
Conformational Heterogeneity and Cooperative Effects of Mammalian ALOX15

 ,
,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Results
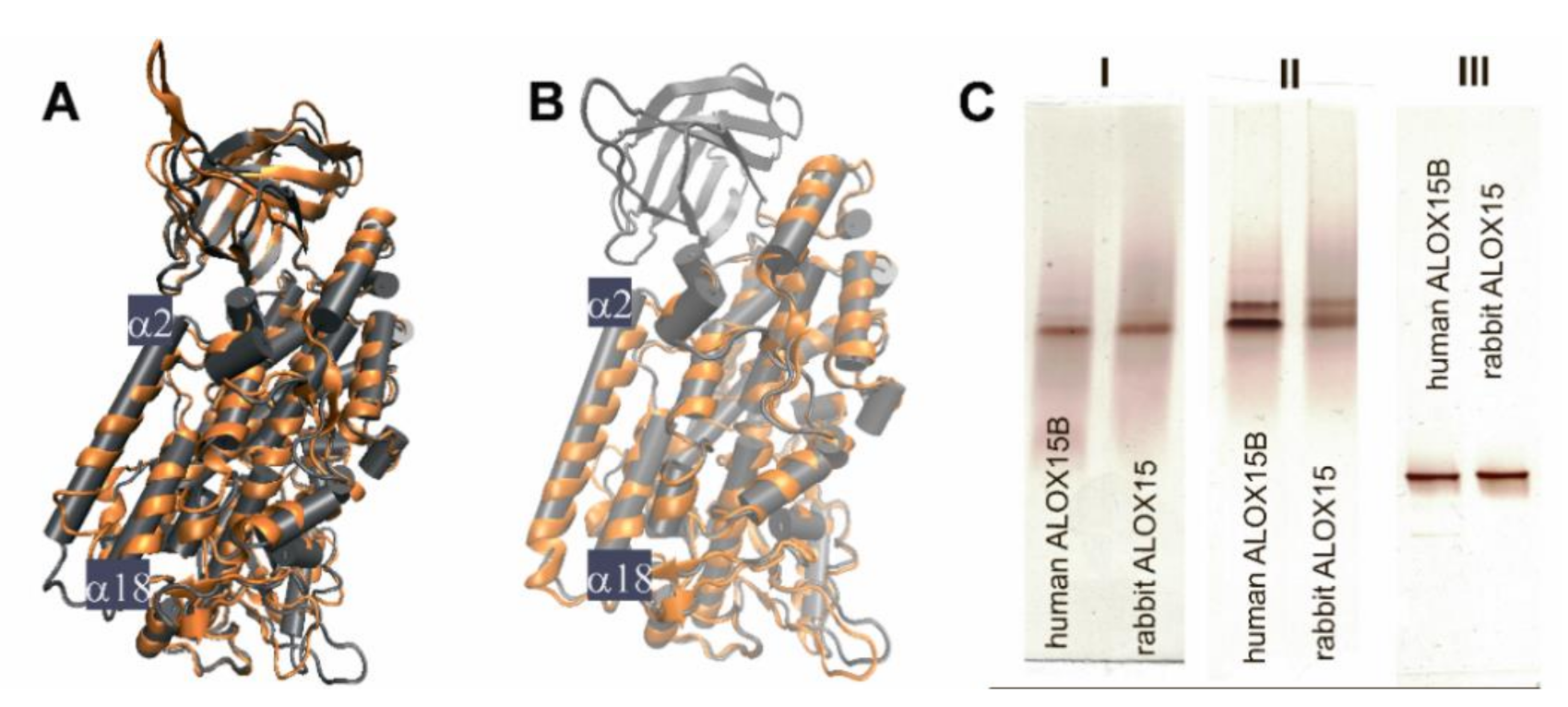
