
Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl

 , ,
, ,  and
and 
Abstract
:1. Introduction
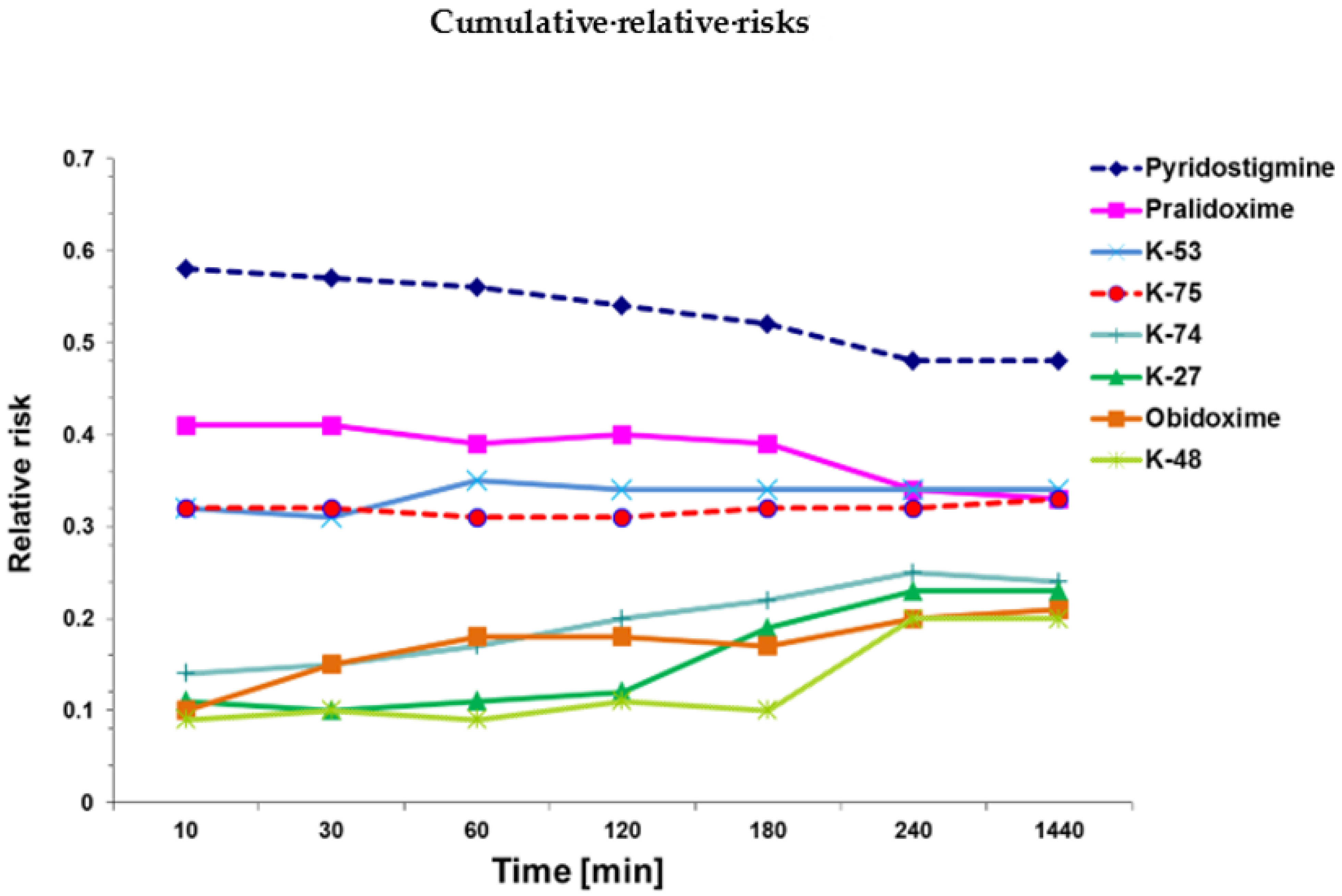
2. Results
2.1. Mortality Rates
2.2. Cox Survival Analysis
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Experimental Animals
4.2.1. Choice of Dosage for Pretreatment
- Reference group: only azinphos-methyl exposure.
