1. Introduction
Basic elemental signaling molecules offer significant potential as novel therapeutics. The most frequently studied is hydrogen sulfide that, alongside carbon monoxide and nitric oxide, constitutes a small group of gaseous molecules, the ‘gasotransmitters’ [
1]. The ability of sulfide gas (H
2S) inhalation to induce a ‘suspended animation-like state’ in mice [
2] generated substantial interest in the use of sulfide-liberating agents to modulate oxidative pathologies by reduction of global- or organ-specific metabolism [
3]. Initially led by basic sulfur salts and, subsequently, by more complex organic and inorganic molecules, this approach has been a major research focus for more than a decade [
4].
Such metabolic modulators can confer protection against reperfusion injury, a consequence of the necessary urgent revascularization of ischemic organs. While underlying pathophysiological mechanisms have yet to be definitively determined, a strong evidence base indicates that rapid restoration of oxygen and substrate to the ischemic tissue drives mitochondrial respiration to generate large quantities of damaging reactive oxygen species (ROS) [
5]. This overproduction may be ameliorated with a short-term inhibitor of oxidative phosphorylation while maintaining a level of metabolism that supports cell viability and functionality [
6]. This approach extends beyond traditional gasotransmitters, with gases and basic salts of both non-sulfur chalcogens (selenides) [
7] and halogens (iodides, bromides) [
8] impacting on metabolism and/or exhibiting pre-clinical efficacy in treating oxidative pathologies.
Owing to their proximity in the periodic table, sulfur and selenium share several chemical and biological characteristics. We [
9] and others [
10] have postulated that the physiological derivative of selenium, hydrogen selenide (as gaseous H
2Se and anionic HSe
−), could be an additional member of the gasotransmitter class. Less contentiously, selenium is considered an essential micronutrient [
11] that supports diverse physiological roles, most notably as a structural and functional component of antioxidant ‘selenoproteins’ [
12]. Thus, selenium and its reduced forms could offer multiple antioxidant mechanisms; as an elemental reducing agent, a modulator of metabolism, and as the catalytic component of specialized enzymes. Here we assessed the impact of sodium hydroselenide (NaHSe) on metabolism and explored a mitochondrial mechanism of action. We also gained novel insights on the pharmacological manipulation of selenoprotein expression, and herein discuss the utility of NaHSe as a research tool and prospective therapeutic.
3. Discussion
Elemental signaling molecules such as hydrogen sulfide, selenides [
7], iodides and bromides [
8] are being investigated as the bioactive component of novel therapeutics. One indication is reperfusion injury, where a transient reduction of mitochondrial respiration could temper the initial burst in mitochondrial activity (and pathologically excessive ROS production) occurring on abrupt restoration of necessary oxygen and substrate provision during revascularization [
15].
Gasotransmitters are endogenously generated and act across diverse physiological systems, including modulation of oxidative metabolism [
1]. When delivered exogenously, and usually in excess of their normal physiological range(s), aerobic respiration is decreased by either competitive or non-competitive inhibition of mitochondrial cytochrome C oxidase [
16]. The most compelling demonstration was the ability of inhaled H
2S to induce a ‘suspended animation-like state’ in mice [
2]. The same group later showed that inhalation of gaseous selenides and iodides could induce similar effects [
7,
8].
Mindful of the chemical and biological similarities between sulfur and its chalcogen homologue, selenium [
17], we [
9] and others [
10] postulated that its reduced form, hydrogen selenide, could be an additional member of the gasotransmitter class. To explore this hypothesis, we began experimental work using the basic selenide salt, NaHSe. Our approach has been analogous to early studies on delivery of exogenous sulfide, where the basic salts NaHS and Na
2S became important research tools and the basis of novel (albeit unsuccessful) therapies [
18]. With necessary safety precautions and handling, synthesis of NaHSe is straightforward and inexpensive, and can be used in a range of chemical or biological matrices. It should be noted that reduced selenides are particularly prone to oxidation [
17], hence, their handling should always be carried out under anoxic conditions, and all diluents/adjuvants should be purified to remove trace metals that can act as oxidants.
We extended the observation that H
2Se gas modulates metabolism in mice [
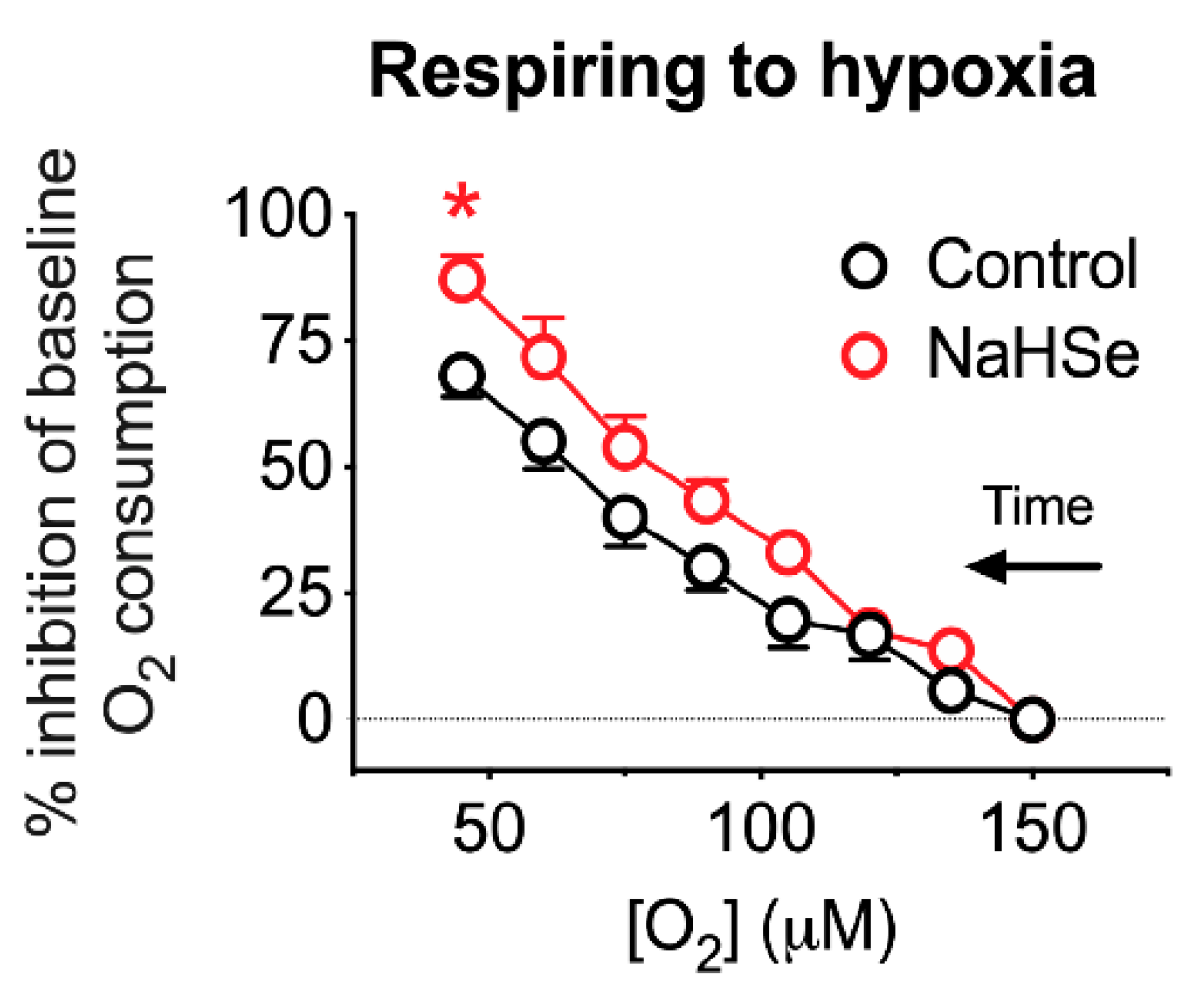
7], showing dose-dependent reduction of oxygen consumption with NaHSe in two tissue types (skeletal muscle and liver) taken from rats. The known inhibition of mitochondrial complex IV by similar molecules suggested that this would be a strong candidate as the underlying mechanism and, indeed, this inhibition was demonstrated with NaHSe. Of note, inhibition of oxidative phosphorylation observed with selenide differed from sulfide and cyanide with regard to potency and duration of action. Significantly more selenide was required to inhibit oxygen consumption ex vivo, and its effects on cytochrome c oxidase activity were, perhaps desirably, more transient. With respect to its biological chemistry, selenium is more readily oxidized and more kinetically labile than sulfur [
17]; this is likely to explain its transient nature. The greater effectiveness observed during hypoxia, where less selenide is deactivated by oxidation, is supportive of this notion.
