, J.; Lipin, A.; Pablo, L.; Green, R.; Simister, N.E. A major histocompatibility complex class I-related Fc receptor for IgG on rat hepatocytes. J. Clin. Investig. 1995, 95, 2397–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, B.L.; Badizadegan, K.; Wu, Z.; Ahouse, J.C.; Zhu, X.; Simister, N.E.; Blumberg, R.S.; Lencer, W.I. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J. Clin. Investig. 1999, 104, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Israel, E.J.; Taylor, S.; Wu, Z.; Mizoguchi, E.; Blumberg, R.S.; Bhan, A.; Simister, N.E. Expression of the neonatal Fc receptor, FcRn, on human intestinal epithelial cells. Immunology 1997, 92, 69–74. [Google Scholar] [CrossRef]
- Kim, K.J.; Fandy, T.E.; Lee, V.H.; Ann, D.K.; Borok, Z.; Crandall, E.D. Net absorption of IgG via FcRn-mediated transcytosis across rat alveolar epithelial cell monolayers. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L616–L622. [Google Scholar] [CrossRef]
- Sachs, U.J.; Socher, I.; Braeunlich, C.G.; Kroll, H.; Bein, G.; Santoso, S. A variable number of tandem repeats polymorphism influences the transcriptional activity of the neonatal Fc receptor alpha-chain promoter. Immunology 2006, 119, 83–89. [Google Scholar] [CrossRef]
- Challa, D.K.; Wang, X.; Montoyo, H.P.; Velmurugan, R.; Ober, R.J.; Ward, E.S. Neonatal Fc receptor expression in macrophages is indispensable for IgG homeostasis. mAbs 2019, 11, 848–860. [Google Scholar] [CrossRef] [Green Version]
- Rossing, N. Intra- and extravascular distribution of albumin and immunoglobulin in man. Lymphology 1978, 11, 138–142. [Google Scholar] [PubMed]
- Antohe, F.; Rădulescu, L.; Gafencu, A.; Gheţie, V.; Simionescu, M. Expression of functionally active FcRn and the differentiated bidirectional transport of IgG in human placental endothelial cells. Hum. Immunol. 2001, 62, 93–105. [Google Scholar] [CrossRef]
- Kiskova, T.; Mytsko, Y.; Schepelmann, M.; Helmer, H.; Fuchs, R.; Miedl, H.; Wadsack, C.; Ellinger, I. Expression of the neonatal Fc-receptor in placental-fetal endothelium and in cells of the placental immune system. Placenta 2019, 78, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.; Sager, R.; Schneider, H. Maternal-fetal transport of immunoglobulin G and its subclasses during the third trimester of human pregnancy. Am. J. Reprod. Immunol. 1994, 32, 8–14. [Google Scholar] [CrossRef]
- Clements, T.; Rice, T.F.; Vamvakas, G.; Barnett, S.; Barnes, M.; Donaldson, B.; Jones, C.E.; Kampmann, B.; Holder, B. Update on Transplacental Transfer of IgG Subclasses: Impact of Maternal and Fetal Factors. Front. Immunol. 2020, 11, 1920. [Google Scholar] [CrossRef]
- Malek, A.; Sager, R.; Zakher, A.; Schneider, H. Transport of immunoglobulin G and its subclasses across the in vitro-perfused human placenta. Am. J. Obstet. Gynecol. 1995, 173, 760–767. [Google Scholar] [CrossRef]
- Firan, M.; Bawdon, R.; Radu, C.; Ober, R.J.; Eaken, D.; Antohe, F.; Ghetie, V.; Ward, E.S. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int. Immunol. 2001, 13, 993–1002. [Google Scholar] [CrossRef]
- Jennewein, M.F.; Abu-Raya, B.; Jiang, Y.; Alter, G.; Marchant, A. Transfer of maternal immunity and programming of the newborn immune system. Semin. Immunopathol. 2017, 39, 605–613. [Google Scholar] [CrossRef]
- Wilcox, C.R.; Holder, B.; Jones, C.E. Factors affecting the FcRn-mediated transplacental transfer of antibodies and implications for vaccination in pregnancy. Front. Immunol. 2017, 8, 1294. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, C.; Lang, S.; Zhu, G.; Reheman, A.; Spring, C.M.; Freedman, J.; Ni, H. Animal model of fetal and neonatal immune thrombocytopenia: Role of neonatal Fc receptor in the pathogenesis and therapy. Blood 2010, 116, 3660–3668. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.H.; Morrison, S.L. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 1989, 143, 2595–2601. [Google Scholar] [PubMed]
- Martinez, D.R.; Fong, Y.; Li, S.H.; Yang, F.; Jennewein, M.F.; Weiner, J.A.; Harrell, E.A.; Mangold, J.F.; Goswami, R.; Seage, G.R., 3rd; et al. Fc Characteristics Mediate Selective Placental Transfer of IgG in HIV-Infected Women. Cell 2019, 178, 190–201. [Google Scholar] [CrossRef]
- Jennewein, M.F.; Goldfarb, I.; Dolatshahi, S.; Cosgrove, C.; Noelette, F.J.; Krykbaeva, M.; Das, J.; Sarkar, A.; Gorman, M.J.; Fischinger, S.; et al. Fc Glycan-Mediated Regulation of Placental Antibody Transfer. Cell 2019, 178, 202–215. [Google Scholar] [CrossRef] [Green Version]
- Borghi, S.; Bournazos, S.; Thulin, N.K.; Li, C.; Gajewski, A.; Sherwood, R.W.; Zhang, S.; Harris, E.; Jagannathan, P.; Wang, L.X.; et al. FcRn, but not FcγRs, drives maternal-fetal transplacental transport of human IgG antibodies. Proc. Natl. Acad. Sci. USA 2020, 117, 12943–12951. [Google Scholar] [CrossRef]
- Einarsdottir, H.K.; Selman, M.H.; Kapur, R.; Scherjon, S.; Koeleman, C.A.; Deelder, A.M.; van der Schoot, C.E.; Vidarsson, G.; Wuhrer, M. Comparison of the Fc glycosylation of fetal and maternal immunoglobulin G. Glycoconj. J. 2013, 30, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.K.; Betts, A.M. Antibody biodistribution coefficients: Inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. mAbs 2013, 5, 297–305. [Google Scholar] [CrossRef]
- Eigenmann, M.J.; Fronton, L.; Grimm, H.P.; Otteneder, M.B.; Krippendorff, B.F. Quantification of IgG monoclonal antibody clearance in tissues. mAbs 2017, 9, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- van der Flier, A.; Liu, Z.; Tan, S.; Chen, K.; Drager, D.; Liu, T.; Patarroyo-White, S.; Jiang, H.; Light, D.R. FcRn Rescues Recombinant Factor VIII Fc Fusion Protein from a VWF Independent FVIII Clearance Pathway in Mouse Hepatocytes. PLoS ONE 2015, 10, e0124930. [Google Scholar] [CrossRef] [Green Version]