- Pyridostigmine: 1 µmol/rat = 0.26 mg/rat (= 1.0 mg/kg average body weight).
- Pralidoxime: 30 µmol/rat = 5.2 mg/rat (= 20 mg/kg average body weight).
- Obidoxime: 25 µmol/rat = 9.0 mg/rat (=35 mg/kg average body weight).
- K-27: 60 µmol/rat = 26.8 mg/rat (=103 mg/kg average body weight).
- K-48: 25 µmol/rat = 11.5 mg/rat (=44 mg/kg average body weight).
- K-53: 3 µmol/rat = 1.37 mg/rat (=5.3 mg/kg average body weight).
- K-74: 3 µmol/rat = 1.38 mg/rat (=5.3 mg/kg average body weight).
- K-75: 3 µmol/rat = 1.37 mg/rat (=5.3 mg/kg average body weight).
4.2.2. Pretreatment and Azinphos-Methyl Exposure

{kind=link}
| Molecular Weight | Injected Dose (µmol/rat) | Injected Dose (mg/rat) | Injected Dose (mg/kg Average Body Weight) | IC50 Human (µM] | IC50 Rat (µM) | LD50/LD01 (µmol/rat) | |
|---|---|---|---|---|---|---|---|
| Azinphos-methyl | 317.3 | 5, 10, 15 | 1.59, 3.18, 4.77 | 6.14, 12.28, 18.42 | 189 * | NA | 3.2/0.4 * |
| Pyridostigmine | 172.60 | 30 | 0.26 | 1.0 | 0.33 | NA | 7.2/3.7 |
| Pralidoxime | 172.60 | 30 | 5.2 | 20 | 592 | 412 | 180/117 |
| Obidoxime | 359.21 | 25 | 9.0 | 35 | 702 | 193 | 132/107 |
| K-27 | 446.16 | 60 | 26.8 | 103 | 414 | 1054 | 350/250 |
| K-45 | 460.16 | 25 | 11.5 | 44 | 461 | 643 | 140/110 |
| K-53 | 458.15 | 3 | 1.37 | 5.3 | 115 | 83 | 21/13 |
| K-74 | 460.16 | 3 | 1.38 | 5.3 | 103 | 66 | 28/13 |
| K-75 | 458.15 | 3 | 1.37 | 5.3 | 63 | 101 | 51/13 |
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Casida, J.E.; Quistad, G.B. Golden Age of Insecticide Research: Past, Present, or Future? Annu. Rev. Èntomol. 1998, 43, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.A.; Watson, A. Organophosphate nerve agents. In Handbook of Toxicology of Chemical Warfare Agents, 3rd ed.; Gupta, R.C., Ed.; Academic Press: Boston, MA, USA, 2020; Chapter 8; pp. 97–126. [Google Scholar] [CrossRef]
- Dworkin, J.; Prescott, M.; Jamal, R.; Hardawan, S.A.; Abdullah, A.; Galea, S. The Long-Term Psychosocial Impact of a Surprise Chemical Weapons Attack on Civilians in Halabja, Iraqi Kurdistan. J. Nerv. Ment. Dis. 2008, 196, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Macilwain, C. Study proves Iraq used nerve gas. Nature 1993, 363, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, B. The toxicology and treatment of injuries from chemical warfare agents. Curr. Anaesth. Crit. Care 2003, 14, 149–154. [Google Scholar] [CrossRef]
- Balali-Mood, M.; Balali-Mood, K. Neurotoxic disorders of organophosphorus compounds and their managements. Arch. Iran. Med. 2008, 11, 65–89. [Google Scholar]
- Delfino, R.T.; Ribeiro, T.S.; Figueroa-Villar, J.D. Organophosphorus compounds as chemical warfare agents: A review. J. Braz. Chem. Soc. 2009, 20, 407–428. [Google Scholar] [CrossRef]
- Yanagisawa, N.; Morita, H.; Nakajima, T. Sarin experiences in Japan: Acute toxicity and long-term effects. J. Neurol. Sci. 2006, 249, 76–85. [Google Scholar] [CrossRef]
- Kostadinov, R.; Kanev, K.; Dimov, D. Chemical Terrorism, History and Threat Assessment. Med. Manag. Chem. Biol. Casualties 2010, 8, 77–84. [Google Scholar]
- Brooks, J.; Erickson, T.B.; Kayden, S.; Ruiz, R.; Wilkinson, S.; Burklejr, F.M., Jr. Responding to chemical weapons violations in Syria: Legal, health, and humanitarian recommendations. Confl. Health 2018, 12, 12. [Google Scholar] [CrossRef] [Green Version]