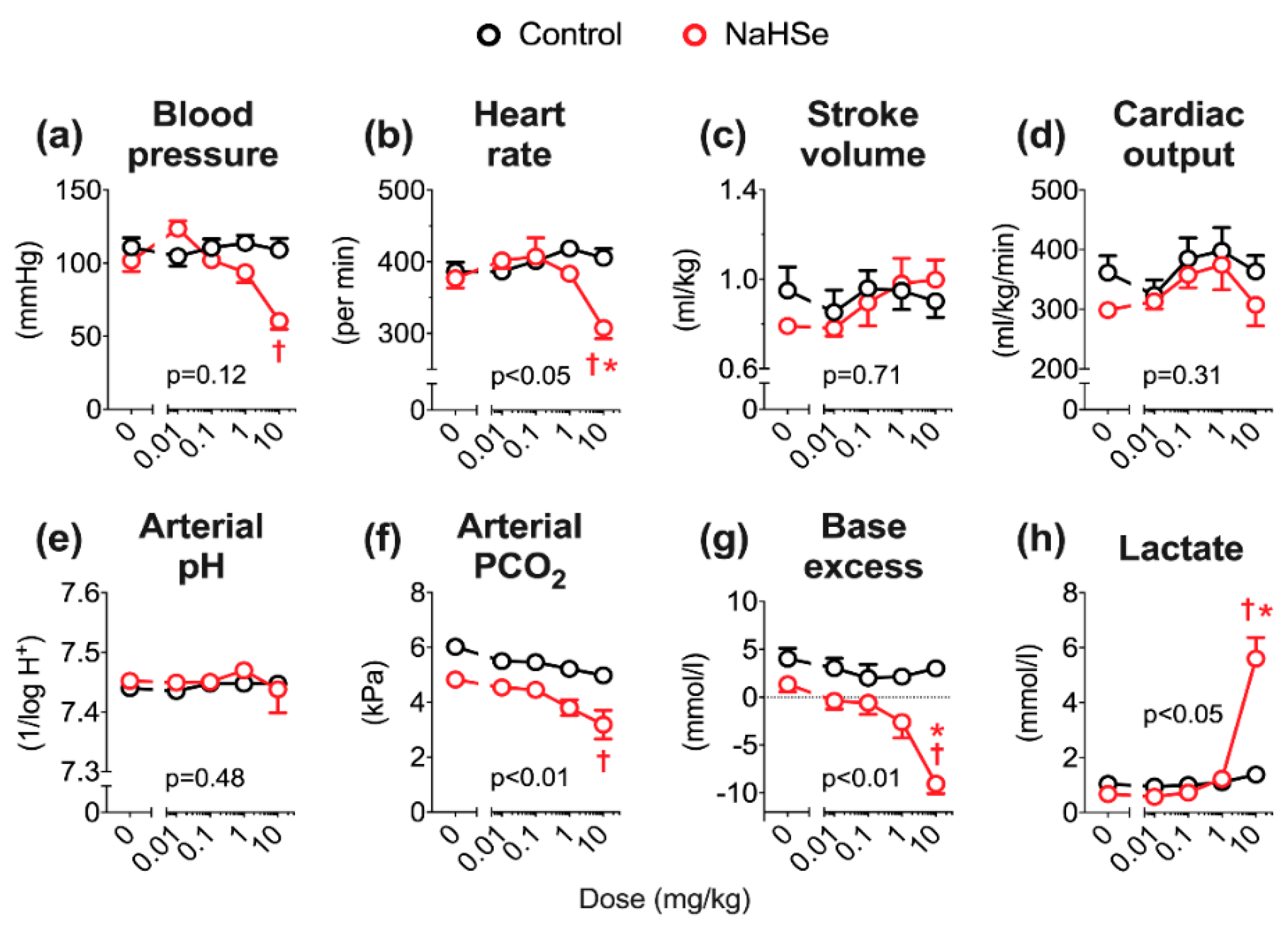
In vivo, we previously reported that administration of NaHS is dose-limiting, with a maximum tolerated (intravenous bolus) dose (MTD) of 1 mg/kg [
15]. Using the same anesthetized rat model, the MTD of NaHSe was 10 mg/kg; this 10-fold difference is broadly in line with our ex vivo findings. NaHSe could replicate some of the hemodynamic effects of basic sulfur salts, most notably the impact on blood pressure and heart rate. Aside from an initial rise in blood pressure (that could reflect interaction with other vasoactive molecules), NaHSe caused dose-dependent hypotension. Sulfide-induced vasodilation is well recognized, with diverse potential mechanisms including interaction with ion channels, smooth muscle ATP depletion, intracellular acidification, release of lipid mediators and modulation of calcium signaling [
19]. While an abrupt decrease in systemic arterial pressure would be expected to induce a baroreceptor-mediated tachycardia [
20], our findings here (notably in normothermic rats), and in reports using basic sulfur salts, suggest the opposite [
20,
21,
22]. Sulfide-induced bradycardia occurs despite no changes in QT-interval, vagal tone, or enablement by ion channels [
20,
22]. The precise molecular mode(s) of action underpinning the hemodynamic effects of selenide, and comparability to sulfide, require further investigation.
Contrary to its hemodynamic effects, the impact of selenide on acid-base balance is more readily explained. No impact was seen on arterial pH, as in these anesthetized but spontaneously breathing animals, a respiratory alkalosis compensated for the metabolic acidosis. We previously observed metabolic acidosis in mechanically ventilated rats receiving a slow-release sulfide donor, and postulated an in vivo mechanism of action at the level of the mitochondrion [
15]. When oxidative metabolism is unsustainable, either due to decreased delivery and/or utilization of O
2, cells increase ATP hydrolysis and their dependency on glycolysis. The observed rise in lactate levels with NaHSe treatment supports a greater reliance on (aerobic) glycolysis following partial inhibition of oxidative phosphorylation, with diversion of pyruvate towards lactate.
Inclusion of any prospective candidate into the gasotransmitter class requires satisfaction of five criteria [
1]. Hydrogen selenide can fulfil the first three criteria, being present as a small molecule of gas [
17], freely permeable to cell membranes [
23], and endogenously generated under regulatory (enzymatic) conditions [
9]. The currently accepted function of hydrogen selenide at physiologically relevant concentrations relates entirely to its incorporation into selenoproteins, and there are no recognized cellular effects that operate via second messengers. To date, there are 25 known human selenoproteins, most of which operate as oxidoreductase enzymes [
24]. These include families of glutathione peroxidases, thioredoxin reductases and deiodinases. Their dysfunction and/or dysregulation is implicated in diverse pathologies including diabetes, Alzheimer’s disease, cardiovascular diseases and cancer [
24,
25]. Incorporation of selenium into selenoproteins requires a uniquely adapted translational machinery that utilizes hydrogen selenide derived either from Se-oxidation products, or enzymatically from selenocysteine [
9].
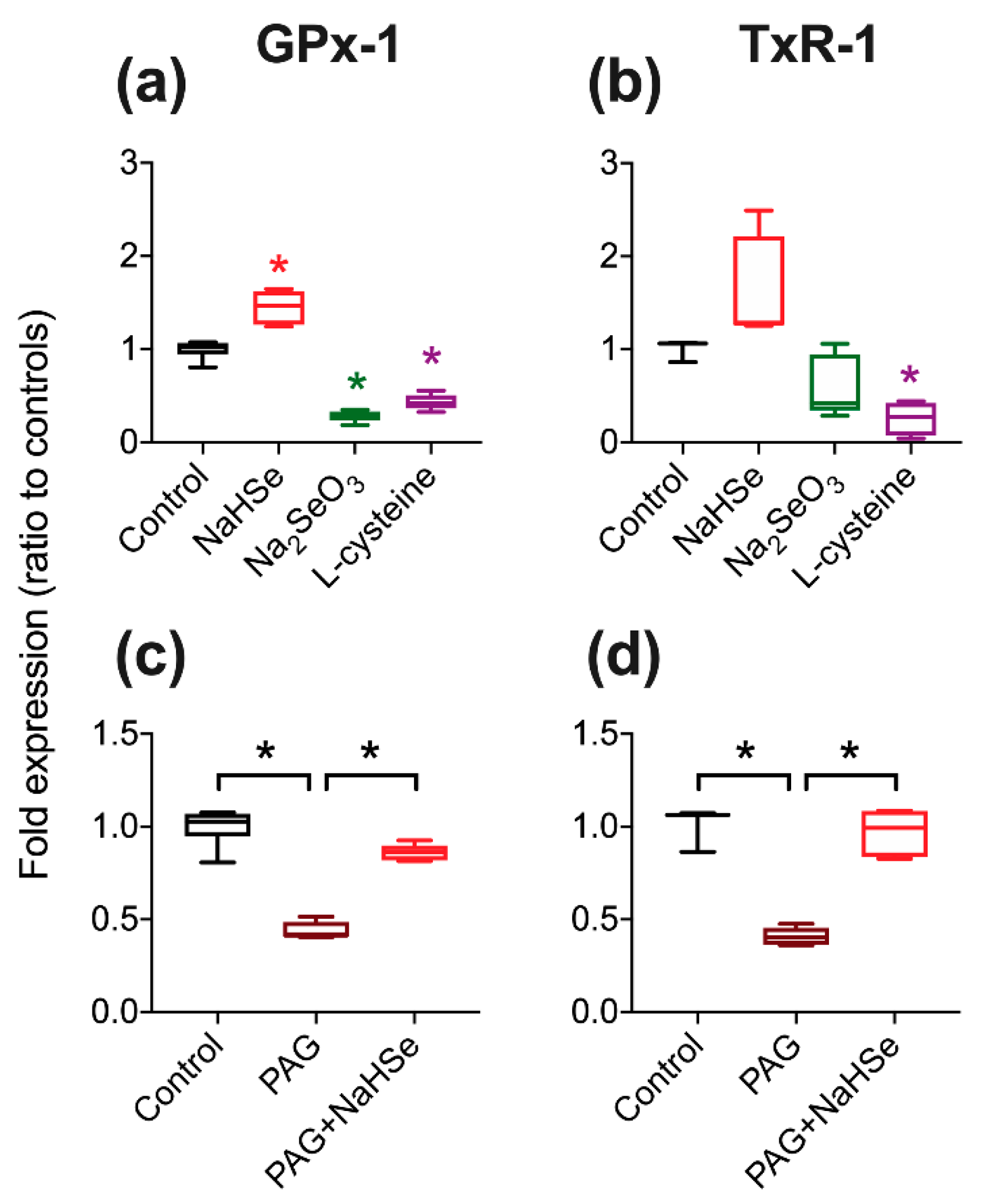
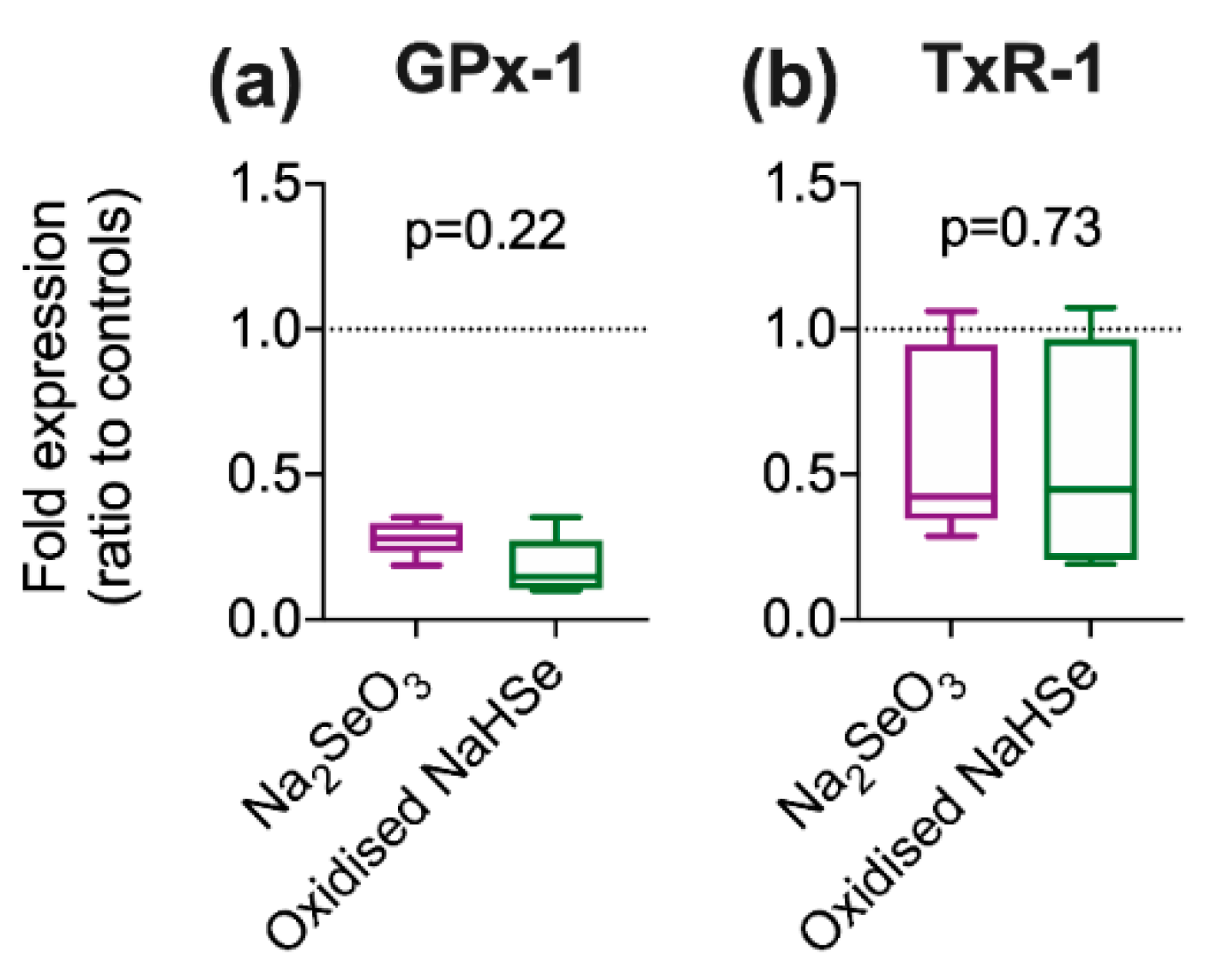
