
Recent Advances in Oligonucleotide Therapeutics in Oncology



Abstract
:1. Introduction
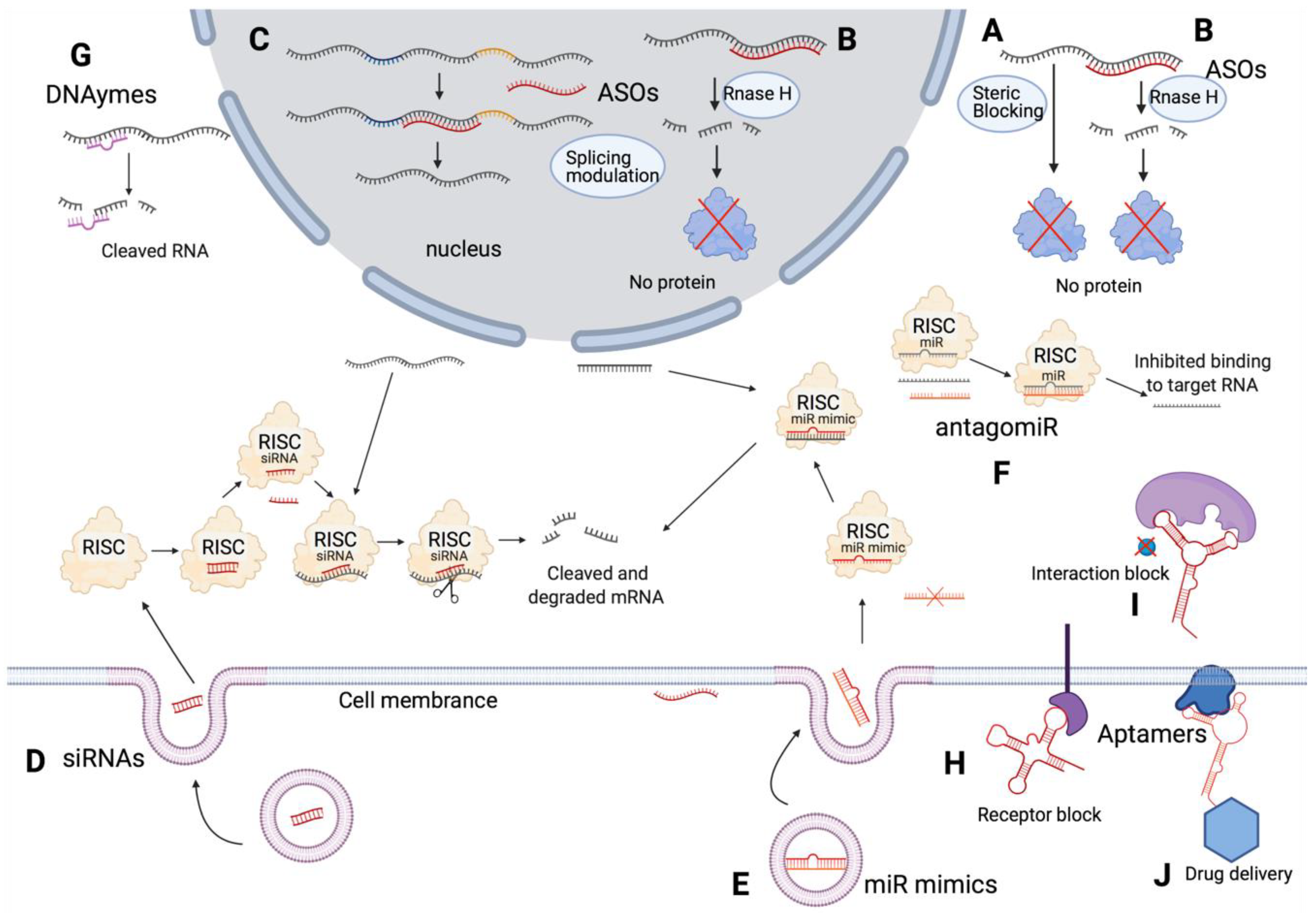
2. Types of Oligonucleotide Therapeutics
2.1. Antisense Oligonucleotides (ASOs)
2.2. CpG Oligonucleotides
2.3. miRNAs
2.4. siRNAs
2.5. Aptamers
2.6. DNAzymes
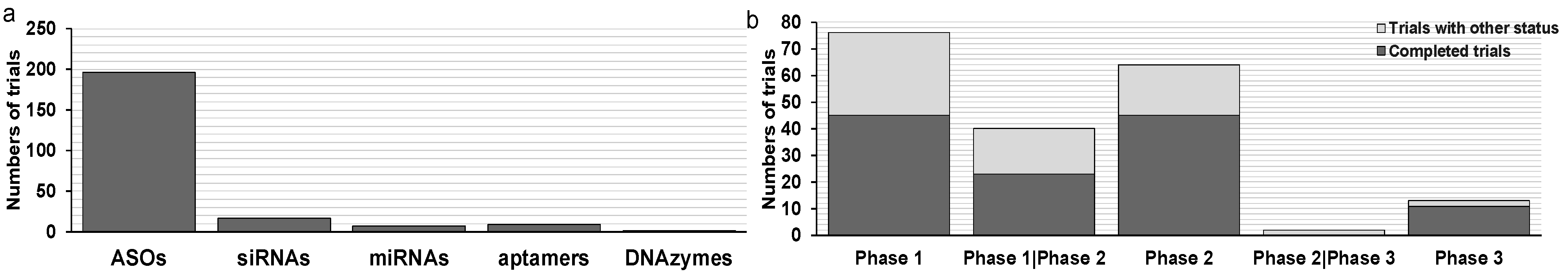
3. Clinical Trials for Oligonucleotide Therapeutics in Oncology
3.1. Clinical Trials for ASOs in Oncology
3.1.1. First ASO in Cancer Clinical Trials, G4460
3.1.2. ASOs in Phase 3 Trials
B-Cell Lymphoma 2 (BCL2)
Protein Kinase C Alpha (PKCα)
Transforming Growth Factor Beta (TGFβ)
Apolipoprotein J (ApoJ)
3.1.3. Additional ASOs in Phase 2 Trials
X-Linked Inhibitor of Apoptosis (XIAP)
Signal Transducer and Activator of Transcription 3 (STAT3)
Heat Shock Protein 27 (HSP27)
3.2. CpG Oligonucleotides
3.3. miRNAs
3.4. siRNAs
3.5. Aptamers
3.6. DNAzymes
4. Benefits of Oligonucleotide Therapeutics
5. Overcoming Challenges of Oligonucleotide Therapeutics
5.1. Drug Delivery Efficiency
5.2. Complexity of Cancer
5.3. Drug Interactions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASO | Antisense Oligonucleotide |
| PN | Phosphoramidate |
| PS | Phosphorothioate |
| MP | Methyl-phosphonate |
| OS | Overall survival |
| PFS | Progression-free survival |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Stewart, B.W.; Wild, C.P. World Cancer Report 2014; International Agency for Research on Cancer: Lyon, France, 2014. [Google Scholar]
- Brill, J.V. Screening for cancer: The economic, medical, and psychosocial issues. Am. J. Manag. Care 2020, 26, S300–S306. [Google Scholar]
- Qiao, J.; Liu, Z.; Fu, Y.-X. Adapting conventional cancer treatment for immunotherapy. J. Mol. Med. 2016, 94, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.-N.; Mei, Y.; Zhang, J. Cancer metastasis: Issues and challenges. Chin. J. Cancer 2017, 36, 38. [Google Scholar] [CrossRef] [PubMed]
- Editore, S. Management of Adverse Effects of Radiotherapy; SICS Editore: Milan, Italy, 2014. [Google Scholar]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef]
- Ribas, A.; Hu-Lieskovan, S. What does PD-L1 positive or negative mean? J. Exp. Med. 2016, 213, 2835–2840. [Google Scholar] [CrossRef]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.-R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Hou, B.; Tang, Y.; Li, W.; Zeng, Q.; Chang, D. Efficiency of CAR-T Therapy for Treatment of Solid Tumor in Clinical Trials: A Meta-Analysis. Dis. Markers 2019, 2019, 291. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Stacy, A.E.; Porter, G.M.; Merlot, A.M. Clinical development of targeted and immune based anti-cancer therapies. J. Exp. Clin. Cancer Res. 2019, 38, 156. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Lacouture, M.; Sibaud, V. Toxic Side Effects of Targeted Therapies and Immunotherapies Affecting the Skin, Oral Mucosa, Hair, and Nails. Am. J. Clin. Dermatol. 2018, 19, 31–39. [Google Scholar] [CrossRef]