2.1. Structural Characterization of ALOX15 Isoenzymes by Native PAGE
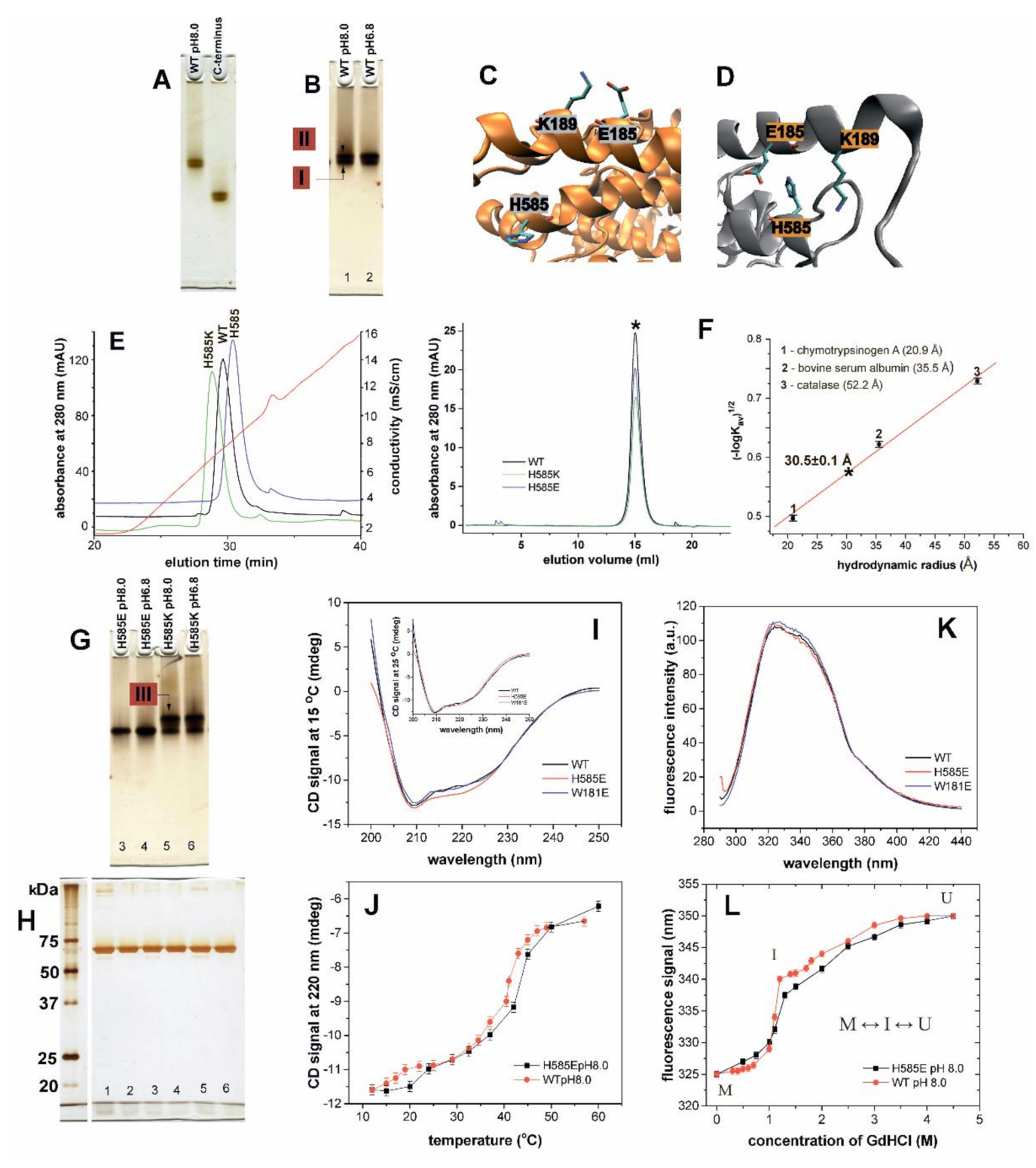
2.2. Consequences of N-terminal Truncation and Point Mutations of His585 on Conformational Heterogeneity of Rabbit ALOX15