Using an in vitro model, the expected increase in selenoprotein expression was noted on exposure to hydrogen selenide. We were however surprised to observe a significant decrease in selenoprotein expression upon incubation with the oxidized Se-product, sodium selenite. The impact of oxidation status of basic elemental salts has been previously noted whereby reperfusion injury (following myocardial ischemia) in mice could be moderated by selenide and iodide (reduced forms), but not by the oxidation product(s) selenite and iodate [
7,
8]. The oxidation status of Se supplements could have implications for current clinical practice but this has been overlooked [
9]. A recent meta-analysis examined 19 clinical trials where intravenous sodium selenite (the oxidized form) was used in acute or critically ill patients [
26]; although a modest reduction in overall mortality was recorded, numerous secondary endpoints, including 28-day all-cause mortality and length of stay in intensive care, showed no effect. The oxidized product was selected for use in these studies as it shows lower toxicity, far greater stability and, when used in (healthy) animals, can increase expression of selenoproteins [
27]. However, conversion to bioactive hydrogen selenide requires reduction by endogenous antioxidant proteins such as glutathione and thioredoxin. This may not be achievable in patients in whom antioxidant defenses are already strained.
The synthesis and metabolism of (hydrogen) sulfide and selenide show several similarities. Hydrogen sulfide is synthesized by three enzymes; CSE, cystathionine b-synthase (CBS), and 3-mercaptopyruvate sulfur-transferase (3MST), reviewed in detail elsewhere [
28]. CSE catalyzes production of hydrogen sulfide from L-cysteine using pyridoxal 5′-phosphate (PLP) as a cofactor. The analogous generation of hydrogen selenide, catalyzed by SCLY from selenocysteine, is also PLP-dependent [
29]. PAG, commonly used as a (broadly specific) CSE inhibitor, is reported to act by targeting the PLP binding site of the enzyme [
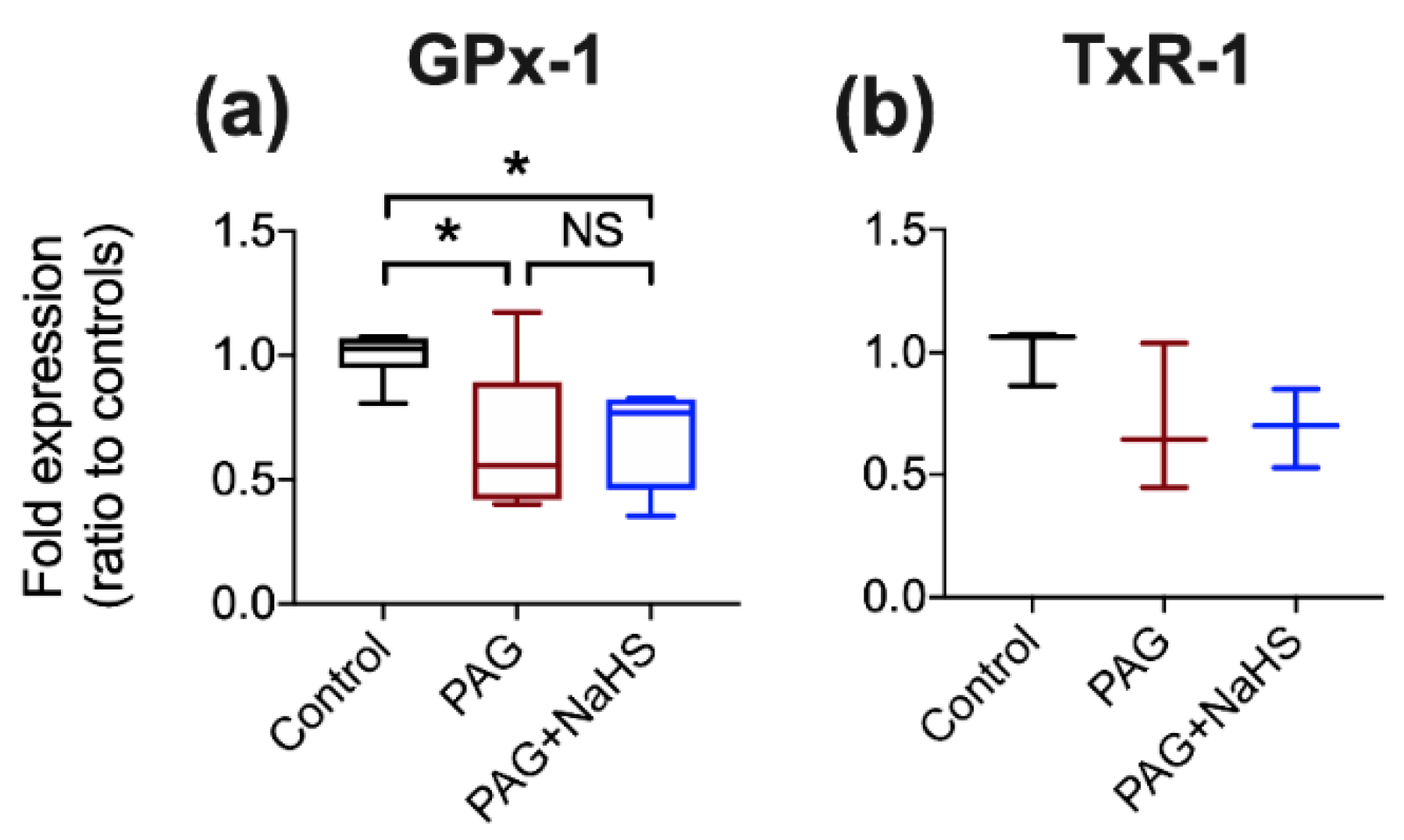
30]. Consequently, we demonstrated that PAG could inhibit selenoprotein synthesis, potentially by PAG/PLP disruption of SCLY. Selenoprotein expression could be rescued by exogenous addition of selenide as this provides substrate for selenoprotein synthesis downstream of SCLY inhibition. However, the concentration of PAG used would also inhibit CSE and decrease endogenous hydrogen sulfide production. This is relevant as (supra)physiological concentrations of sulfide impact on selenoprotein expression [
31]. However, using sulfide concentrations that reflect physiological conditions (mid-nanomolar), we found the impact of PAG-induced SCLY inhibition on selenoprotein expression was sulfide-independent. Since PAG also impacts on other enzymes, either PLP- or non-PLP-dependent [
30,
32], involvement of other pathways cannot be completely excluded. However, given the complete restoration of the abundance of two selenoproteins with selenide in PAG-treated cells, off-target effects appear unlikely. Many studies have used PAG to elucidate the (patho)physiological and/or protective role(s) of endogenous hydrogen sulfide, for example, on cardio-protection [
33]. Although the impact of PAG on selenoprotein expression in vivo is yet to be determined, our results suggest the possibility of an unrecognized manipulation of selenoproteins.