- Mehta, M.; Deeksha, D.; Tewari, D.; Gupta, G.; Awasthi, R.; Singh, H.; Pandey, P.; Chellappan, D.K.; Wadhwa, R.; Collet, T.; et al. Oligonucleotide therapy: An emerging focus area for drug delivery in chronic inflammatory respiratory diseases. Chem. Biol. Interact. 2019, 308, 206–215. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, C.; Larcher, L.M.; Barrero, R.A.; Veedu, R.N. Three decades of nucleic acid aptamer technologies: Lessons learned, progress and opportunities on aptamer development. Biotechnol. Adv. 2019, 37, 28–50. [Google Scholar] [CrossRef] [PubMed]
- Lipi, F.; Chen, S.; Chakravarthy, M.; Rakesh, S.; Veedu, R.N. In vitro evolution of chemically-modified nucleic acid aptamers: Pros and cons, and comprehensive selection strategies. RNA Biol. 2016, 13, 1232–1245. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Corey, D.R. The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2020, 30, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Pescador, R.; Capuzzi, L.; Mantovani, M.; Fulgenzi, A.; Ferrero, M.E. Defibrotide: Properties and clinical use of an old/new drug. Vascul. Pharmacol. 2013, 59, 1–10. [Google Scholar] [CrossRef]
- Wan, J.; Bauman, J.A.; Graziewicz, M.A.; Sazani, P.; Kole, R. Oligonucleotide therapeutics in cancer. Cancer Treat. Res. 2013, 158, 213–233. [Google Scholar]
- Juliano, R.L.; Carver, K. Cellular uptake and intracellular trafficking of oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.-D. The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef] [PubMed]
- Deleavey, G.F.; Damha, M.J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. Res. 2020, 9, 4. [Google Scholar] [CrossRef]
- Thurston, D.E. Chemistry and Pharmacology of Anticancer Drugs; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Younis, H.S.; Templin, M.; Whiteley, L.O.; Kornbrust, D.; Kim, T.W.; Henry, S.P. Overview of the Nonclinical Development Strategies and Class-Effects of Oligonucleotide-Based Therapeutics. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development; Academic Press: Boston, MA, USA, 2017; pp. 737–754. [Google Scholar]
- Crooke, S.T.; Vickers, T.A.; Liang, X.-H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef]
- Le, B.T.; Raguraman, P.; Kosbar, T.R.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Antisense Oligonucleotides Targeting Angiogenic Factors as Potential Cancer Therapeutics. Mol. Ther. Nucleic Acids 2019, 14, 142–157. [Google Scholar] [CrossRef]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Østergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2019, 47, 1110–1122. [Google Scholar] [CrossRef]
- Frazier, K.S. Antisense oligonucleotide therapies: The promise and the challenges from a toxicologic pathologist’s perspective. Toxicol. Pathol. 2015, 43, 78–89. [Google Scholar] [CrossRef]
- Krieg, A.M.; Stein, C.A. Phosphorothioate oligodeoxynucleotides: Antisense or anti-protein? Antisense Res. Dev. 1995, 5, 241. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.-H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Miller, C.M.; Harris, E.N. Antisense Oligonucleotides: Treatment Strategies and Cellular Internalization. RNA Dis. 2016, 3, 93. [Google Scholar] [CrossRef]