- Pyzik, M.; Rath, T.; Kuo, T.T.; Win, S.; Baker, K.; Hubbard, J.J.; Grenha, R.; Gandhi, A.; Krämer, T.D.; Mezo, A.R.; et al. Hepatic FcRn regulates albumin homeostasis and susceptibility to liver injury. Proc. Natl. Acad. Sci. USA 2017, 114, E2862–E2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarav, M.; Wang, Y.; Hack, B.K.; Chang, A.; Jensen, M.; Bao, L.; Quigg, R.J. Renal FcRn reclaims albumin but facilitates elimination of IgG. J. Am. Soc. Nephrol. 2009, 20, 1941–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akilesh, S.; Huber, T.B.; Wu, H.; Wang, G.; Hartleben, B.; Kopp, J.B.; Miner, J.H.; Roopenian, D.C.; Unanue, E.R.; Shaw, A.S. Podocytes use FcRn to clear IgG from the glomerular basement membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 967–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaru, F.; Luo, W.; Suleiman, H.; St John, P.L.; Ge, L.; Mezo, A.R.; Shaw, A.S.; Abrahamson, D.R.; Miner, J.H.; Borza, D.B. Neonatal Fc receptor promotes immune complex-mediated glomerular disease. J. Am. Soc. Nephrol. 2014, 25, 918–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, R.; Maeda, K.; Tsokos, M.G.; Pavlakis, M.; Stillman, I.E.; Tsokos, G.C. N-glycosylated IgG in patients with kidney transplants increases calcium/calmodulin kinase IV in podocytes and causes injury. Am. J. Transplant. 2020, 21, 148–160. [Google Scholar] [CrossRef]
- Tonsawan, P.; Dylewski, J.; Lewis, L.; Blaine, J. Knockout of the neonatal Fc receptor in cultured podocytes alters IL-6 signaling and the actin cytoskeleton. Am. J. Physiol. Cell Physiol. 2019, 317, C1048–C1060. [Google Scholar] [CrossRef]
- Cerutti, A.; Chen, K.; Chorny, A. Immunoglobulin responses at the mucosal interface. Annu. Rev. Immunol. 2011, 29, 273–293. [Google Scholar] [CrossRef] [Green Version]
- Claypool, S.M.; Dickinson, B.L.; Wagner, J.S.; Johansen, F.E.; Venu, N.; Borawski, J.A.; Lencer, W.I.; Blumberg, R.S. Bidirectional transepithelial IgG transport by a strongly polarized basolateral membrane Fcgamma-receptor. Mol. Biol. Cell. 2004, 15, 1746–1759. [Google Scholar] [CrossRef]
- Hornby, P.J.; Cooper, P.R.; Kliwinski, C.; Ragwan, E.; Mabus, J.R.; Harman, B.; Thompson, S.; Kauffman, A.L.; Yan, Z.; Tam, S.H.; et al. Human and non-human primate intestinal FcRn expression and immunoglobulin G transcytosis. Pharm. Res. 2014, 31, 908–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Dopico, T.; Clatworthy, M.R. IgG and Fcγ Receptors in Intestinal Immunity and Inflammation. Front. Immunol. 2019, 10, 805. [Google Scholar] [CrossRef]
- Yoshida, M.; Claypool, S.M.; Wagner, J.S.; Mizoguchi, E.; Mizoguchi, A.; Roopenian, D.C.; Lencer, W.I.; Blumberg, R.S. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 2004, 20, 769–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, M.; Kobayashi, K.; Kuo, T.T.; Bry, L.; Glickman, J.N.; Claypool, S.M.; Kaser, A.; Nagaishi, T.; Higgins, D.E.; Mizoguchi, E.; et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J. Clin. Investig. 2006, 116, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.T.; Hodgkinson, A.J.; Prosser, C.G.; Davis, S.R. Immune Components of Colostrum and Milk—A Historical Perspective. J. Mammary Gland Biol. Neoplasia 2007, 12, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, X.; He, J.; Diraviyam, T.; Zhang, X. Quantitative Investigation on Correlation Between IgG and FcRn During Gestation and Lactating Periods in Rat. Am. J. Reprod. Immunol. 2016, 75, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.E. Characteristics of bovine immunoglobulins and related molecules. Review of the bovine immunoglobulins. J. Dairy Sci. 1971, 54, 1315–1316. [Google Scholar]
- Shah, U.; Dickinson, B.L.; Blumberg, R.S.; Simister, N.E.; Lencer, W.I.; Walker, W.A. Distribution of the IgG Fc receptor, FcRn, in the human fetal intestine. Pediatr. Res. 2003, 53, 295–301. [Google Scholar] [CrossRef]
- Li, Z.; Palaniyandi, S.; Zeng, R.; Tuo, W.; Roopenian, D.C.; Zhu, X. Transfer of IgG in the female genital tract by MHC class I-related neonatal Fc receptor (FcRn) confers protective immunity to vaginal infection. Proc. Natl. Acad. Sci. USA 2011, 108, 4388–4393. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.Y.; Pegu, A.; Rudicell, R.S.; Yang, Z.Y.; Joyce, M.G.; Chen, X.; Wang, K.; Bao, S.; Kraemer, T.D.; Rath, T.; et al. Enhanced neonatal Fc receptor function improves protection against primate SHIV infection. Nature 2014, 514, 642–645. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Balthasar, J.P. FcRn Expression in Wildtype Mice, Transgenic Mice, and in Human Tissues. Biomolecules 2018, 8, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cauza, K.; Hinterhuber, G.; Dingelmaier-Hovorka, R.; Brugger, K.; Klosner, G.; Horvat, R.; Wolff, K.; Foedinger, D. Expression of FcRn, the MHC class I-related receptor for IgG, in human keratinocytes. J. Investig. Dermatol. 2005, 124, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Cianga, P.; Cianga, C.; Plamadeala, P.; Branisteanu, D.; Carasevici, E. The neonatal Fc receptor (FcRn) expression in the human skin. Virchows Arch. 2007, 451, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chernyavsky, A.; Webber, R.J.; Grando, S.A.; Wang, P.H. Critical Role of the Neonatal Fc Receptor (FcRn) in the Pathogenic Action of Antimitochondrial Autoantibodies Synergizing with Anti-desmoglein Autoantibodies in Pemphigus Vulgaris. J. Biol. Chem. 2015, 290, 23826–23837. [Google Scholar] [CrossRef] [Green Version]