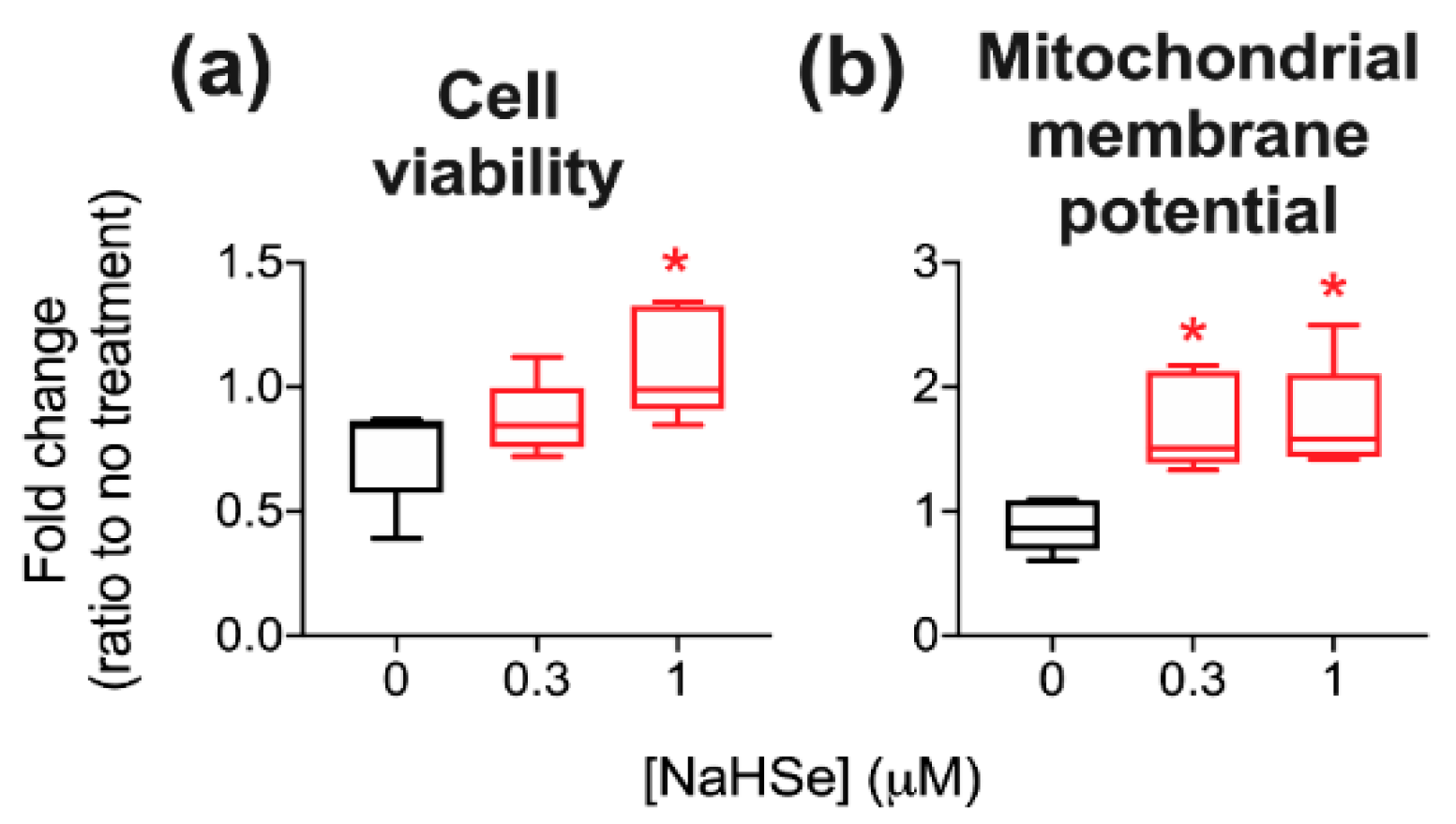
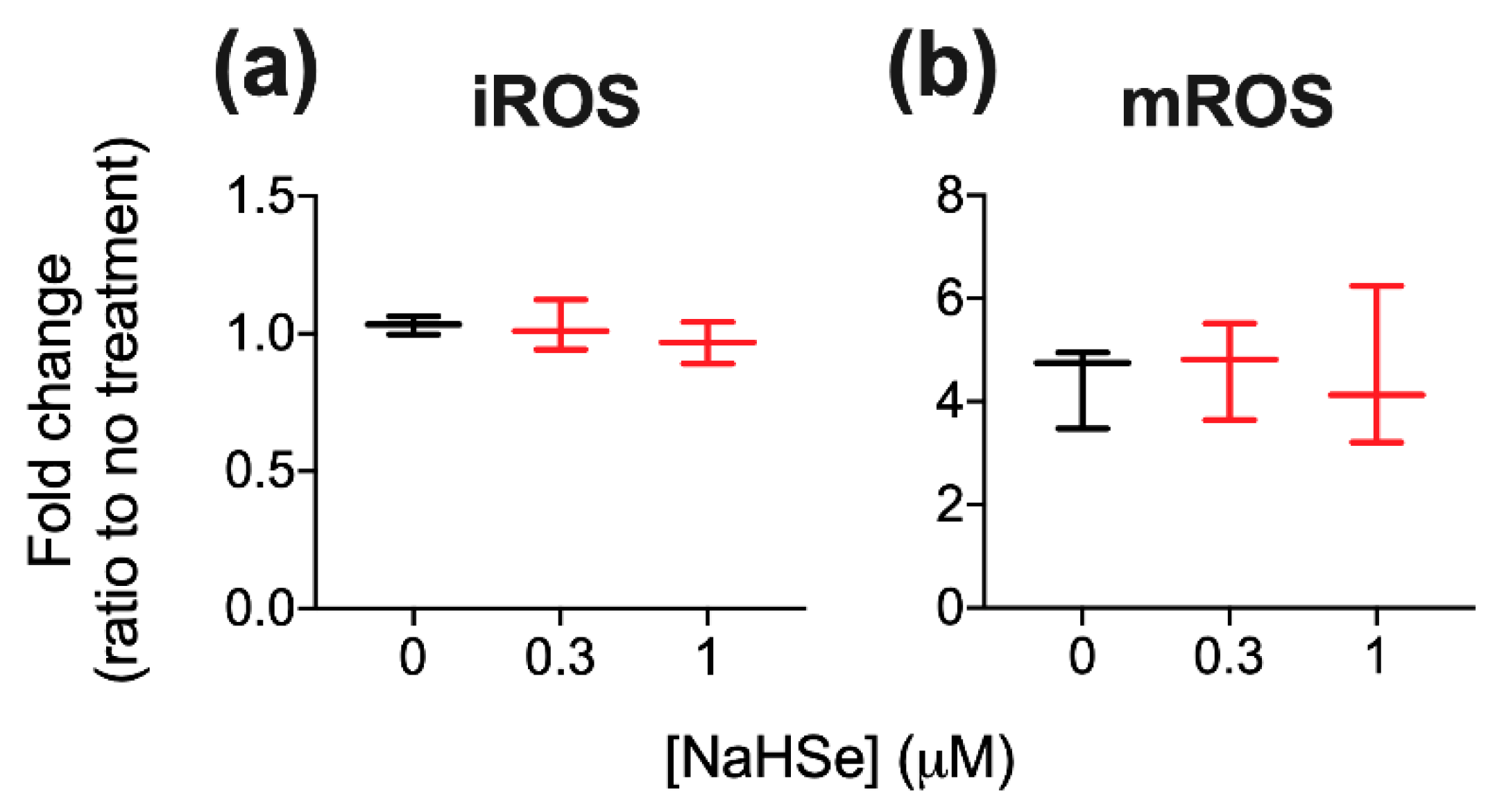
A role for NaHSe as a cytoprotective agent was examined using a H
2O
2-induced oxidative stress model where a marked improvement in viability was noted in NaHSe-treated cells. Unexpectedly, no difference was seen in intracellular ROS (±NaHSe treatment), since a similar model, albeit in a different cell type, showed a two–three-fold rise [
31]. NaHSe could confer cytoprotection by up to three potential mechanisms: as a reducing agent, a metabolic modulator and as the catalytic component of selenoproteins. Our studies do not yet allow robust identification of the precise molecular mode(s) of action. Mitochondrial membrane potential (MMP), with the accumulation of protons in the intermembrane space, drives phosphorylation of ADP by ATP synthase, and is frequently used as a marker of cell functionality. MMP is also subject to regulation by ROS, such that superoxide can activate mitochondrial uncoupling proteins to dissipate the membrane potential and temper ROS production [
34]. While we observed a five-fold increase in mitochondrial ROS in H
2O
2-treated cells, this was not modified by NaHSe treatment, perhaps due to preservation of MMP by NaHSe. A higher membrane potential for the same quantity of mitochondrial ROS suggests an improvement in ROS handling; however, this requires further study for full clarification.