- Faria, M.; Spiller, D.G.; Dubertret, C.; Nelson, J.S.; White, M.R.; Scherman, D.; Hélène, C.; Giovannangeli, C. Phosphoramidate oligonucleotides as potent antisense molecules in cells and in vivo. Nat. Biotechnol. 2001, 19, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Krisztina, P.S.G. Oligonucleotide N3′→P5′ thiophosphoramidates: Synthesis and properties. Tetrahedron Lett. 1999, 40, 7661–7664. [Google Scholar]
- Nair, P.R. Delivering Combination Chemotherapies and Targeting Oncogenic Pathways via Polymeric Drug Delivery Systems. Polymers 2019, 11, 630. [Google Scholar] [CrossRef]
- Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef]
- Pallan, P.S.; Egli, M. Insights into RNA/DNA hybrid recognition and processing by RNase H from the crystal structure of a non-specific enzyme-dsDNA complex. Cell Cycle 2008, 7, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Vitravene Study Group. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am. J. Ophthalmol. 2002, 133, 467–474. [Google Scholar]
- Prakash, T.P.; Kawasaki, A.M.; Wancewicz, E.V.; Shen, L.; Monia, B.P.; Ross, B.S.; Bhat, B.; Manoharan, M. Comparing in vitro and in vivo activity of 2′-O-[2-(methylamino)-2-oxoethyl]- and 2′-O-methoxyethyl-modified antisense oligonucleotides. J. Med. Chem. 2008, 51, 2766–2776. [Google Scholar] [CrossRef]
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acids Res. 2020, 48, 2853–2865. [Google Scholar] [CrossRef]
- Liang, X.-H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Braasch, D.A.; Corey, D.R. Locked nucleic acid (LNA): Fine-tuning the recognition of DNA and RNA. Chem. Biol. 2001, 8, 1–7. [Google Scholar] [CrossRef]
- Levin, J.D.; Fiala, D.; Samala, M.F.; Kahn, J.D.; Peterson, R.J. Position-dependent effects of locked nucleic acid (LNA) on DNA sequencing and PCR primers. Nucleic Acids Res. 2006, 34, e142. [Google Scholar] [CrossRef]
- Iwamoto, N.; Butler, D.C.D.; Svrzikapa, N.; Mohapatra, S.; Zlatev, I.; Sah, D.W.Y.; Meena, D.; Standley, S.M.; Lu, G.; Apponi, L.H.; et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Sardone, V.; Zhou, H.; Muntoni, F.; Ferlini, A.; Falzarano, M.S. Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease. Molecules 2017, 22, 563. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Ju, Y.; Park, H. A highly effective and long-lasting inhibition of miRNAs with PNA-based antisense oligonucleotides. Mol. Cells 2009, 28, 341–345. [Google Scholar] [CrossRef]
- Debacker, A.J.; Voutila, J.; Catley, M.; Blakey, D.; Habib, N. Delivery of oligonucleotides to the liver with GalNAc: From research to registered therapeutic drug. Mol. Ther. 2020, 28, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Wei, Z.; Wang, M.; Hoff, A.; Bao, Y.; Tian, L. Oligonucleotide-polymer conjugates: From molecular basics to practical application. Top. Curr. Chem. J. 2020, 378, 24. [Google Scholar] [CrossRef]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef] [PubMed]