- Chowdhary, S.; Bhattacharyya, R.; Banerjee, D. Acute organophosphorus poisoning. Clin. Chim. Acta 2014, 431, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Petroianu, G.; Toomes, L.M.; Petroianu, A.; Bergler, W.; Rüfer, R. Control of blood pressure, heart rate and haematocrit during high-dose intravenous paraoxon exposure in mini pigs. J. Appl. Toxicol. 1998, 18, 293–298. [Google Scholar] [CrossRef]
- Eddleston, M.; Buckley, N.A.; Eyer, P.; Dawson, A.H. Management of acute organophosphorus pesticide poisoning. Lancet 2008, 371, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Masson, P.; Nachon, F. Cholinesterase reactivators and bioscavengers for pre- and post-exposure treatments of organophosphorus poisoning. J. Neurochem. 2017, 142 (Suppl. 2), 26–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorke, D.E.; Petroianu, G.A. Reversible cholinesterase inhibitors as pretreatment for exposure to organophosphates. A review. J. Appl. Toxicol. 2019, 39, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Petroianu, G.A.; Nurulain, S.M.; Hasan, M.Y.; Kuca, K.; Lorke, D.E. Reversible cholinesterase inhibitors as pre-treatment for exposure to organophosphates: Assessment using azinphos-methyl. J. Appl. Toxicol. 2015, 35, 493–499. [Google Scholar] [CrossRef]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuca, K.; Petroianu, G.A. Optimal Pre-treatment for Acute Exposure to the Organophosphate Dicrotophos. Curr. Pharm. Des. 2017, 23, 3432–3439. [Google Scholar] [CrossRef]
- Lorke, D.E.; Hasan, M.Y.; Nurulain, S.M.; Shafiullah, M.; Kuca, K.; Petroianu, G.A. Pretreatment for acute exposure to diisopropylfluorophosphate: In vivo efficacy of various acetylcholinesterase inhibitors. J. Appl. Toxicol. 2011, 31, 515–523. [Google Scholar] [CrossRef]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuča, K.; Petroianu, G.A. Combined Pre- and Posttreatment of Paraoxon Exposure. Molecules 2020, 25, 1521. [Google Scholar] [CrossRef] [Green Version]
- Petroianu, G.A.; Nurulain, S.M.; Shafiullah, M.; Hasan, M.Y.; Kuča, K.; Lorke, D.E. Usefulness of administration of non-organophosphate cholinesterase inhibitors before acute exposure to organophosphates: Assessment using paraoxon. J. Appl. Toxicol. 2013, 33, 894–900. [Google Scholar] [CrossRef]
- Lorke, D.E.; Hasan, M.Y.; Nurulain, S.M.; Shafiullah, M.; Kuča, K.; Petroianu, G.A. Acetylcholinesterase inhibitors as pretreatment before acute exposure to organophosphates: Assessment using methyl-paraoxon. CNS Neurol. Disord. Drug Targets 2012, 11, 1052–1060. [Google Scholar] [CrossRef]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuca, K.; Petroianu, G.A. Prophylactic administration of non-organophosphate cholinesterase inhibitors before acute exposure to organophosphates: Assessment using terbufos sulfone. J. Appl. Toxicol. 2014, 34, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration FDA. Approves Pyridostigmine Bromide as Pretreatment against Nerve Gas. Available online: http://www.fda.gov/Drugs/EmergencyPreparedness/BioterrorismandDrugPreparedness/ucm130342.htm (accessed on 15 February 2021).