Our study has several limitations. First, due to the chemical similarities of gaseous sulfide and selenide, we used a H2S detector to measure H2Se. Notably, the bespoke H2Se detector(s) we trialed lacked sensitivity. Consequently, our methodology does not allow for a fully quantitative assessment of H2Se, but it could be used in future to assess relative changes with NaHSe as a reference standard. Second, the method used to synthesize NaHSe generates sodium tetraborate as a by-product. However, we are unaware of any studies or biological mechanism(s) where this could impact upon the outcome measures assessed. Third, NaHSe was used as pre-treatment in our cytoprotection study; while this precludes translation to a more clinically relevant scenario (where a treatment is generally applied after the insult), we were mindful that we did not want to quench H2O2 with the reducing capacity of NaHSe. Future work, in addition to enabling a more complete understanding of the molecular mode of action, should focus on post-treatment (in non-chemical models) and longitudinal testing, preferably with measurement of real-time ROS production. Lastly, we acknowledge that our in vivo assessment of NaHSe is limited; conscious, long-term safety, and efficacy testing of seleno-mimetics are still required, but this should ideally be performed with more promising clinical candidates.
In summary, we provide a significant contribution to selenium pharmacology, demonstrating that the metabolic effects of NaHSe are due to transient inhibition of cytochrome C oxidase. The importance of oxidation status of selenium therapies is further illustrated, with significant implications for clinical practice. As noted, the work performed here is analogous to the first reports examining basic sulfur salts. These were eventually superseded by superior molecules that allowed better control of sulfide delivery [
15,
35,
36], improved targeting to its intended site of action, and at concentrations that better reflect those derived from endogenous sources [
4,
37]. We postulate the same will hold true for NaHSe; while it will remain an important research tool, its utility as a therapy would likely be hindered by poor pharmacokinetic and safety profiles. While alternative approaches to treat oxidative pathologies remain elusive, intelligent seleno-mimetic drug design could yield more sophisticated pharmacological agents as prospective future medicines.
4. Materials and Methods
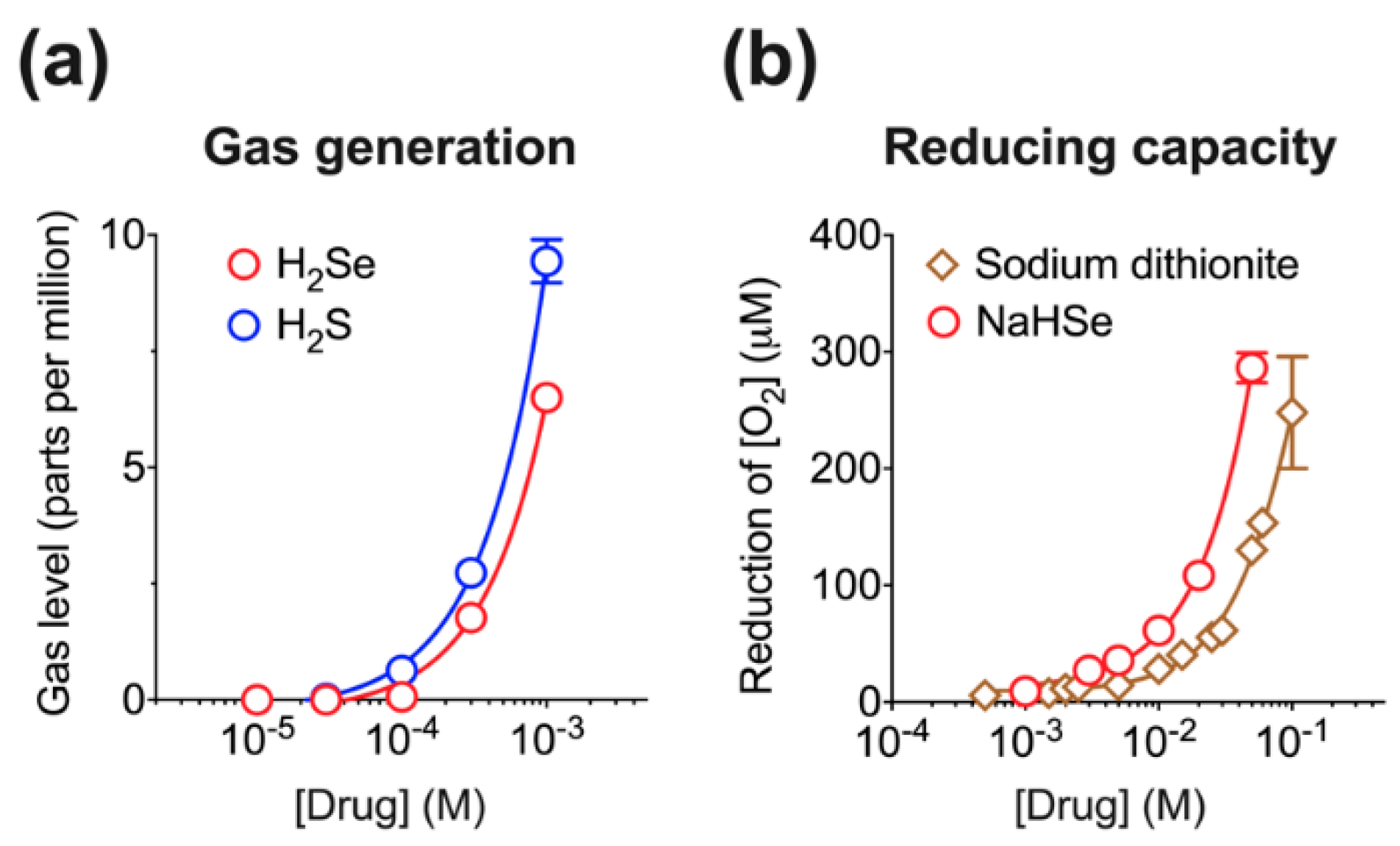
4.1. Synthesis and Characterization of NaHSe
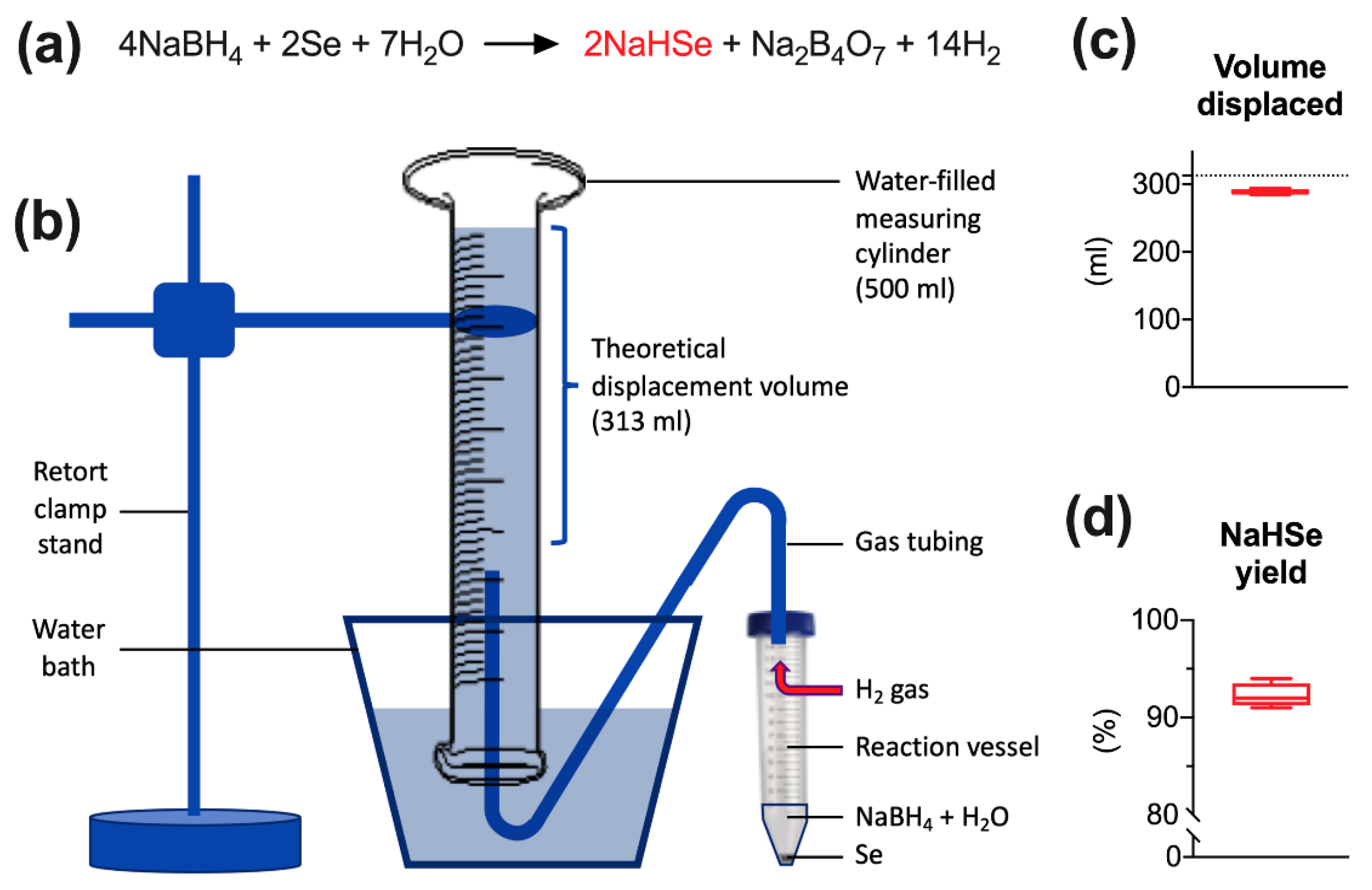
NaHSe was synthesized as previously described [
38] on each day of use (
Figure 1a). The reaction uses aqueous sodium borohydride (NaBH
4) to reduce elemental selenium. These precursors and all other reagents were purchased from Sigma-Aldrich (Gillingham, Kent, UK) unless stated otherwise. Briefly, under a fume hood, and in a large Perspex box filled with (inert) argon gas, NaBH
4 (151.32 mg) was dissolved in (nitrogen-purged) deionized water (2 mL) and added to an open 50 mL Falcon tube containing an excess (>200 mg) of selenium. The reaction visibly generates 313 mL hydrogen gas over approximately 5 s and is mildly exothermic. Once reacted, 1 mL of product was transferred to an Eppendorf tube and centrifuged (14000 rpm for 1 min). This separates unreacted selenium from the supernatant (1M NaHSe) that was transferred to a fresh tube, kept on ice, and diluted (as required) under argon gas using nitrogen-purged (1×) phosphate buffer saline (pH 7.4).