- Bequignon, E.; Dhommée, C.; Angely, C.; Thomas, L.; Bottier, M.; Escudier, E.; Isabey, D.; Coste, A.; Louis, B.; Papon, J.F.; et al. FcRn-Dependent Transcytosis of Monoclonal Antibody in Human Nasal Epithelial Cells In Vitro: A Prerequisite for a New Delivery Route for Therapy? Int. J. Mol. Sci. 2019, 20, 1379. [Google Scholar] [CrossRef] [Green Version]
- Deissler, H.L.; Lang, G.K.; Lang, G.E. Internalization of bevacizumab by retinal endothelial cells and its intracellular fate: Evidence for an involvement of the neonatal Fc receptor. Exp. Eye Res. 2016, 143, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Dithmer, M.; Hattermann, K.; Pomarius, P.; Aboul Naga, S.H.; Meyer, T.; Mentlein, R.; Roider, J.; Klettner, A. The role of Fc-receptors in the uptake and transport of therapeutic antibodies in the retinal pigment epithelium. Exp. Eye Res. 2016, 145, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Deissler, H.L.; Lang, G.K.; Lang, G.E. Neonatal Fc receptor FcRn is involved in intracellular transport of the Fc fusion protein aflibercept and its transition through retinal endothelial cells. Exp. Eye Res. 2017, 154, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Amour, I.; Paré, I.; Alata, W.; Coulombe, K.; Ringuette-Goulet, C.; Drouin-Ouellet, J.; Vandal, M.; Soulet, D.; Bazin, R.; Calon, F. Brain bioavailability of human intravenous immunoglobulin and its transport through the murine blood-brain barrier. J. Cereb. Blood Flow Metab. 2013, 33, 1983–1992. [Google Scholar] [CrossRef] [Green Version]
- Schlachetzki, F.; Zhu, C.; Pardridge, W.M. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J. Neurochem. 2002, 81, 203–206. [Google Scholar] [CrossRef]
- Villaseñor, R.; Ozmen, L.; Messaddeq, N.; Grüninger, F.; Loetscher, H.; Keller, A.; Betsholt, C.; Freskgård, P.O.; Collin, L. Trafficking of Endogenous Immunoglobulins by Endothelial Cells at the Blood-Brain Barrier. Sci. Rep. 2016, 6, 25658. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.R.; Ciambrone, G.J.; Kliwinski, C.M.; Maze, E.; Johnson, L.; Li, Q.; Feng, Y.; Hornby, P.J. Efflux of monoclonal antibodies from rat brain by neonatal Fc receptor, FcRn. Brain Res. 2013, 1534, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pardridge, W.M. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J. Neuroimmunol. 2001, 114, 168–272. [Google Scholar] [CrossRef]
- Chen, N.; Wang, W.; Fauty, S.; Fang, Y.; Hamuro, L.; Hussain, A.; Prueksaritanont, T. The effect of the neonatal Fc receptor on human IgG biodistribution in mice. mAbs 2014, 6, 502–508. [Google Scholar] [CrossRef] [Green Version]
- Abuqayyas, L.; Balthasar, J.P. Investigation of the role of FcγR and FcRn in mAb distribution to the brain. Mol. Pharm. 2013, 10, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Ruano-Salguero, J.S.; Lee, K.H. Antibody transcytosis across brain endothelial-like cells occurs nonspecifically and independent of FcRn. Sci. Rep. 2020, 28, 3685. [Google Scholar] [CrossRef] [Green Version]
- Castaneda, D.C.; Brachet, G.; Goupille, C.; Ouldamer, L.; Gouilleux-Gruart, V. The neonatal Fc receptor in cancer FcRn in cancer. Cancer Med. 2020, 9, 4736–4742. [Google Scholar] [CrossRef] [PubMed]
- Dalloneau, E.; Baroukh, N.; Mavridis, K.; Maillet, A.; Gueugnon, F.; Courty, Y.; Petit, A.; Kryza, T.; Del Rio, M.; Guyetant, S.; et al. Downregulation of the neonatal Fc receptor expression in non-small cell lung cancer tissue is associated with a poor prognosis. Oncotarget 2016, 7, 54415–54429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Zhang, W.; Zou, F.; Mei, L.; Wu, G.; Teng, Y. KLHL21, a novel gene that contributes to the progression of hepatocellular carcinoma. BMC Cancer 2016, 16, 815. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bae, J.S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediators Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passot, C.; Azzopardi, N.; Renault, S.; Baroukh, N.; Arnoult, C.; Ohresser, M.; Boisdron-Celle, M.; Gamelin, E.; Watier, H.; Paintaud, G.; et al. Influence of FCGRT gene polymorphisms on pharmacokinetics of therapeutic antibodies. mAbs 2013, 5, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Gouilleux-Gruart, V.; Chapel, H.; Chevret, S.; Lucas, M.; Malphettes, M.; Fieschi, C.; Patel, S.; Boutboul, D.; Marson, M.N.; Gérard, L.; et al. Efficiency of immunoglobulin G replacement therapy in common variable immunodeficiency: Correlations with clinical phenotype and polymorphism of the neonatal Fc receptor. Clin. Exp. Immunol. 2013, 171, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Cejas, R.B.; Ferguson, D.C.; Quiñones-Lombraña, A.; Bard, J.E.; Blanco, J.G. Contribution of DNA methylation to the expression of FCGRT in human liver and myocardium. Sci. Rep. 2019, 9, 8674. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.C.; Blanco, J.G. Regulation of the Human Fc-Neonatal Receptor alpha-Chain Gene FCGRT by MicroRNA-3181. Pharm. Res. 2018, 35, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, L.; Christianson, G.J.; Yang, J.Q.; Roopenian, D.C.; Zhu, X. NF-kappaB signaling regulates functional expression of the MHC class I-related neonatal Fc receptor for IgG via intronic binding sequences. J. Immunol. 2007, 179, 2999–3011. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ye, L.; Bai, Y.; Mojidi, H.; Simister, N.E.; Zhu, X. Activation of the JAK/STAT-1 signaling pathway by IFN-gamma can down-regulate functional expression of the MHC class I-related neonatal Fc receptor for IgG. J. Immunol. 2008, 181, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Weflen, A.W.; Baier, N.; Tang, Q.J.; Van den Hof, M.; Blumberg, R.S.; Lencer, W.I.; Massol, R.H. Multivalent immune complexes divert FcRn to lysosomes by exclusion from recycling sorting tubules. Mol. Biol. Cell. 2013, 24, 2398–2405. [Google Scholar] [CrossRef]