- Lorke, D.E.; Hasan, M.Y.; Nurulain, S.M.; Sheen, R.; Kuča, K.; Petroianu, G.A. Entry of two new asymmetric bispyridinium oximes (K-27 and K-48) into the rat brain: Comparison with obidoxime. J. Appl. Toxicol. 2007, 27, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Kalasz, H.; Petroianu, G.A.; Tekes, K. Entry of Oximes into the Brain: A Review. Curr. Med. Chem. 2008, 15, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Petroianu, G.A. The Experimental Oxime K027—A Promising Protector from Organophosphate Pesticide Poisoning. A Review Comparing K027, K048, Pralidoxime, and Obidoxime. Front. Neurosci. 2019, 13, 427. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Cabal, J. In vitro reactivation of tabun-inhibited acetylcholinesterase using new oximes—K027, K005, K033 and K048. Central Eur. J. Public Health 2004, 12, S59–S61. [Google Scholar]
- Kuca, K.; Cabal, J.; Kassa, J. A Comparison of the Potency of Newly Developed Oximes (K005, K027, K033, K048) and Currently Used Oximes (Pralidoxime, Obidoxime, HI-6) to Reactivate Sarin-Inhibited Rat Brain Acetylcholinesterase by In Vitro Methods. J. Toxicol. Environ. Health Part A 2005, 68, 677–686. [Google Scholar] [CrossRef]
- Kuca, K.; Kassa, J. In vitro reactivation of acetylcholinesterase using the oxime K027. Vet. Hum. Toxicol. 2004, 46, 15–18. [Google Scholar]
- Kuca, K.; Musilek, K.; Jun, D.; Pohanka, M.; Ghosh, K.K.; Hrabinova, M. Oxime K027: Novel low-toxic candidate for the universal reactivator of nerve agent- and pesticide-inhibited acetylcholinesterase. J. Enzym. Inhib. Med. Chem. 2010, 25, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Musilek, K.; Petroianu, G.A. Eight new bispyridinium oximes in comparison with the conventional oximes pralidoxime and obidoxime:in vivoefficacy to protect from diisopropylfluorophosphate toxicity. J. Appl. Toxicol. 2008, 28, 920–928. [Google Scholar] [CrossRef]
- Petroianu, G.A.; Lorke, D.E.; Kalász, H. Comparison of the Ability of Pyridinium Aldoximes to Reactivate Human Red Blood Cell Acetylcholinesterases Inhibited by ethyl- and methyl-paraoxon. Curr. Org. Chem. 2012, 16, 1359–1369. [Google Scholar] [CrossRef]
- Petroianu, G.A.; Lorke, D.E. Pyridinium oxime reactivators of cholinesterase inhibited by diisopropyl-fluorophosphate (DFP): Predictive value of in-vitro testing for in-vivo efficacy. Mini Rev. Med. Chem. 2008, 8, 1328–1342. [Google Scholar] [CrossRef]
- Kayouka, M.; Houzé, P.; Lejay, M.; Baud, F.J.; Kuca, K. Safety and Efficacy of New Oximes to Reverse Low Dose Diethyl-Paraoxon-Induced Ventilatory Effects in Rats. Molecules 2020, 25, 3056. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Petroianu, G.A. Treatment of Organophosphate Poisoning with Experimental Oximes: A Review. Curr. Org. Chem. 2019, 23, 628–639. [Google Scholar] [CrossRef]