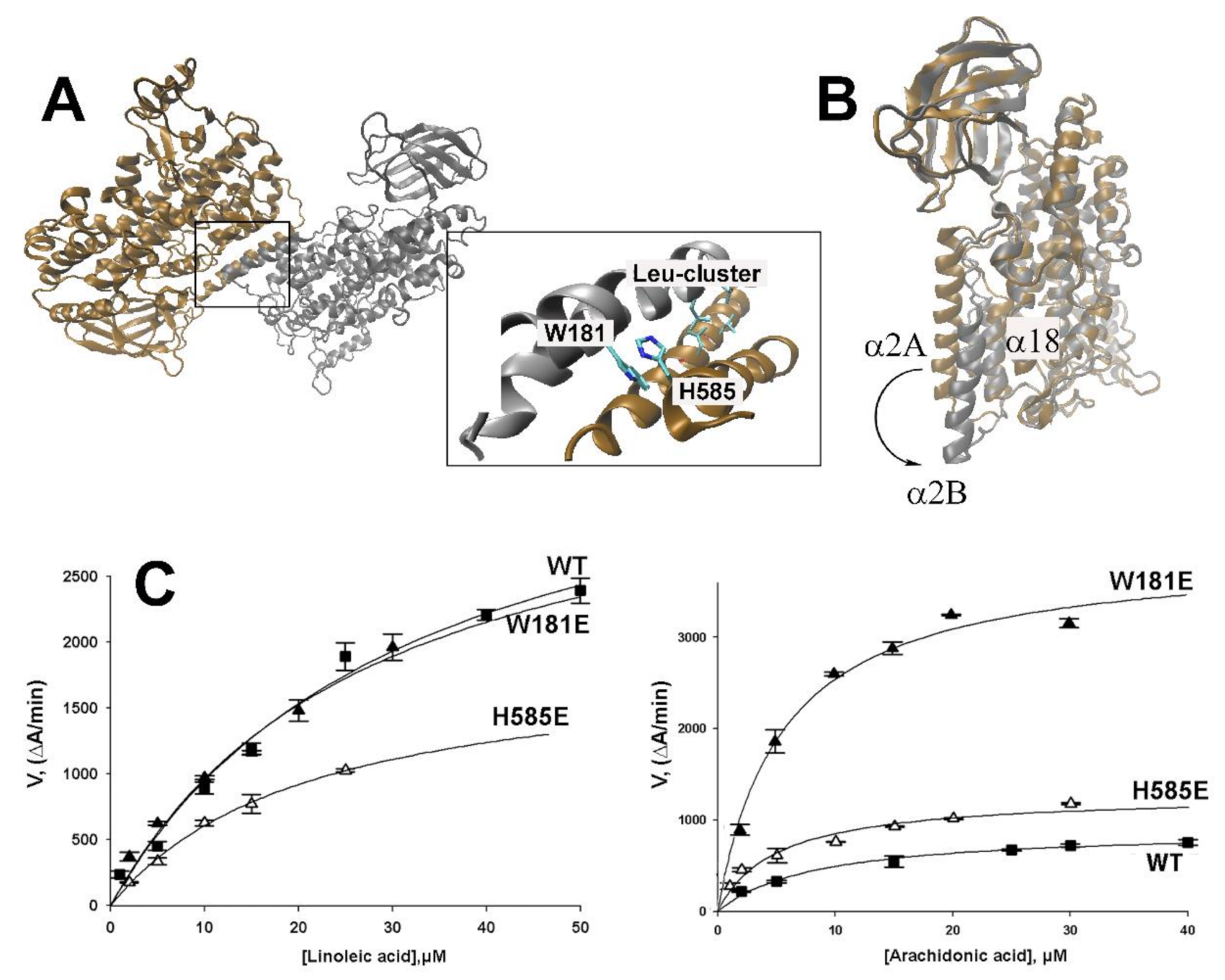
2.3. Impact of Point Mutations of Amino Acids Localized at the Inter-Monomer Contact Site on the Reaction Kinetics of Rabbit ALOX15