- Qiao, S.W.; Kobayashi, K.; Johansen, F.E.; Sollid, L.M.; Andersen, J.T.; Milford, E.; Roopenian, D.C.; Lencer, W.I.; Blumberg, R.S. Dependence of antibody-mediated presentation of antigen on FcRn. Proc. Natl. Acad. Sci. USA 2008, 105, 9337–9342. [Google Scholar] [CrossRef] [Green Version]
- Vidarsson, G.; Stemerding, A.M.; Stapleton, N.M.; Spliethoff, S.E.; Janssen, H.; Rebers, F.E.; de Haas, M.; van de Winkel, J.G. FcRn: An IgG receptor on phagocytes with a novel role in phagocytosis. Blood 2006, 108, 3573–3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, W.H.; Louber, J.; Mahmoud, I.S.; Chia, J.; Bass, G.T.; Dower, S.K.; Verhagen, A.M.; Gleeson, P.A. FcRn mediates fast recycling of endocytosed albumin and IgG from early macropinosomes in primary macrophages. J. Cell Sci. 2019, 133, jcs235416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molfetta, R.; Quatrini, L.; Gasparrini, F.; Zitti, B.; Santoni, A.; Paolini, R. Regulation of Fc receptor endocytic trafficking by ubiquitination. Front. Immunol. 2014, 5, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, J.J.; Pyzik, M.; Rath, T.; Kozicky, L.K.; Sand, K.M.K.; Gandhi, A.K.; Grevys, A.; Foss, S.; Menzies, S.C.; Glickman, J.N.; et al. FcRn is a CD32a coreceptor that determines susceptibility to IgG immune complex-driven autoimmunity. J. Exp. Med. 2020, 217, e20200359. [Google Scholar] [CrossRef]
- Castaneda, D.C.; Dhommée, C.; Baranek, T.; Dalloneau, E.; Lajoie, L.; Valayer, A.; Arnoult, C.; Demattéi, M.V.; Fouquenet, D.; Parent, C.; et al. Lack of FcRn Impairs Natural Killer Cell Development and Functions in the Tumor Microenvironment. Front. Immunol. 2018, 9, 2259. [Google Scholar] [CrossRef] [Green Version]
- Tulp, A.; Verwoerd, D.; Dobberstein, B.; Ploegh, H.L.; Pieters, J. Isolation and characterization of the intracellular MHC class II compartment. Nature 1994, 369, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.; Qiao, S.W.; Kuo, T.T.; Aveson, V.G.; Platzer, B.; Andersen, J.T.; Sandlie, I.; Chen, Z.; de Haar, C.; Lencer, W.I.; et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9927–9932. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.; Rath, T.; Flak, M.B.; Arthur, J.C.; Chen, Z.; Glickman, J.N.; Zlobec, I.; Karamitopoulou, E.; Stachler, M.D.; Odze, R.D.; et al. Neonatal Fc receptor expression in dendritic cells mediates protective immunity against colorectal cancer. Immunity 2013, 39, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Culina, S.; Meslier, Y.; Dimitrov, J.; Arnoult, C.; Delignat, S.; Gangadharan, B.; Lecerf, M.; Justesen, S.; Gouilleux-Gruart, V.; et al. Regulation of immune responses to protein therapeutics by transplacental induction of T cell tolerance. Sci. Transl. Med. 2015, 7, 275ra21. [Google Scholar] [CrossRef]
- Culina, S.; Gupta, N.; Boisgard, R.; Afonso, G.; Gagnerault, M.C.; Dimitrov, J.; Østerbye, T.; Justesen, S.; Luce, S.; Attias, M.; et al. Materno-Fetal Transfer of Preproinsulin Through the Neonatal Fc Receptor Prevents Autoimmune Diabetes. Diabetes 2015, 64, 3532–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsaki, A.; Venturelli, N.; Buccigrosso, T.M.; Osganian, S.K.; Lee, J.; Blumberg, R.S.; Oyoshi, M.K. Maternal IgG immune complexes induce food allergen-specific tolerance in offspring. J. Exp. Med. 2018, 215, 91–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, E.; Rekima, A.; Seitz-Polski, B.; Kanda, A.; Fleury, S.; Tissandie, E.; Monteiro, R.; Dombrowicz, D.D.; Julia, V.; Glaichenhaus, N.; et al. Breast milk immune complexes are potent inducers of oral tolerance in neonates and prevent asthma development. Mucosal Immunol. 2010, 3, 461–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhasselt, V.; Milcent, V.; Cazareth, J.; Kanda, A.; Fleury, S.; Dombrowicz, D.; Glaichenhaus, N.; Julia, V. Breast milk-mediated transfer of an antigen induces tolerance and protection from allergic asthma. Nat. Med. 2008, 14, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Kobayashi, K.; Ishikawa, Y.; Yamamoto, M.; Funada, Y.; Kotani, Y.; Blumberg, R.S.; Karasuyama, H.; Yoshida, M.; Nishimura, Y. The transfer of maternal antigen-specific IgG regulates the development of allergic airway inflammation early in life in an FcRn-dependent manner. Biochem. Biophys. Res. Commun. 2010, 395, 238–243. [Google Scholar] [CrossRef] [Green Version]
- Bundhoo, A.; Paveglio, S.; Rafti, E.; Dhongade, A.; Blumberg, R.S.; Matson, A.P. Evidence that FcRn mediates the transplacental passage of maternal IgE in the form of IgG anti-IgE/IgE immune complexes. Clin. Exp. Allergy 2015, 45, 1085–1098. [Google Scholar] [CrossRef]
- Wilcox, C.R.; Jones, C.E. Beyond Passive Immunity: Is There Priming of the Fetal Immune System Following Vaccination in Pregnancy and What Are the Potential Clinical Implications? Front. Immunol. 2018, 9, 1548. [Google Scholar] [CrossRef] [Green Version]
- Hinton, P.R.; Johlfs, M.G.; Xiong, J.M.; Hanestad, K.; Ong, K.C.; Bullock, C.; Keller, S.; Tang, M.T.; Tso, J.Y.; Vásquez, M.; et al. Engineered human IgG antibodies with longer serum half-lives in primates. J. Biol. Chem. 2004, 279, 6213–6216. [Google Scholar] [CrossRef] [Green Version]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.Q.; Robbie, G.J.; Wu, Y.; Esser, M.T.; Jensen, K.; Schwartz, H.I.; Bellamy, T.; Hernandez-Illas, M.; Jafri, H.S. Safety, Tolerability, and Pharmacokinetics of MEDI4893, an Investigational, Extended-Half-Life, Anti-Staphylococcus aureus Alpha-Toxin Human Monoclonal Antibody, in Healthy Adults. Antimicrob. Agents Chemother. 2016, 61, e01020-e16. [Google Scholar] [CrossRef] [Green Version]