The yield of product was assessed by measuring the quantity of water displaced by hydrogen gas, a by-product generated during NaHSe synthesis. This was performed on four separate occasions as described above. As no other gases (save for small quantities of hydrogen selenide gas) should be evolved during the reaction, this method would give a good approximation of yield. A 500 mL measuring cylinder was filled with water, inverted in a large water-filled container, and held in place with a retort clamp stand. A Falcon tube lid was modified to incorporate air-tight tubing that was fed from the reaction vessel to the measuring cylinder and used to transfer H2 gas.
Given the chemical similarities between gaseous sulfide and selenide, we used an assay previously optimized for measurement of hydrogen sulfide [
13] to assess hydrogen selenide gas (H
2Se) generation. This assay relies on the detection of free headspace H
2Se, measured using a commercially available H
2S detector (Z900XP, Environmental Sensors, Boca Raton, FL, USA). Briefly, either NaHS (as a positive control) or NaHSe was dissolved in PBS to 10× stock solutions and rapidly diluted (0.5 into 4.5 mL) into airtight 50 mL Falcon tubes containing nitrogen-purged PBS. This was incubated in a water bath for 1 h at 37 °C. Five mL of headspace gas withdrawn over 10 s was passed through the detector. Peak H
2S values were recorded and used as a non-quantitative surrogate of H
2Se generation.
The ability of NaHSe to act as a chemical reducing agent was assessed in air-tight chambers using a polarographic Clark-type electrode (Rank Brothers, Bottisham, UK). This was connected to a sealed chamber and maintained at 37 °C with an external, jacketed water heater. Increasing concentrations of either NaHSe (1–50 mM), or the standard laboratory reducing agent, sodium dithionite (Na2S2O4; 0.5–100 mM), were added to the chamber, and the decrease in oxygen concentration was recorded.
Particular care needs to be taken when preparing and using hydrogen selenide. Good safety practices should be maintained, and work should only commence following liaison with divisional safety officers. In particular, fume cupboards or hoods must operate efficiently, and researchers should avoid working alone in the laboratory. These rules are essentially the same as those described for use of hydrogen sulfide [
39].
4.2. Ethics, Animals and Husbandry
Male Wistar rats (300–400 g body weight) were used to harvest tissues for ex vivo studies, and for in vivo experimentation. All experiments were performed in the UK according to local ethics committee (University College London) and UK Home Office guidelines under the Animals (Scientific Procedures) Act 1986. Animals were purchased from Charles River Laboratories (Margate, UK). All animals were healthy and certified pathogen-free and housed in cages of four individuals on a 12-h light/dark cycle, with food and water ad libitum prior to experimentation. Standard cages and bedding were used with additional materials (tissue paper, cardboard tubes) for comfort and cage enrichment. The ‘actual severity’ classification for all studies is ‘non-recovery’. All animals received peri-operative analgesia (subcutaneous buprenorphine, 0.05 mg/kg; Reckitt Benckiser, Slough, UK). Euthanasia at experiment end was performed either by cessation of the circulation (cardiac biopsy) or intravenous sodium pentobarbitone (Pentoject; Animalcare, York, UK).
4.3. Modulation of Metabolism
4.3.1. Oxygen Consumption Experiments
For ex vivo oxygen consumption experiments, soleus muscle or liver samples were obtained from anaesthetized rats. Soleus muscle was dissected and immediately transferred to plastic Petri dishes containing ice-cold biopsy preserving solution (BIOPS; isolation medium). This constituted CaK
2EGTA (2.77 mM), K
2EGTA (7.23 mM), Na
2ATP (5.7 mM), MgCl
26H
2O (6.56 mM), taurine (20 mM), Na
2 phosphocreatine (15 mM), imidazole (20 mM), dithiothreitol (0.5 mM) and MES monohydrate (50 mM), adjusted to pH 7.4. This method enables storage of tissue with no significant impairment of mitochondrial integrity [
40]. The muscle was subsequently dissected with fine forceps and resulting fibres permeabilized with 50 µg saponin in 2 mL isolation medium. Fibres were gently stirred on ice for 20 min. Saponin and metabolites were then removed by washing the fibres three times with ice-cold respiratory medium containing EGTA (0.5 mM), MgCl
2·6H
2O (3 mM), K-lactobionate (60 mM), taurine (20 mM), KH
2PO
4 (10 mM), HEPES (
N-(2-hydroxyethyl)piperazine-
N′-(2-ethanesulfonic acid; 20 mM), sucrose (110 mM) and bovine serum albumin (BSA; 1 mg/mL), adjusted to pH 7.4. Following midline laparotomy, a sample of liver (~1 g) was removed, washed briefly with ice-cold PBS and snap-frozen in liquid nitrogen. The tissue was then pulverized under liquid nitrogen using a pestle and mortar, and kept at −80 °C until use. Sixty mg was then added to 1 mL BIOPS; the homogenate was centrifuged at 7800 rpm for two minutes, and the supernatant was kept on ice until use.
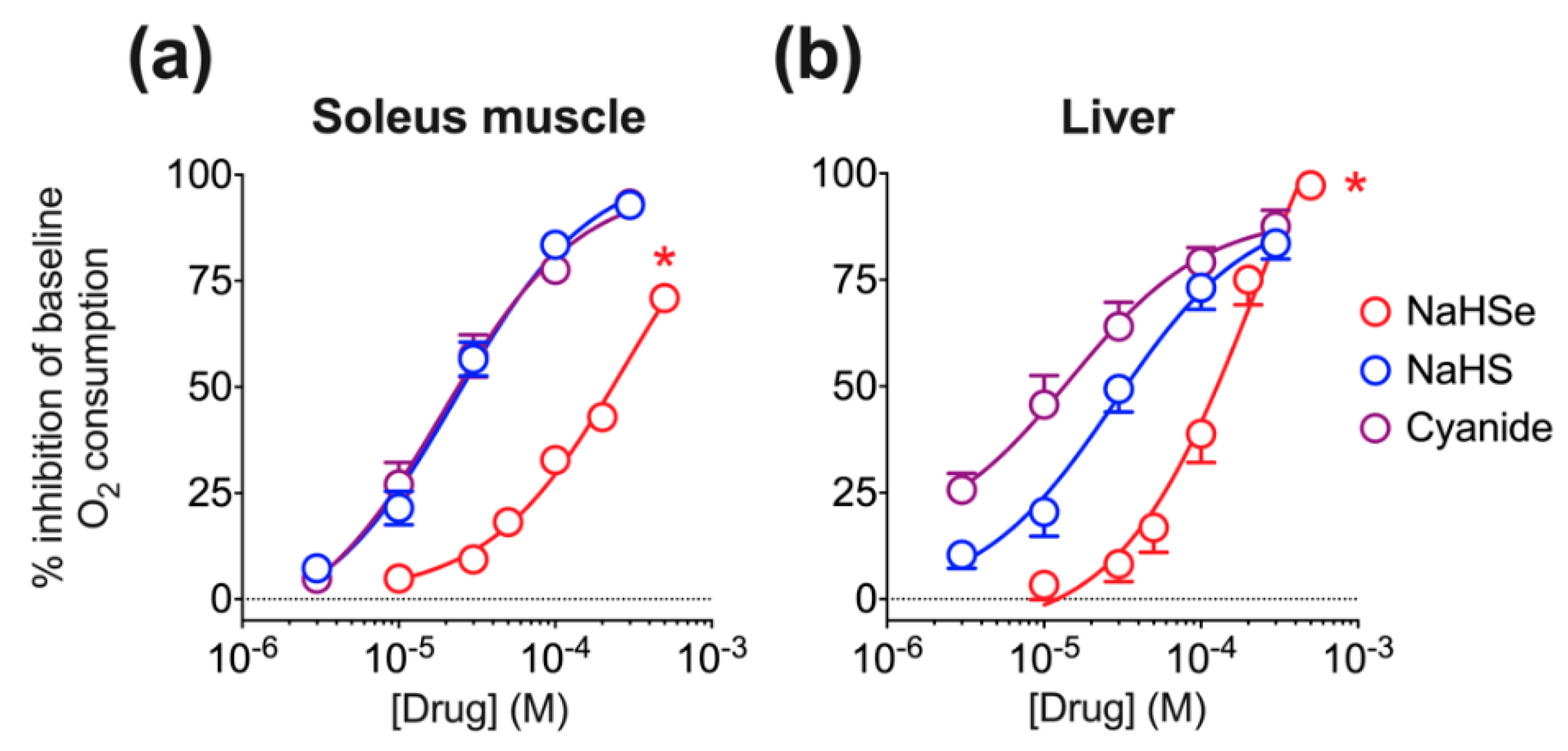
Oxygen consumption was determined using the above Clark electrode. The fall in oxygen concentration within the closed chamber over time was recorded; this was subsequently corrected for drift (oxygen consumption by the electrode), and either the dry weight of each muscle sample or protein content of the liver supernatant. The respiratory medium in the chamber was agitated using a magnetic stirrer. Substrates for mitochondrial respiratory chain complexes I (glutamate, 10 mM; malate, 5 mM) and II (succinate, 5 mM) were added to each chamber by injection through the chamber lid seal. Small bundles (approximately 5 mg) of muscle tissue or 30 µL liver supernatant were then added and the medium oxygenated to approximately 250 μM O2 by injecting 3 mL of 100% O2 gas.
Baseline oxygen consumption within the chamber was assessed over two minutes followed by addition of increasing concentrations of NaHSe (10–500 μM), NaHS or K-cyanide (both 3–300 μM). Concentrations were increased at 2-min intervals, with oxygen consumption measured in the last minute of each interval. All ‘normoxia’ experiments were performed between 250 and 150 μM O2, below which the level of oxygen consumption becomes dependent on the O2 concentration within the chamber.