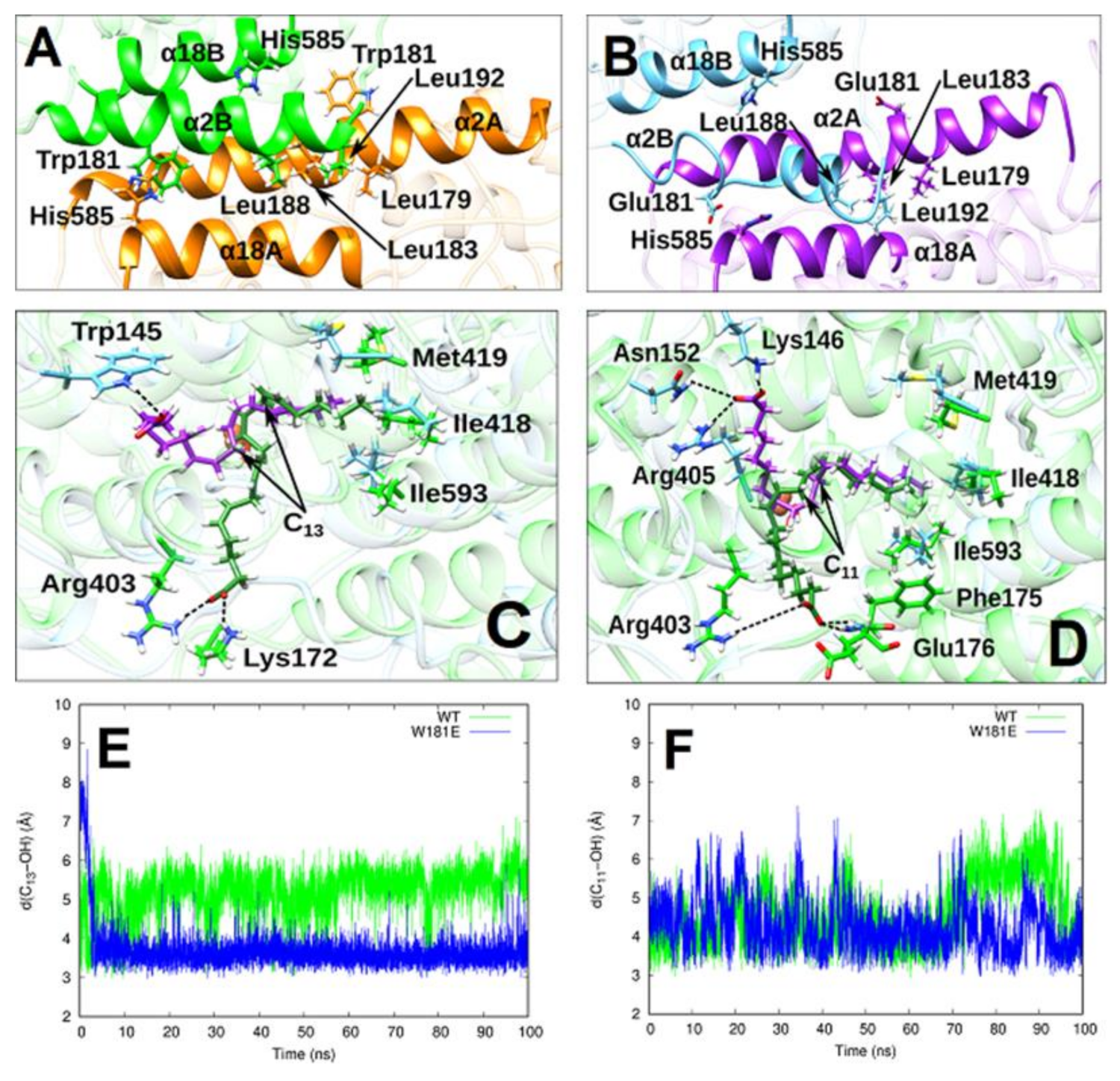
2.4. MD Simulation of the Structural Alterations of Rabbit ALOX15 Induced by Trp181Glu Exchange
2.5. AA and LA Binding Modes
2.6. Analysis of Pre-Catalytic Structures
3. Discussion
3.1. Conformational Heterogeneity of ALOX15
3.2. Cooperativity of Rabbit ALOX15 Monomers during Lipoxygenase Reaction
4. Materials and Methods
4.1. Chemicals
4.2. Expression of Rabbit ALOX15
4.3. Expression of Human ALOX15B
4.4. Purification of Rabbit ALOX15 and Human ALOX15B
4.5. Estimation of the Hydrodynamic Radii for Wild-Type and Mutant ALOX15
4.6. Native PAGE Electrophoresis
4.7. CD Measurements
4.8. Guanidine Denaturation Studies.
4.9. Rabbit ALOX15 Activity Assay (Purified Enzyme)
4.10. Molecular Docking Simulations
4.11. Molecular Dynamics (MD) Simulations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 15-H(p)ETE | 15-Hydro(pero)xy-5Z,8Z,11Z,13E-eicosatetraenoic acid |
| 12-H(p)ETE | 12-Hydro(pero)xy-5Z,8Z,10E,14Z-eicosatetraenoic acid |
| 13-H(p)ODE | 13-Hydro(pero)xy-9Z,12E-octadecadienoic acid |
| GdnHCl | Guanidine hydrochloride |
References
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 2015, 1851, 308–330. [Google Scholar] [CrossRef] [Green Version]
- Brash, A.R. Lipoxygenases: Occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 1999, 274, 23679–23682. [Google Scholar] [CrossRef] [Green Version]
- Haeggstrom, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Kuhn, H.; Humeniuk, L.; Kozlov, N.; Roigas, S.; Adel, S.; Heydeck, D. The evolutionary hypothesis of reaction specificity of mammalian ALOX15 orthologs. Prog. Lipid Res. 2018, 72, 55–74. [Google Scholar] [CrossRef]
- Singh, N.K.; Rao, G.N. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog. Lipid Res. 2019, 73, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Wecksler, A.T.; Kenyon, V.; Deschamps, J.D.; Holman, T.R. Substrate specificity changes for human reticulocyte and epithelial 15-lipoxygenases reveal allosteric product regulation. Biochemistry 2008, 47, 7364–7375. [Google Scholar] [CrossRef] [Green Version]
- Mikulska-Ruminska, K.; Shrivastava, I.; Krieger, J.; Zhang, S.; Li, H.; Bayir, H.; Wenzel, S.E.; VanDemark, A.P.; Kagan, V.E.; Bahar, I. Characterization of Differential Dynamics, Specificity, and Allostery of Lipoxygenase Family Members. J. Chem. Inf. Model. 