- Robbie, G.J.; Criste, R.; Dall’acqua, W.F.; Jensen, K.; Patel, N.K.; Losonsky, G.A.; Griffin, M.P. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob. Agents Chemother. 2013, 57, 6147–6153. [Google Scholar] [CrossRef] [Green Version]
- Yeung, Y.A.; Leabman, M.K.; Marvin, J.S.; Qiu, J.; Adams, C.W.; Lien, S.; Starovasnik, M.A.; Lowman, H.B. Engineering human IgG1 affinity to human neonatal Fc receptor: Impact of affinity improvement on pharmacokinetics in primates. J. Immunol. 2009, 182, 7663–7671. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; McLellan, J.S.; Kallewaard, N.L.; Ulbrandt, N.D.; Palaszynski, S.; Zhang, J.; Moldt, B.; Khan, A.; Svabek, C.; McAuliffe, J.M.; et al. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci. Transl. Med. 2017, 9, eaaj1928. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, W.F.; Kiener, P.A.; Wu, H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 2006, 281, 23514–23524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grevys, A.; Bern, M.; Foss, S.; Bratlie, D.B.; Moen, A.; Gunnarsen, K.S.; Aase, A.; Michaelsen, T.E.; Sandlie, I.; Andersen, J.T. Fc Engineering of Human IgG1 for Altered Binding to the Neonatal Fc Receptor Affects Fc Effector Functions. J. Immunol. 2015, 194, 5497–5508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Kang, T.H.; Godon, O.; Watanabe, M.; Delidakis, G.; Gillis, C.M.; Sterlin, D.; Hardy, D.; Cogné, M.; Macdonald, L.E.; et al. An engineered human Fc domain that behaves like a pH-toggle switch for ultra-long circulation persistence. Nat. Commun. 2019, 10, 5031. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Iwayanagi, Y.; Haraya, K.; Tachibana, T.; Nakamura, G.; Nambu, T.; Esaki, K.; Hattori, K.; Igawa, T. Identification of human IgG1 variant with enhanced FcRn binding and without increased binding to rheumatoid factor autoantibody. mAbs 2017, 9, 844–853. [Google Scholar] [CrossRef] [Green Version]
- Yeung, Y.A.; Wu, X.; Reyes, A.E., 2nd; Vernes, J.M.; Lien, S.; Lowe, J.; Maia, M.; Forrest, W.F.; Meng, Y.G.; Damico, L.A.; et al. A therapeutic anti-VEGF antibody with increased potency independent of pharmacokinetic half-life. Cancer Res. 2010, 70, 3269–3277. [Google Scholar] [CrossRef] [Green Version]
- Deng, R.; Loyet, K.M.; Lien, S.; Iyer, S.; DeForge, L.E.; Theil, F.P.; Lowman, H.B.; Fielder, P.J.; Prabhu, S. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-{alpha} antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug. Metab. Dispos. 2010, 38, 600–605. [Google Scholar] [CrossRef] [Green Version]
- Borrok, M.J.; Wu, Y.; Beyaz, N.; Yu, X.Q.; Oganesyan, V.; Dall’Acqua, W.F.; Tsui, P. pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J. Biol. Chem. 2015, 290, 4282–4290. [Google Scholar] [CrossRef] [Green Version]
- Goulet, D.R.; Watson, M.J.; Tam, S.H.; Zwolak, A.; Chiu, M.L.; Atkins, W.M.; Nath, A. Toward a Combinatorial Approach for the Prediction of IgG Half-Life and Clearance. Drug. Metab. Dispos. 2018, 46, 1900–1907. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chen, Y.; Pelletier, M.; Cvitkovic, R.; Bonnell, J.; Chang, C.Y.; Koksal, A.C.; O’Connor, E.; Gao, X.; Yu, X.Q.; et al. Enhancement of antibody functions through Fc multiplications. mAbs 2017, 9, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Goulet, D.R.; Zwolak, A.; Williams, J.A.; Chiu, M.L.; Atkins, W.M. Design and characterization of novel dual Fc antibody with enhanced avidity for Fc receptors. Proteins 2020, 88, 689–697. [Google Scholar] [CrossRef]
- O’Hear, C.E.; Foote, J. Antibody buffering of a ligand in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 40–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, L.T.; Neta, R.; Moldawer, L.L.; Kenney, J.S.; Patel, K.; Sehgal, P.B. Antibodies chaperone circulating IL-6. Paradoxical effects of anti-IL-6 “neutralizing” antibodies in vivo. J. Immunol. 1993, 151, 3225–3236. [Google Scholar] [PubMed]
- Igawa, T.; Ishii, S.; Tachibana, T.; Maeda, A.; Higuchi, Y.; Shimaoka, S.; Moriyama, C.; Watanabe, T.; Takubo, R.; Doi, Y.; et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat. Biotechnol. 2010, 28, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Henne, K.R.; Ason, B.; Howard, M.; Wang, W.; Sun, J.; Higbee, J.; Tang, J.; Matsuda, K.C.; Xu, R.; Zhou, L.; et al. Anti-PCSK9 antibody pharmacokinetics and low-density lipoprotein-cholesterol pharmacodynamics in nonhuman primates are antigen affinity-dependent and exhibit limited sensitivity to neonatal Fc receptor-binding enhancement. J. Pharmacol. Exp. Ther. 2015, 353, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Devanaboyina, S.C.; Lynch, S.M.; Ober, R.J.; Ram, S.; Kim, D.; Puig-Canto, A.; Breen, S.; Kasturirangan, S.; Fowler, S.; Peng, L.; et al. The effect of pH dependence of antibody-antigen interactions on subcellular trafficking dynamics. mAbs 2013, 5, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Heo, Y.A. Satralizumab: First Approval. Drugs 2020, 80, 1477–1482. [Google Scholar] [CrossRef]
- Chaparro-Riggers, J.; Liang, H.; DeVay, R.M.; Bai, L.; Sutton, J.E.; Chen, W.; Geng, T.; Lindquist, K.; Casas, M.G.; Boustany, L.M.; et al. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J. Biol. Chem. 2012, 287, 11090–11097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuzawa, T.; Sampei, Z.; Haraya, K.; Ruike, Y.; Shida-Kawazoe, M.; Shimizu, Y.; Gan, S.W.; Irie, M.; Tsuboi, Y.; Tai, H.; et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci. Rep. 2017, 7, 1080. [Google Scholar] [CrossRef]