In a second experiment, a single concentration of NaHSe (10 μM) was used. This concentration was ineffective during normoxia, with soleus muscle tissue allowed to respire to hypoxia. Respiratory medium was used as a control. Oxygen consumption was determined at 150 μM (baseline) and after the addition of NaHSe, at 15 μM [O2] gradations until the concentration fell to 45 μM O2.
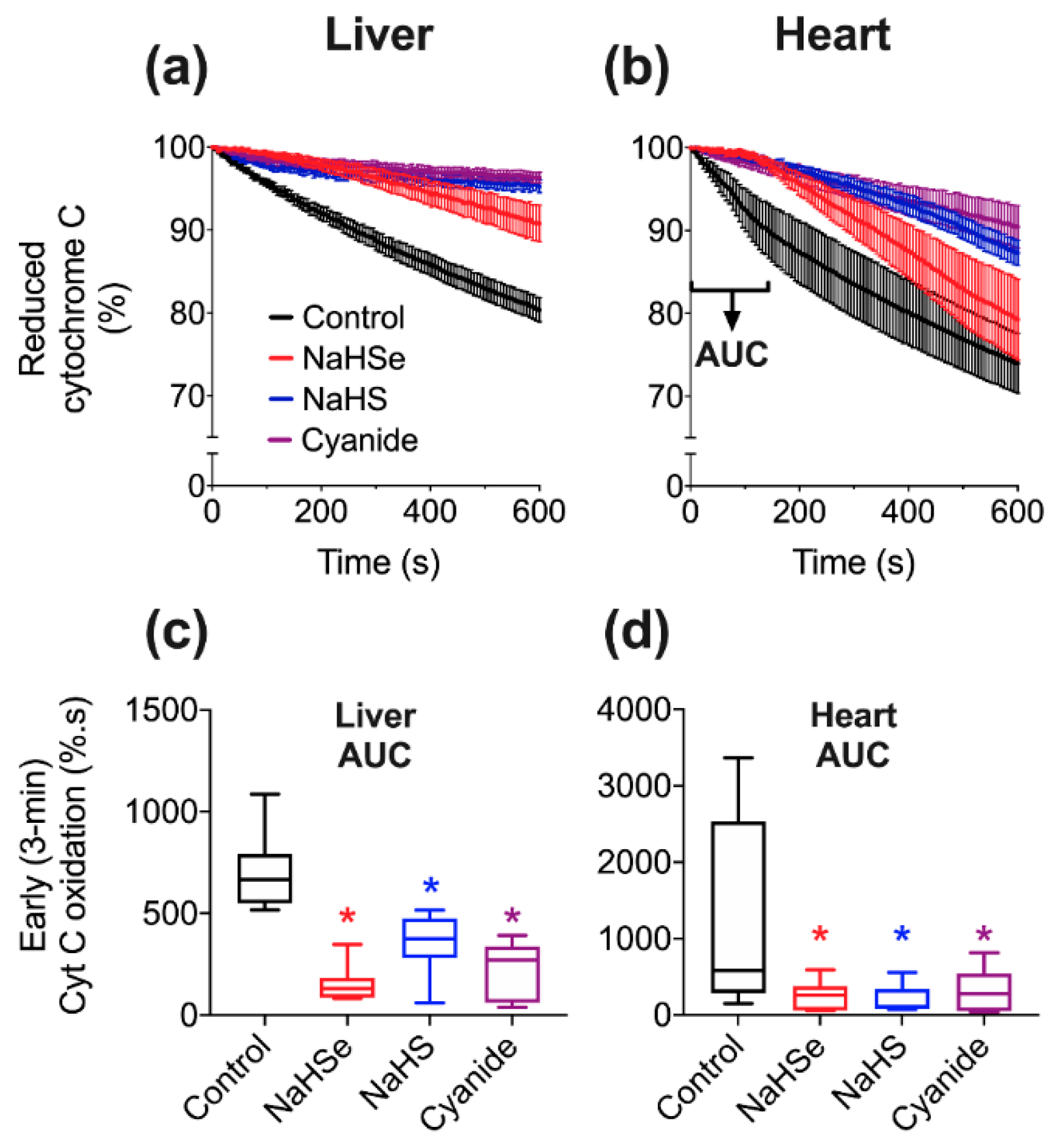
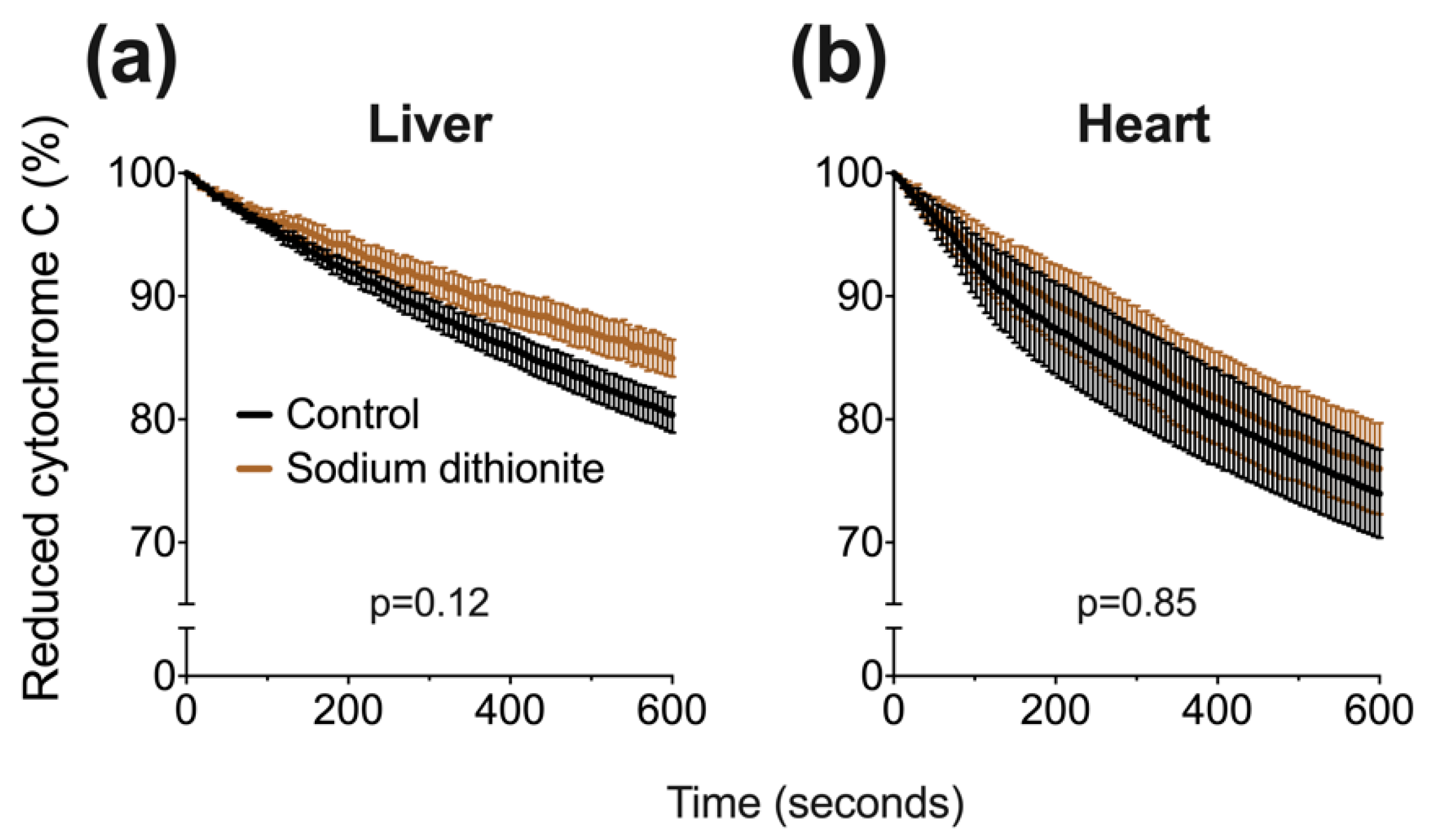
4.3.2. Cytochrome C Oxidase Activity
A well-established assay [
14] was used (with modifications) to assess CCO activity in liver and heart tissue. Briefly, 12.5 mg cytochrome C was added to 1 mL of 20 mM potassium phosphate buffer, followed by 1 mg of sodium dithionite to fully reduce cytochrome c. The reduction of cytochrome C was assessed by absorbance spectrophotometry using a microplate reader and BioTek (Gen5) software (Synergy 2, North Star Scientific, Sandy, UK). The ratio of absorbance at 550 and 565 nm was confirmed to be above 6, signifying an acceptable proportion of reduced cytochrome C. Next, a master-mix was assembled, comprising 5 parts potassium phosphate buffer (100 mM), 3.6 parts distilled water and either 0.4 parts tissue or BIOPS solution. 80 μL of either tissue-positive or negative mix was added to a 96-well plate followed by 10 μL of drugs. We used the IC
50 value from our oxygen consumption experiments in liver tissue (
Figure 2) for either NaHSe, NaHS or K-cyanide. BIOPS (10 μL) served as a negative control. We additionally tested sodium dithionite (at twice the NaHSe concentration; 458 μM) to ensure CCO activity was not artificially stalled by the reducing capacity of these drugs. Enzyme activity was initiated with the addition of 10 μL reduced cytochrome C, with (decreasing) absorbance (550 nm) measured every 6 s for 10 min.
4.4. In Vitro Studies
Human hepatoma (HepG2) cells were cultured to 80% confluence in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (37 °C, 5% CO2). Cells were seeded on 24-well plates in triplicate at a density of 1.5 × 106 cells/mL. Cells were incubated for a further 24h before being assigned to separate studies.