2019, 59, 2496–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wecksler, A.T.; Jacquot, C.; van der Donk, W.A.; Holman, T.R. Mechanistic investigations of human reticulocyte 15- and platelet 12-lipoxygenases with arachidonic acid. Biochemistry 2009, 48, 6259–6267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, D.S.; Liu, W.; Gu, Z.; Langevine, C.; Ngu, K.; Fadnis, L.; Combs, D.W.; Sitkoff, D.; Ahmad, S.; Zhuang, S.; et al. Tryptamine and homotryptamine-based sulfonamides as potent and selective inhibitors of 15-lipoxygenase. Bioorg. Med. Chem. Lett. 2005, 15, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, D.S.; Liu, W.; Ngu, K.; Langevine, C.; Combs, D.W.; Zhuang, S.; Chen, C.; Madsen, C.S.; Harper, T.W.; Robl, J.A. Discovery of selective imidazole-based inhibitors of mammalian 15-lipoxygenase: Highly potent against human enzyme within a cellular environment. Bioorg. Med. Chem. Lett. 2007, 17, 5115–5120. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; McClendon, C.L.; Dai, Z.; Li, K.; Zhang, X.; He, S.; Shang, E.; Liu, Y.; Lai, L. Discovery of Novel 15-Lipoxygenase Activators To Shift the Human Arachidonic Acid Metabolic Network toward Inflammation Resolution. J. Med. Chem. 2016, 59, 4202–4209. [Google Scholar] [CrossRef]
- Meng, H.; Dai, Z.; Zhang, W.; Liu, Y.; Lai, L. Molecular mechanism of 15-lipoxygenase allosteric activation and inhibition. Phys. Chem. Chem. Phys. 2018, 20, 14785–14795. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Shang, W.; Toledo, L.; Masgrau, L.; Svergun, D.I.; Stehling, S.; Gomez, H.; Di Venere, A.; Mei, G.; Lluch, J.M.; et al. Ligand-induced formation of transient dimers of mammalian 12/15-lipoxygenase: A key to allosteric behavior of this class of enzymes? Proteins 2012, 80, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Chon, J.K.; Kim, S.; Shin, W. Conformational flexibility in mammalian 15S-lipoxygenase: Reinterpretation of the crystallographic data. Proteins 2008, 70, 1023–1032. [Google Scholar] [CrossRef]
- Shang, W.; Ivanov, I.; Svergun, D.I.; Borbulevych, O.Y.; Aleem, A.M.; Stehling, S.; Jankun, J.; Kuhn, H.; Skrzypczak-Jankun, E. Probing dimerization and structural flexibility of mammalian lipoxygenases by small-angle X-ray scattering. J. Mol. Biol. 2011, 409, 654–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, A.K.; Cernescu, M.; Hofmann, B.; Ermisch, M.; Hornig, M.; Metzner, J.; Schneider, G.; Brutschy, B.; Steinhilber, D. Dimerization of human 5-lipoxygenase. Biol. Chem. 2011, 392, 1097–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, A.; Di Venere, A.; Mei, G.; Zhuravlev, A.; Golovanov, A.; Stehling, S.; Heydeck, D.; Lluch, J.M.; González-Lafont, À.; Kuhn, H.; et al. A role of Gln596 in fine-tuning mammalian ALOX15 specificity, protein stability and allosteric properties. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158680. [Google Scholar] [CrossRef]