- Bonvin, P.; Venet, S.; Fontaine, G.; Ravn, U.; Gueneau, F.; Kosco-Vilbois, M.; Proudfoot, A.E.; Fischer, N. De novo isolation of antibodies with pH-dependent binding properties. mAbs 2015, 7, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hironiwa, N.; Ishii, S.; Kadono, S.; Iwayanagi, Y.; Mimoto, F.; Habu, K.; Igawa, T.; Hattori, K. Calcium-dependent antigen binding as a novel modality for antibody recycling by endosomal antigen dissociation. mAbs 2016, 8, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Igawa, T.; Haraya, K.; Hattori, K. Sweeping antibody as a novel therapeutic antibody modality capable of eliminating soluble antigens from circulation. Immunol. Rev. 2016, 270, 132–151. [Google Scholar] [CrossRef] [PubMed]
- Igawa, T.; Maeda, A.; Haraya, K.; Tachibana, T.; Iwayanagi, Y.; Mimoto, F.; Higuchi, Y.; Ishii, S.; Tamba, S.; Hironiwa, N.; et al. Engineered monoclonal antibody with novel antigen-sweeping activity in vivo. PLoS ONE 2013, 8, e63236. [Google Scholar] [CrossRef]
- Yang, D.; Giragossian, C.; Castellano, S.; Lasaro, M.; Xiao, H.; Saraf, H.; Hess Kenny, C.; Rybina, I.; Huang, Z.F.; Ahlberg, J.; et al. Maximizing in vivo target clearance by design of pH-dependent target binding antibodies with altered affinity to FcRn. mAbs 2017, 9, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab. Available online: https://www.fda.gov/media/143603/download (accessed on 7 February 2021).
- Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Casirivimab and Imdevimab. Available online: https://www.fda.gov/media/143892/download (accessed on 7 February 2021).
- Perez, E.E.; Orange, J.S.; Bonilla, F.; Chinen, J.; Chinn, I.K.; Dorsey, M.; El-Gamal, Y.; Harville, T.O.; Hossny, E.; Mazer, B.; et al. Update on the use of immunoglobulin in human disease: A review of evidence. J. Allergy Clin. Immunol. 2017, 139, S1–S46. [Google Scholar] [CrossRef] [Green Version]
- Schiff, R.I.; Rudd, C. Alterations in the half-life and clearance of IgG during therapy with intravenous gamma-globulin in 16 patients with severe primary humoral immunodeficiency. J. Clin. Immunol. 1986, 6, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Datta-Mannan, A.; Croy, J.E.; Schirtzinger, L.; Torgerson, S.; Breyer, M.; Wroblewski, V.J. Aberrant bispecific antibody pharmacokinetics linked to liver sinusoidal endothelium clearance mechanism in cynomolgus monkeys. mAbs 2016, 8, 969–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regula, J.T.; Lundh von Leithner, P.; Foxton, R.; Barathi, V.A.; Cheung, C.M.; Bo Tun, S.B.; Wey, Y.S.; Iwata, D.; Dostalek, M.; Moelleken, J.; et al. Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol. Med. 2016, 8, 1265–1288. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Brown, R.M.; Fitchett, J.; Heng, A.R.; Balasubramaniam, D.; Pereira, J.; Croy, J.E. Modulation of the Biophysical Properties of Bifunctional Antibodies as a Strategy for Mitigating Poor Pharmacokinetics. Biochemistry 2019, 58, 3116–3132. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Su, W.; Zhang, W.; Wang, P.; Sattler, M.; Zou, P. Recent Advances in Half-life Extension Strategies for Therapeutic Peptides and Proteins. Curr. Pharm. Des. 2018, 24, 4932–4946. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug. Deliv. Rev. 2015, 91, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Huh, K.Y.; Cho, Y.G.; Cha, J.E.; Kim, S.J.; Yoon, S.H.; Park, S.S.; Yoon, H.; Lee, J.; Lee, H. Safety, tolerability and pharmacokinetics and pharmacodynamics of HL2351, a novel hybrid fc-fused interleukin-1 receptor antagonist, in healthy subjects: A first-in-human study. Br. J. Clin. Pharmacol. 2020, 86, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, E.A.; Wang, J.; Strome, S.E. Engineering of Fc Multimers as a Protein Therapy for Autoimmune Disease. Front. Immunol. 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Ying, T.; Wang, Y.; Feng, Y.; Prabakaran, P.; Gong, R.; Wang, L.; Crowder, K.; Dimitrov, D.S. Engineered antibody domains with significantly increased transcytosis and half-life in macaques mediated by FcRn. mAbs 2015, 7, 922–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, O.S.; Rowley, T.F.; Junker, F.; Peters, S.J.; Crilly, S.; Compson, J.; Eddleston, A.; Björkelund, H.; Greenslade, K.; Parkinson, M.; et al. Multivalent Fcγ-receptor engagement by a hexameric Fc-fusion protein triggers Fcγ-receptor internalisation and modulation of Fcγ-receptor functions. Sci. Rep. 2017, 7, 17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sockolosky, J.T.; Tiffany, M.R.; Szoka, F.C. Engineering neonatal Fc receptor-mediated recycling and transcytosis in recombinant proteins by short terminal peptide extensions. Proc. Natl. Acad. Sci. USA 2012, 109, 16095–16100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davé, E.; Adams, R.; Zaccheo, O.; Carrington, B.; Compson, J.E.; Dugdale, S.; Airey, M.; Malcolm, S.; Hailu, H.; Wild, G.; et al. Fab-dsFv: A bispecific antibody format with extended serum half-life through albumin binding. mAbs 2016, 8, 1319–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuercher, A.W.; Spirig, R.; Baz Morelli, A.; Rowe, T.; Käsermann, F. Next-generation Fc receptor-targeting biologics for autoimmune diseases. Autoimmun. Rev. 2019, 18, 102366. [Google Scholar] [CrossRef] [PubMed]
- Norris, P.A.A.; Kaur, G.; Lazarus, A.H. New insights into IVIg mechanisms and alternatives in autoimmune and inflammatory diseases. Curr. Opin. Hematol. 2020, 27, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.K.; Rosenberg, A.S.; Kishnani, P.S. The potential impact of timing of IVIG administration on the efficacy of rituximab for immune tolerance induction for patients with Pompe disease. Clin. Immunol. 2020, 219, 108541. [Google Scholar] [CrossRef]
- Vaccaro, C.; Zhou, J.; Ober, R.J.; Ward, E.S. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat. Biotechnol. 2005, 23, 1283–1288. [Google Scholar] [CrossRef]