- Kassa, J.; Kuca, K.; Cabal, J.; Paar, M. A Comparison of the Efficacy of New Asymmetric Bispyridinium Oximes (K027, K048) with Currently Available Oximes against Tabun by In Vivo Methods. J. Toxicol. Environ. Health Part A 2006, 69, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Bielavsky, J.; Cabal, J.; Bielavska, M. Synthesis of a potential reactivator of acetylcholinesterase—1-(4-hydroxyiminomethylpyridinium)-3-(carbamoylpyridinium)propane dibromide. Tetrahedron Lett. 2003, 44, 3123–3125. [Google Scholar] [CrossRef]
- Kuca, K.; Musilova, L.; Paleček, J.; Církva, V.; Paar, M.; Musilek, K.; Hrabinová, M.; Pohanka, M.; Karasová, J.Ž.; Jun, D. Novel Bisquaternary Oximes—Reactivation of Acetylcholinesterase and Butyrylcholinesterase Inhibited by Paraoxon. Molecules 2009, 14, 4915–4921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musilek, K.; Kuca, K.; Dohnal, V.; Jun, D.; Marek, J.; Koleckar, V. Two Step Synthesis of a Non-symmetric Acetylcholinesterase Reactivator. Molecules 2007, 12, 1755–1761. [Google Scholar] [CrossRef] [Green Version]
- Lorke, D.E.; Hasan, M.Y.; Arafat, K.; Kuča, K.; Musilek, K.; Schmitt, A.; Petroianu, G.A. In vitro oxime protection of human red blood cell acetylcholinesterase inhibited by diisopropyl-fluorophosphate. J. Appl. Toxicol. 2008, 28, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuča, K.; Petroianu, G.A. Oximes as pretreatment before acute exposure to paraoxon. J. Appl. Toxicol. 2019, 39, 1506–1515. [Google Scholar] [CrossRef]
- Cox, D.R. Regression models and life tables. J. R. Stat. Soc. B 1972, 34, 189–220. [Google Scholar] [CrossRef]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuca, K.; Petroianu, G.A. Five Experimental Bispyridinium Oximes in Comparison with the Conventional Oximes Pralidoxime and Obidoxime: In Vivo Efficacy to Protect from Azinphos-methyl-induced Toxicity. J. Environ. Immunol. Toxicol. 2013, 1, 44. [Google Scholar] [CrossRef]
- Lewis, C.M. Azinphos-Methyl (Guthion) Risk Characterization Document (Revision No. 1). Available online: http://www.cdpr.ca.gov/docs/risk/rcd/azmrcdre.pdf (accessed on 4 March 2021).
- Belenguer, V.; Martinez-Capel, F.; Masiá, A.; Picó, Y. Patterns of presence and concentration of pesticides in fish and waters of the Júcar River (Eastern Spain). J. Hazard. Mater. 2014, 265, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Schulz, R. Field Studies on Exposure, Effects, and Risk Mitigation of Aquatic Nonpoint-Source Insecticide Pollution: A Review. J. Environ. Qual. 2004, 33, 419–448. [Google Scholar] [CrossRef]
- Stoner, K.A.; Eitzer, B.D. Using a Hazard Quotient to Evaluate Pesticide Residues Detected in Pollen Trapped from Honey Bees (Apis mellifera) in Connecticut. PLoS ONE 2013, 8, e77550. [Google Scholar] [CrossRef]
- Buratti, F.M.; Volpe, M.T.; Fabrizi, L.; Meneguz, A.; Vittozzi, L.; Testai, E. Kinetic parameters of OPT pesticide desulfuration by c-DNA expressed human CYPs. Environ. Toxicol. Pharmacol. 2002, 11, 181–190. [Google Scholar] [CrossRef]