- Suardíaz, R.; Masgrau, L.; Lluch, J.M.; González-Lafont, À. Regio- and stereospecificity in the oxygenation of arachidonic acid catalyzed by Leu597 mutants of rabbit 15-lipoxygenase: A QM/MM study. Chemphyschem 2014, 15, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Borngraber, S.; Browner, M.; Gillmor, S.; Gerth, C.; Anton, M.; Fletterick, R.; Kuhn, H. Shape and specificity in mammalian 15-lipoxygenase active site. The functional interplay of sequence determinants for the reaction specificity. J. Biol. Chem. 1999, 274, 37345–37350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.; Di Venere, A.; Horn, T.; Scheerer, P.; Nicolai, E.; Stehling, S.; Richter, C.; Skrzypczak-Jankun, E.; Mei, G.; Maccarrone, M.; et al. Tight association of N-terminal and catalytic subunits of rabbit 12/15-lipoxygenase is important for protein stability and catalytic activity. Biochim. Biophys. Acta 2011, 1811, 1001–1010. [Google Scholar] [CrossRef]
- Droege, K.D.; Keithly, M.E.; Sanders, C.R.; Armstrong, R.N.; Thompson, M.K. Structural Dynamics of 15-Lipoxygenase-2 via Hydrogen-Deuterium Exchange. Biochemistry 2017, 56, 5065–5074. [Google Scholar] [CrossRef] [PubMed]
- Kobe, M.J.; Neau, D.B.; Mitchell, C.E.; Bartlett, S.G.; Newcomer, M.E. The structure of human 15-lipoxygenase-2 with a substrate mimic. J. Biol. Chem. 2014, 289, 8562–8569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammel, M.; Walther, M.; Prassl, R.; Kuhn, H. Structural flexibility of the N-terminal beta-barrel domain of 15-lipoxygenase-1 probed by small angle X-ray scattering. Functional consequences for activity regulation and membrane binding. J. Mol. Biol. 2004, 343, 917–929. [Google Scholar] [CrossRef]
- Pace, C.N. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986, 131, 266–280. [Google Scholar] [CrossRef]
- Di Venere, A.; Horn, T.; Stehling, S.; Mei, G.; Masgrau, L.; González-Lafont, À.; Kuhn, H.; Ivanov, I. Role of Arg403 for thermostability and catalytic activity of rabbit 12/15-lipoxygenase. Biochim. Biophys. Acta 2013, 1831, 1079–1088. [Google Scholar] [CrossRef]
- Walther, M.; Roffeis, J.; Jansen, C.; Anton, M.; Ivanov, I.; Kuhn, H. Structural basis for pH-dependent alterations of reaction specificity of vertebrate lipoxygenase isoforms. Biochim. Biophys. Acta 2009, 1791, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Vogel, R.; Jansen, C.; Roffeis, J.; Reddanna, P.; Forsell, P.; Claesson, H.E.; Kuhn, H.; Walther, M. Applicability of the triad concept for the positional specificity of mammalian lipoxygenases. J. Biol. Chem. 2010, 285, 5369–5376. [Google Scholar] [CrossRef] [Green Version]