4.4.1. Pharmacological Manipulation of Selenoprotein Expression
Cells were treated with either standard medium (control), NaHSe (30 nM), sodium selenite (Na2SeO3; 30 nM), L-cysteine (0.1 mM) or DL-propargylglycine (PAG; 2 mM) for 1h. In a subset, cells were pre-treated (for 30 min) with PAG before addition of either NaHSe or NaHS (30 nM). Cells were then washed, the medium replaced, and allowed to incubate under standard conditions for a further 24 h. Protein was extracted in radio-immunoprecipitation assay (RIPA) buffer and concentrations determined using bicinchoninic acid assay (Pierce™ BCA Protein Assay Kit). Western blot was performed on 12% SDS-PAGE gel with 40 μg protein per well. Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare, Amersham, UK) at 10V for 45 min, blocked with 5% BSA, and incubated overnight at 4 °C with primary antibodies (GPx-1; 1:1000, Bio-Techne, Abingdon, UK) or thioredoxin reductase-1 (TxR-1; 1:1000, Cell Signalling, London, UK) and alpha-tubulin (1:3000; Cell SignalFling Technology, Danvers, MA, USA). Secondary antibodies were incubated for 1 h at room temperature (1:3000; Agilent Technologies, Santa Clara, CA, USA). The blots were then detected using an enhanced chemiluminescence detection system (GE Healthcare, Chicago, IL, USA) and ratio-metric expression determined using Image Studio Lite (LI-COR, Cambridge, UK).
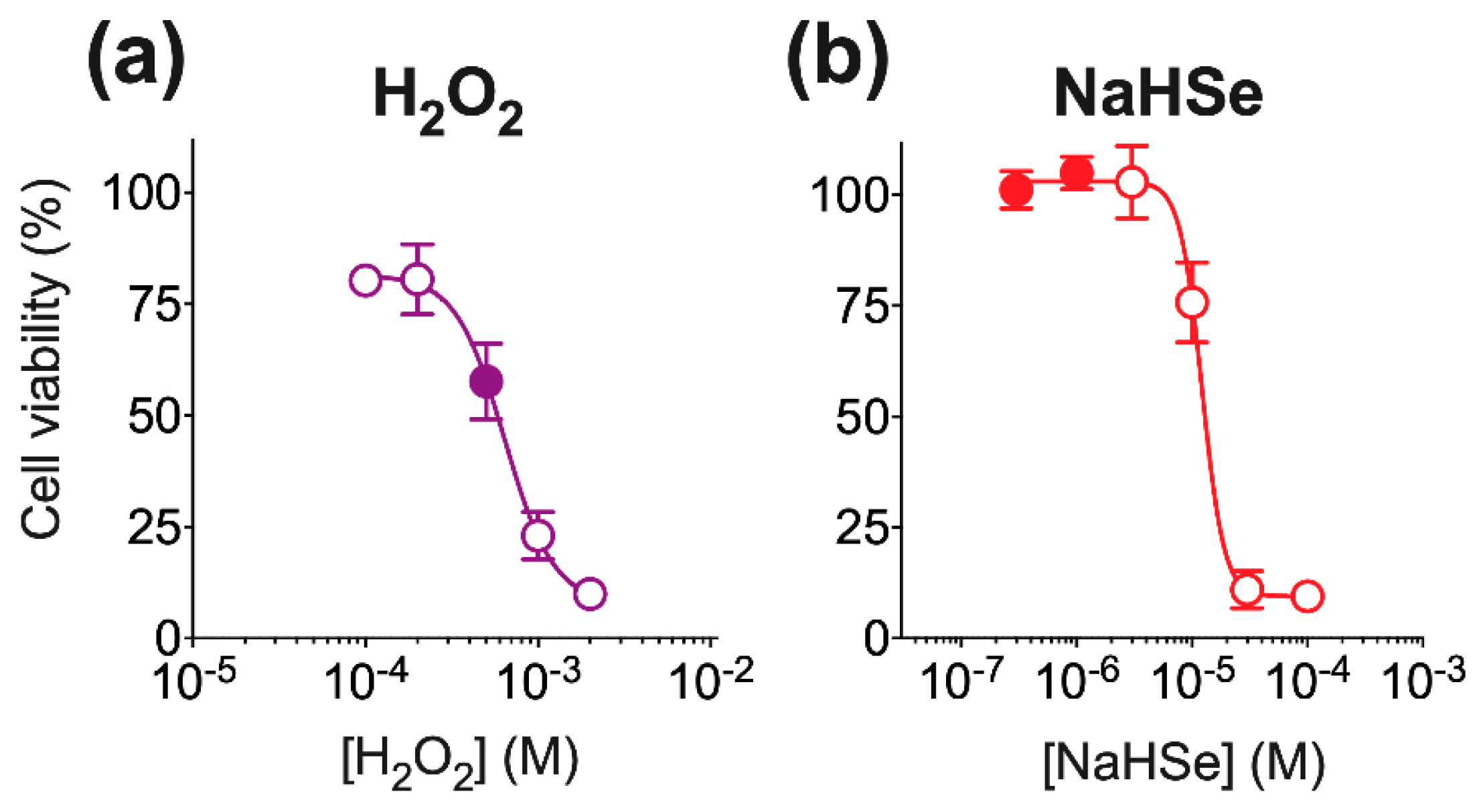
4.4.2. Viability
Cell viability was determined in tetrazolium chloride (TTC; 1%
w/
v)-stained cells [
41]. Cells were treated for 24 h with cell medium (DMEM; negative control), or different concentrations of either NaHSe (0.3–100 μM) or hydrogen peroxide (50–2000 μM). These pilot studies aimed to establish a non-toxic concentration range for NaHSe (for use in all of our in vitro studies), and for optimization of an oxidative insult with H
2O
2. A positive control was provided by heat-killing the cells at 65 °C for 30-min. Cells were incubated for 1h with colourless TTC, which is enzymatically converted to a red formazan product by living cells. Cells were then lysed using ice-cold (1×) RIPA buffer agitated on an orbital shaker for 1.5 h, then centrifuged (12000 rpm for 10-min). The supernatant (300 μL) was transferred to a 96-well plate and absorbance measured at 494 nm using the above microplate reader. The results were normalized for protein content (using a BCA assay kit; Abcam, Cambridge, UK) and calibrated using internal positive and negative controls.
4.4.3. Oxidative Stress Model
Oxidative stress was induced by 500 μM H
2O
2, a concentration that produced ~40% cell death in pilot studies (
Figure A6). Cells receiving NaHSe were pre-treated with either 0.3 or 1 μM for 60 min. The cells were then washed, H
2O
2 added and incubated for 24 h under standard cell culture conditions. Viability was then measured as above. A subset of cells was allocated to flow cytometry experiments.
4.4.4. Flow Cytometry
Cells were incubated with either tetra-methyl-rhodamine methyl ester (TMRM; 25 nM; Thermo Fischer Scientific, Waltham, MA, USA), 2′,7′-dichlorofluorescein diacetate (H
2DCFDA; 5 μM; Thermo Fischer Scientific, Waltham, MA, USA) or MitoSox
TM (2.5 μM; ThermoFisher Scientific, Waltham, MA, USA) to determine mitochondrial membrane potential [
42], intracellular ROS [
43] or mitochondrial [
44] ROS, respectively. The contents of each well were then transferred to FACS (fluorescence-activated cell sorting) tubes and fluorescence measured using a flow cytometer (LSR Fortessa, BDBiosciences, San Jose, CA, USA). Data were collected and analysed using FlowJo v10.6.1 software (FloJo, Ashland, OR, USA).
4.5. In Vivo Studies
Ten animals were anaesthetized as above. They were placed on a heated mat (Harvard Apparatus, Cambridge, UK) to maintain rectal temperature at 37 °C. The left common carotid artery and right internal jugular vein were cannulated using 0.96 mm outside diameter PVC tubing catheter (Scientific Commodities Inc., Lake Havasu City, AZ, USA). The arterial line was connected to a pressure transducer (Powerlab; AD Instruments, Chalgrove, UK) for continuous monitoring of mean arterial blood pressure and intermittent blood sampling, and the venous line was used for administration of fluids and drugs.
Following surgery and a 30-min stabilization period, animals received increasing intravenous bolus doses of NaHSe (0.01–10 mg/kg) or an equivalent quantity (1 mL/kg) of PBS. The diluent was purged with nitrogen prior to use and each dose administered (within 2-min of preparation). Measurements were collected at baseline, then as follows after each dose: blood pressure changes at 30-s to 1-min, cardiac function (by echocardiography; see below for details) within 2 min, and subsequent arterial blood gas analyses (to measure partial pressure of CO2 (PCO2), lactate, and acid/base changes) at 13 min. Doses were escalated every 15 min.
Transthoracic echocardiography was performed using a 14 MHz probe scanning at 0–2 cm depth (Vivid 7 Dimension, GE Healthcare, Bedford, UK). Aortic blood flow velocities were determined in the aortic arch using pulsed-wave Doppler. Stroke volume was determined as the product of velocity-time integral (VTI) and vessel cross-sectional area. Heart rate was determined by measuring the time between cardiac cycles. Cardiac output was calculated as the product of stroke volume and heart rate.
4.6. Data and Statistics
Data are presented as mean ± standard error or median, quartiles and range. Parametric data were analyzed using either repeated measures one- or two-way ANOVA followed by Sidak’s post-hoc test, as appropriate. Non-parametric data were analyzed using the Mann-Whitney U-test. Linear regression was performed using three- or four-parameter inhibitor/response models with least squares fitting method. All statistical analyses were two-tailed and performed using Prism 8.4.3 software (GraphPad Software, San Diego, CA, USA). Multiplicity-adjusted p-values < 0.05 were considered statistically significant.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