- Dubois, K.P.; Thursh, D.R.; Murphy, S.D. Studies on the toxicity and pharmacologic actions of the dimethoxy ester of benzotriazine dithiophosphoric acid (DBD, guthion). J. Pharmacol. Exp. Ther. 1957, 119, 208–218. [Google Scholar] [PubMed]
- Pasquet, J.; Mazuret, A.; Fournel, J.; Koenig, F.H. Acute oral and percutaneous toxicity of phosalone in the rat, in comparison with azinphosmethyl and parathion. Toxicol. Appl. Pharmacol. 1976, 37, 85–92. [Google Scholar] [CrossRef]
- Vrdoljak, A.L.; Čalić, M.; Radić, B.; Berend, S.; Jun, D.; Kuča, K.; Kovarik, Z. Pretreatment with pyridinium oximes improves antidotal therapy against tabun poisoning. Toxicology 2006, 228, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Koelle, G.B. Protection of cholinesterase against irreversible inactivation by di-isopropyl fluorophosphate in vitro. J. Pharmacol. Exp. Ther. 1946, 88, 232–237. [Google Scholar] [PubMed]
- Koster, R. Synergisms and antagonisms between physostigmine and di-isopropyl fluorophosphate in cats. J. Pharmacol. Exp. Ther. 1946, 88, 39–46. [Google Scholar]
- Lenina, O.A.; Zueva, I.V.; Zobov, V.V.; Semenov, V.E.; Masson, P.; Petrov, K.A. Slow-binding reversible inhibitor of acetylcholinesterase with long-lasting action for prophylaxis of organophosphate poisoning. Sci. Rep. 2020, 10, 16611. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, D.; Friedman, A.; Seidman, S.; Soreq, H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 1998, 393, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Meshorer, E.; Erb, C.; Gazit, R.; Pavlovsky, L.; Kaufer, D.; Friedman, A.; Glick, D.; Ben-Arie, N.; Soreq, H. Alternative Splicing and Neuritic mRNA Translocation Under Long-Term Neuronal Hypersensitivity. Science 2002, 295, 508–512. [Google Scholar] [CrossRef] [Green Version]
- Soreq, H.; Seidman, S. Acetylcholinesterase—New roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef]
- Prado, A.; Petroianu, G.A.; Lorke, D.E.; Chambers, J.W. A trivalent approach for determining in vitro toxicology: Examination of oxime K027. J. Appl. Toxicol. 2015, 35, 219–227. [Google Scholar] [CrossRef]
- Jun, D.; Kuca, K.; Stodülka, P.; Koleckar, V.; Doležal, B.; Simon, P.; Veverka, M. HPLC Analysis of HI-6 Dichloride and Dimethanesulfonate—Antidotes against Nerve Agents and Organophosphorus Pesticides. Anal. Lett. 2007, 40, 2783–2787. [Google Scholar] [CrossRef]
- Jun, D.; Stodulka, P.; Kuca, K.; Koleckar, V.; Dolezal, B.; Simon, P.; Veverka, M. TLC analysis of intermediates arising during the preparation of oxime HI-6 dimethanesulfonate. J. Chromatogr. Sci. 2008, 46, 316–319. [Google Scholar] [CrossRef] [Green Version]
- Lorke, D.E.; Petroianu, G.A. Minireview: Does in-vitro testing of oximes help predict their in-vivo action after paraoxon exposure? J. Appl. Toxicol. 2009, 29, 459–469. [Google Scholar] [CrossRef] [PubMed]