- Gable, K.L.; Guptill, J.T. Antagonism of the Neonatal Fc Receptor as an Emerging Treatment for Myasthenia Gravis. Front. Immunol. 2020, 10, 3052. [Google Scholar] [CrossRef]
- Ling, L.E.; Hillson, J.L.; Tiessen, R.G.; Bosje, T.; van Iersel, M.P.; Nix, D.J.; Markowitz, L.; Cilfone, N.A.; Duffner, J.; Streisand, J.B.; et al. M281, an Anti-FcRn Antibody: Pharmacodynamics, Pharmacokinetics, and Safety Across the Full Range of IgG Reduction in a First-in-Human Study. Clin. Pharmacol. Ther. 2019, 105, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Robak, T.; Kaźmierczak, M.; Jarque, I.; Musteata, V.; Treliński, J.; Cooper, N.; Kiessling, P.; Massow, U.; Woltering, F.; Snipes, R.; et al. Phase 2 multiple-dose study of an FcRn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood Adv. 2020, 4, 4136–4146. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, L.J.; Humphries, J.E.; Jones, S.D.; Pearce, L.B.; Holgate, R.; Hearn, A.; Cheung, J.; Mahmood, A.; Del Tito, B.; Graydon, J.S.; et al. Blocking FcRn in humans reduces circulating IgG levels and inhibits IgG immune complex-mediated immune responses. Sci. Adv. 2019, 5, eaax9586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newland, A.C.; Sánchez-González, B.; Rejtő, L.; Egyed, M.; Romanyuk, N.; Godar, M.; Verschueren, K.; Gandini, D.; Ulrichts, P.; Beauchamp, J.; et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am. J. Hematol. 2020, 95, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Liu, F.; Zheng, N.S.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse Functional Autoantibodies in Patients with COVID-19. medRxiv. (Preprint). Available online: https://www.medrxiv.org/content/10.1101/2020.12.10.20247205v4.full.pdf (accessed on 11 February 2021).
- Kasprick, A.; Hofrichter, M.; Smith, B.; Ward, P.; Bieber, K.; Shock, A.; Ludwig, R.J.; Schmidt, E. Treatment with anti-neonatal Fc receptor (FcRn) antibody ameliorates experimental epidermolysis bullosa acquisita in mice. Br. J. Pharmacol. 2020, 177, 2381–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.A.; Puig-Canto, A.; Challa, D.K.; Perez Montoyo, H.; Ober, R.J.; Ward, E.S. Neonatal Fc receptor blockade by Fc engineering ameliorates arthritis in a murine model. J. Immunol. 2011, 187, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Challa, D.K.; Bussmeyer, U.; Khan, T.; Montoyo, H.P.; Bansal, P.; Ober, R.J.; Ward, E.S. Autoantibody depletion ameliorates disease in murine experimental autoimmune encephalomyelitis. mAbs 2013, 5, 655–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.; Christodoulou, L.; Clargo, A.; Eddleston, A.; Greenslade, K.; Lightwood, D.; Shock, A.; Tyson, K.; Brennan, F.R. Generation of two high affinity anti-mouse FcRn antibodies: Inhibition of IgG recycling in wild type mice and effect in a mouse model of immune thrombocytopenia. Int. Immunopharmacol. 2019, 66, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Seijsing, J.; Yu, S.; Frejd, F.Y.; Höiden-Guthenberg, I.; Gräslund, T. In vivo depletion of serum IgG by an affibody molecule binding the neonatal Fc receptor. Sci. Rep. 2018, 8, 5141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03502954 (accessed on 20 January 2021).
- Alashkar, F.; Rottinghaus, S.; Vance, C.; Herich-Terhürne, D.; Dührsen, U.; Assert, R.; Röth, A. No evidence for hypogammaglobulinemia in patients with paroxysmal nocturnal hemoglobinuria (PNH) chronically treated with ravulizumab. PLoS ONE 2020, 15, e0230869. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Zaitsev, S.; Rauova, L.; Rux, A.H.; Stepanova, V.; Krishnaswamy, S.; Sarkar, A.; Kowalska, M.A.; Zhao, G.; Mast, A.E.; et al. FcRn augments induction of tissue factor activity by IgG-containing immune complexes. Blood 2020, 135, 2085–2093. [Google Scholar] [CrossRef]
- Ipe, T.S.; Marques, M.B. Vascular access for therapeutic plasma exchange. Transfusion 2018, 58, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Valente, D.; Mauriac, C.; Schmidt, T.; Focken, I.; Beninga, J.; Mackness, B.; Qiu, H.; Vicat, P.; Kandira, A.; Radošević, K.; et al. Pharmacokinetics of novel Fc-engineered monoclonal and multispecific antibodies in cynomolgus monkeys and humanized FcRn transgenic mouse models. mAbs 2020, 12, 1829337. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Gurbaxani, B.; Dostalek, M.; Gardner, I. Are endosomal trafficking parameters better targets for improving mAb pharmacokinetics than FcRn binding affinity? Mol. Immunol. 2013, 56, 660–674. [Google Scholar] [CrossRef] [PubMed]
- Proetzel, G.; Wiles, M.V.; Roopenian, D.C. Genetically engineered humanized mouse models for preclinical antibody studies. BioDrugs 2014, 28, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Roopenian, D.C.; Christianson, G.J.; Sproule, T.J. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol. Biol. 2010, 602, 93–104. [Google Scholar]
- Avery, L.B.; Wang, M.; Kavosi, M.S.; Joyce, A.; Kurz, J.C.; Fan, Y.Y.; Dowty, M.E.; Zhang, M.; Zhang, Y.; Cheng, A.; et al. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. mAbs 2016, 8, 1064–1078. [Google Scholar] [CrossRef] [Green Version]