- Newcomer, M.E.; Brash, A.R. The structural basis for specificity in lipoxygenase catalysis. Protein Sci. 2015, 24, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, P.; Kaganer, I.; Heydeck, D.; Lluch, J.M.; Kuhn, H.; Gonzalez-Lafont, A. Mutagenesis of Sequence Determinants of Truncated Porcine ALOX15 Induces Changes in the Reaction Specificity by Altering the Catalytic Mechanism of Initial Hydrogen Abstraction. Chemistry 2018, 24, 962–973. [Google Scholar] [CrossRef]
- Saura, P.; Suardíaz, R.; Masgrau, L.; González-Lafont, À.; Rosta, E.; Lluch, J.M. Understanding the Molecular Mechanism of the Ala-versus-Gly Concept Controlling the Product Specificity in Reactions Catalyzed by Lipoxygenases: A Combined Molecular Dynamics and QM/MM Study of Coral 8R-Lipoxygenase. ACS Catal. 2017, 7, 4854–4866. [Google Scholar] [CrossRef] [Green Version]
- Saura, P.; Suardíaz, R.; Masgrau, L.; Lluch, J.M.; González-Lafont, À. Unraveling How Enzymes Can Use Bulky Residues To Drive Site-Selective C–H Activation: The Case of Mammalian Lipoxygenases Catalyzing Arachidonic Acid Oxidation. ACS Catal. 2014, 4351–4363. [Google Scholar] [CrossRef]
- Adel, S.; Karst, F.; González-Lafont, À.; Pekarova, M.; Saura, P.; Masgrau, L.; Lluch, J.M.; Stehling, S.; Horn, T.; Kuhn, H.; et al. Evolutionary alteration of ALOX15 specificity optimizes the biosynthesis of antiinflammatory and proresolving lipoxins. Proc. Natl. Acad. Sci. USA 2016, 113, E4266–E4275. [Google Scholar] [CrossRef] [Green Version]
- Gillmor, S.A.; Villasenor, A.; Fletterick, R.; Sigal, E.; Browner, M.F. The structure of mammalian 15-lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat. Struct. Biol. 1997, 4, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.C.; Gerstmeier, J.; Schexnaydre, E.E.; Börner, F.; Garscha, U.; Neau, D.B.; Werz, O.; Newcomer, M.E. Structural and mechanistic insights into 5-lipoxygenase inhibition by natural products. Nat. Chem. Biol. 2020, 16, 783–790. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pK(a)s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, I.T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosco, P. A mechanistic hypothesis for the aspirin-induced switch in lipid mediator production by cyclooxygenase-2. J. Am. Chem. Soc. 2013, 135, 10404–10410. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Golovanov, A.B.; Ferretti, C.; Canyelles-Niño, M.; Heydeck, D.; Stehling, S.; Lluch, J.M.; González-Lafont, À.; Kuhn, H. Mutations of Triad Determinants Changes the Substrate Alignment at the Catalytic Center of Human ALOX5. ACS Chem. Biol. 2019, 14, 2768–2782. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Le Grand, S.; Gotz, A.W.; Walker, R.C. SPFP: Speed without compromise-A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.R. Molecular Modeling: Principles and Applications; Eddison Wesley Longman Limited: Essex, UK, 1996; p. 595. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.; Ciccotti, G.; Berendsen, H. Numerical-integration of cartesian equations of motion of a system with contraints - molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | LA | AA | ||||
|---|---|---|---|---|---|---|
| kcat, s−1 | KM, µM | kcat/KM, s−1µM−1 | kcat, s−1 | KM, µM | kcat/KM, s−1µM−1 | |
| WT | 47.2 ± 2.8 | 21.4 ± 1.3 | 2.2 ± 0.1 | 11.3 ± 0. 6 | 8.1 ± 0.4 | 1.4 ± 0.2 |
| His585Glu | 21.2 ± 3.1 | 19.8 ± 2.9 | 1.1 ± 0.2 | 15.8 ± 0.9 | 4.9 ± 0.3 | 3.2 ± 0.1 |
| Trp181Glu | 39.8 ± 6.2 | 24.7 ± 3.9 | 1.6 ± 0.2 | 54.40 ± 2.01 | 6.6 ± 0.4 | 8.3 ± 0.51 |
| System | d(C13-OH) (Å) | d(H13proS-OH) (Å) | d(H13proR-OH) (Å) | Well-Oriented Structures (%) | Pre-Catalytic Structures With At least One H Well- Oriented (%) |
|---|---|---|---|---|---|
| Trp181Glu | 3.70 | 4.50 | 3.15 | 99.10 | 55.69 |
| WT | 5.19 | 4.85 | 5.47 | 97.78 | 3.40 |
| System | d(C11-OH) (Å) | d(H11proS-OH) (Å) | d(H11proR-OH) (Å) | Well-Oriented Structures (%) | Pre-Catalytic Structures With At least One H Well- Oriented (%) |
|---|---|---|---|---|---|
| Trp181Glu | 4.26 | 4.17 | 4.18 | 87.99 | 22.65 |
| WT | 4.58 | 4.74 | 4.56 | 90.09 | 14.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanov, I.; Cruz, A.; Zhuravlev, A.; Di Venere, A.; Nicolai, E.; Stehling, S.; Lluch, J.M.; González-Lafont, À.; Kuhn, H. Conformational Heterogeneity and Cooperative Effects of Mammalian ALOX15. Int. J. Mol. Sci. 2021, 22, 3285. https://doi.org/10.3390/ijms22063285
Ivanov I, Cruz A, Zhuravlev A, Di Venere A, Nicolai E, Stehling S, Lluch JM, González-Lafont À, Kuhn H. Conformational Heterogeneity and Cooperative Effects of Mammalian ALOX15. International Journal of Molecular Sciences. 2021; 22(6):3285. https://doi.org/10.3390/ijms22063285
Chicago/Turabian StyleIvanov, Igor, Alejandro Cruz, Alexander Zhuravlev, Almerinda Di Venere, Eleonora Nicolai, Sabine Stehling, José M. Lluch, Àngels González-Lafont, and Hartmut Kuhn. 2021. "Conformational Heterogeneity and Cooperative Effects of Mammalian ALOX15" International Journal of Molecular Sciences 22, no. 6: 3285. https://doi.org/10.3390/ijms22063285
APA StyleIvanov, I., Cruz, A., Zhuravlev, A., Di Venere, A., Nicolai, E., Stehling, S., Lluch, J. M., González-Lafont, À., & Kuhn, H. (2021). Conformational Heterogeneity and Cooperative Effects of Mammalian ALOX15. International Journal of Molecular Sciences, 22(6), 3285. https://doi.org/10.3390/ijms22063285