| Substance | Structure |
|---|---|
| Azinphos-methyl |  |
| Pyridostigmine |  |
| Pralidoxime |  |
| Obidoxime |  |
| K-27 |  |
| K-48 |  |
| K-53 |  |
| K-74 |  |
| K-75 |  |
| Groups (G) | 10 min | 30 min | 1 h | 2 h | 3 h | 4 h | 24 h | 48 h |
|---|---|---|---|---|---|---|---|---|
| G1: Azinphos-methyl only | 71/92/92 | 71/92/96 | 71/92/100 | 71/92/100 | 71/92/100 | 71/92/100 | 75/92/100 | 75/92/100 |
| G2: Pyridostigmine pretreatment | 29/50/71 | 42/54/75 | 42/54/79 | 42/58/83 | 42/58/88 | 42/58/88 | 46/58/88 | 46/58/88 |
| G3: Pralidoxime pretreatment | 21/33/54 | 21/50/58 | 21/50/67 | 25/50/71 | 25/63/71 | 25/63/71 | 25/63/71 | 25/63/71 |
| G4: Obidoxime pretreatment | 4/8/21 | 4/8/21 | 4/8/42 | 4/29/42 | 4/29/54 | 4/29/54 | 17/46/63 | 17/46/67 |
| G5: K-27 pretreatment | 21/0/4 | 21/4/13 | 21/4/13 | 21/4/25 | 29/4/29 | 29/42/29 | 50/46/46 | 50/50/46 |
| G6: K-48 pretreatment | 0/8/8 | 8/13/13 | 8/13/21 | 8/13/21 | 8/13/38 | 8/13/38 | 25/50/58 | 25/54/58 |
| G7: K-53 pretreatment | 13/33/29 | 29/33/42 | 29/33/50 | 29/46/63 | 29/50/63 | 29/50/63 | 38/54/71 | 38/54/71 |
| G8: K-74 pretreatment | 0/8/17 | 17/8/25 | 17/8/38 | 17/13/46 | 17/33/50 | 17/50/50 | 33/58/54 | 33/58/54 |
| G9: K-75 pretreatment | 4/17/33 | 33/21/50 | 38/21/58 | 38/25/58 | 38/38/58 | 42/42/58 | 50/50/63 | 50/58/67 |
| Groups | Relative Risk (RR) | 95% CI | p-Value |
|---|---|---|---|
| Azinphos-methyl only | 1 | reference | reference |
| Pyridostigmine + azinphos | 0.52 ± 0.10 | 0.36–0.68 | ≤0. 01 |
| Pralidoxime + azinphos | 0.37 ± 0.03 | 0.33–0.41 | ≤0. 01 a |
| Obidoxime+ azinphos | 0.21 ± 0.09 | 0.06–0.36 | ≤0. 01 a, b |
| K-27 + azinphos | 0.23 ± 0.02 | 0.20–0.25 | ≤0.01 a, b, c |
| K-48 + azinphos | 0.20 ± 0.03 | 15–0.24 | ≤0.01 a, b, c |
| K-53 + azinphos | 0.37 ± 0.14 | 0.14–0.59 | ≤0.01 |
| K-74 + azinphos | 0.26 ± 0.10 | 0.09–0.42 | ≤0.01 a |
| K-75 + azinphos | 0.35 ± 0.03 | 0.30–0.39 | ≤0.01 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuča, K.; Petroianu, G.A. Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl. Int. J. Mol. Sci. 2021, 22, 3072. https://doi.org/10.3390/ijms22063072
Lorke DE, Nurulain SM, Hasan MY, Kuča K, Petroianu GA. Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl. International Journal of Molecular Sciences. 2021; 22(6):3072. https://doi.org/10.3390/ijms22063072
Chicago/Turabian StyleLorke, Dietrich E., Syed M. Nurulain, Mohamed Y. Hasan, Kamil Kuča, and Georg A. Petroianu. 2021. "Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl" International Journal of Molecular Sciences 22, no. 6: 3072. https://doi.org/10.3390/ijms22063072
APA StyleLorke, D. E., Nurulain, S. M., Hasan, M. Y., Kuča, K., & Petroianu, G. A. (2021). Experimental and Established Oximes as Pretreatment before Acute Exposure to Azinphos-Methyl. International Journal of Molecular Sciences, 22(6), 3072. https://doi.org/10.3390/ijms22063072