- Datta-Mannan, A.; Wroblewski, V.J. Application of FcRn binding assays to guide mAb development. Drug Metab. Dispos. 2014, 42, 1867–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta-Mannan, A.; Witcher, D.R.; Tang, Y.; Watkins, J.; Wroblewski, V.J. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J. Biol. Chem. 2007, 282, 1709–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta-Mannan, A.; Witcher, D.R.; Tang, Y.; Watkins, J.; Jiang, W.; Wroblewski, V.J. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: Relationship to pharmacokinetics in mice and primates. Drug Metab. Dispos. 2007, 35, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Ishii-Watabe, A.; Tada, M.; Kobayashi, T.; Kanayasu-Toyoda, T.; Kawanishi, T.; Yamaguchi, T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: A comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J. Immunol. 2010, 184, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Balthasar, J.P. Evaluation of a catenary PBPK model for predicting the in vivo disposition of mAbs engineered for high-affinity binding to FcRn. AAPS J. 2012, 14, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Maas, B.M.; Cao, Y. A minimal physiologically based pharmacokinetic model to investigate FcRn-mediated monoclonal antibody salvage: Effects of Kon, Koff, endosome trafficking, and animal species. mAbs 2018, 10, 1322–1331. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, C.R.; Trowbridge, I.S. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J. Cell. Biol. 1983, 97, 508–521. [Google Scholar] [CrossRef]
- Vaughn, D.E.; Bjorkman, P.J. High-affinity binding of the neonatal Fc receptor to its IgG ligand requires receptor immobilization. Biochemistry 1997, 36, 9374–9380. [Google Scholar] [CrossRef]
- Neuber, T.; Frese, K.; Jaehrling, J.; Jager, S.; Daubert, D.; Felderer, K.; Mechthild, L.; Höhne, A.; Kaden, S.; Kölln, J.; et al. Characterization and screening of IgG binding to the neonatal Fc receptor. mAbs 2014, 6, 928–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grevys, A.; Nilsen, J.; Sand, K.; Daba, M.B.; Øynebråten, I.; Bern, M.; McAdam, M.B.; Foss, S.; Schlothauer, T.; Michaelsen, T.E.; et al. A human endothelial cell-based recycling assay for screening of FcRn targeted molecules. Nat. Commun. 2018, 9, 621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souders, C.A.; Nelson, S.C.; Wang, Y.; Crowley, A.R.; Klempner, M.S.; Thomas, W., Jr. A novel in vitro assay to predict neonatal Fc receptor-mediated human IgG half-life. mAbs 2015, 7, 912–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.; Lin, Y.L.; Nguyen, V.; Kamen, L.; Zheng, K.; Vora, B.; Song, A. Development of a label-free FcRn-mediated transcytosis assay for in vitro characterization of FcRn interactions with therapeutic antibodies and Fc-fusion proteins. J. Immunol. Methods 2018, 462, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Nguyen, V.; Lin, Y.L.; Lafrance-Vanasse, J.; Scales, S.J.; Lin, K.; Deng, R.; Williams, K.; Sperinde, G.; Li, J.J.; et al. An in vitro FcRn- dependent transcytosis assay as a screening tool for predictive assessment of nonspecific clearance of antibody therapeutics in humans. mAbs 2019, 11, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Sager, J.E.; Yu, J.; Ragueneau-Majlessi, I.; Isoherranen, N. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab. Dispos. 2015, 43, 1823–1837. [Google Scholar] [CrossRef]
- Shah, D.K.; Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 2012, 39, 67–86. [Google Scholar] [CrossRef]
- Garg, A.; Balthasar, J.P. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J. Pharmacokinet. Pharmacodyn. 2007, 34, 687–709. [Google Scholar] [CrossRef]
- Jones, H.M.; Tolsma, J.; Zhang, Z.; Jasper, P.; Luo, H.; Weber, G.L.; Wright, K.; Bard, J.; Bell, R.; Messing, D.; et al. A Physiologically-Based Pharmacokinetic Model for the Prediction of “Half-Life Extension” and “Catch and Release” Monoclonal Antibody Pharmacokinetics. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 534–541. [Google Scholar] [CrossRef]
- Ngo, L.; Oh, J.; Kim, A.; Back, H.M.; Kang, W.H.; Chae, J.W.; Yun, H.Y.; Lee, H. Development of a Pharmacokinetic Model Describing Neonatal Fc Receptor-Mediated Recycling of HL2351, a Novel Hybrid Fc-Fused Interleukin-1 Receptor Antagonist, to Optimize Dosage Regimen. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 584–595. [Google Scholar] [CrossRef]
- Deng, R.; Balthasar, J.P. Pharmacokinetic/pharmacodynamic modeling of IVIG effects in a murine model of immune thrombocytopenia. J. Pharm. Sci. 2007, 96, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jusko, W.J. Applications of minimal physiologically-based pharmacokinetic models. J. Pharmacokinet. Pharmacodyn. 2012, 39, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Balthasar, J.P.; Jusko, W.J. Second-generation minimal physiologically-based pharmacokinetic model for monoclonal antibodies. J. Pharmacokinet. Pharmacodyn. 2013, 40, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, D.; Rode, F.; Cao, Y. A Minimal Physiologically Based Pharmacokinetic Model with a Nested Endosome Compartment for Novel Engineered Antibodies. AAPS J. 2018, 20, 48. [Google Scholar] [CrossRef]
- Abdallah, H.M.; Zhu, A.Z.X. A Minimal Physiologically-Based Pharmacokinetic Model Demonstrates Role of the Neonatal Fc Receptor (FcRn) Competition in Drug-Disease Interactions with Antibody Therapy. Clin. Pharmacol. Ther. 2020, 107, 423–434. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, T.; Cao, Y. In Translation: FcRn across the Therapeutic Spectrum. Int. J. Mol. Sci. 2021, 22, 3048. https://doi.org/10.3390/ijms22063048
Qi T, Cao Y. In Translation: FcRn across the Therapeutic Spectrum. International Journal of Molecular Sciences. 2021; 22(6):3048. https://doi.org/10.3390/ijms22063048
Chicago/Turabian StyleQi, Timothy, and Yanguang Cao. 2021. "In Translation: FcRn across the Therapeutic Spectrum" International Journal of Molecular Sciences 22, no. 6: 3048. https://doi.org/10.3390/ijms22063048
APA StyleQi, T., & Cao, Y. (2021). In Translation: FcRn across the Therapeutic Spectrum. International Journal of Molecular Sciences, 22(6), 3048. https://doi.org/10.3390/ijms22063048