- Schleimann, M.H.; Kobberø, M.-L.; Vibholm, L.K.; Kjær, K.; Giron, L.B.; Busman-Sahay, K.; Chan, C.N.; Nekorchuk, M.; Schmidt, M.; Wittig, B.; et al. TLR9 agonist MGN1703 enhances B cell differentiation and function in lymph nodes. EBioMedicine 2019, 45, 328–340. [Google Scholar] [CrossRef]
- Kang, T.H.; Mao, C.-P.; Kim, Y.S.; Kim, T.W.; Yang, A.; Lam, B.; Tseng, S.-H.; Farmer, E.; Park, Y.-M.; Hung, C.-F. TLR9 acts as a sensor for tumor-released DNA to modulate anti-tumor immunity after chemotherapy. J. Immunother. Cancer 2019, 7, 260. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, J.; Krieg, A.M. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv. Drug Deliv. Rev. 2009, 61, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Hager, S.; Fittler, F.J.; Wagner, E.; Bros, M. Nucleic Acid-Based Approaches for Tumor Therapy. Cells 2020, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Rothenfusser, S.; Hornung, V.; Jahrsdörfer, B.; Blackwell, S.; Ballas, Z.K.; Endres, S.; Krieg, A.M.; Hartmann, G. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur. J. Immunol. 2001, 31, 2154–2163. [Google Scholar] [CrossRef]
- Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef]
- Nehete, P.N.; Williams, L.E.; Chitta, S.; Nehete, B.P.; Patel, A.G.; Ramani, M.D.; Wisniewski, T.; Scholtzova, H. Class C CpG Oligodeoxynucleotide Immunomodulatory Response in Aged Squirrel Monkey. Front. Aging Neurosci. 2020, 12, 36. [Google Scholar] [CrossRef]
- Scheiermann, J.; Klinman, D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine 2014, 32, 6377–6389. [Google Scholar] [CrossRef]
- Macfarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Hammond, S.M. Dicing and slicing: The core machinery of the RNA interference pathway. FEBS Lett. 2005, 579, 5822–5829. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Barbarotto, E.; Schmittgen, T.D.; Calin, G.A. MicroRNAs and cancer: Profile, profile, profile. Int. J. Cancer 2008, 122, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Dong, C.; Ji, C. MicroRNA and drug resistance. Cancer Gene Ther. 2010, 17, 523–531. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Mahmoodi, C.G.; Dana, H.; Gharagouzloo, E.; Grijalvo, S.; Eritja, R.; Logsdon, C.D.; Memari, F.; Miri, S.R.; Rad, M.R.; Marmari, V. Small interfering RNAs (siRNAs) in cancer therapy: A nano-based approach. Int. J. Nanomed. 2019, 14, 3111–3128. [Google Scholar] [CrossRef] [PubMed]
- Chernikov, I.V.; Vlassov, V.V.; Chernolovskaya, E.L. Current development of siRNA bioconjugates: From research to the clinic. Front. Pharmacol. 2019, 10, 444. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.L.; Rossi, J.J.; Han, S.-P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Tatiparti, K.; Sau, S.; Kashaw, S.K.; Iyer, A.K. SiRNA delivery strategies: A comprehensive review of recent developments. Nanomaterials 2017, 7, 77. [Google Scholar] [CrossRef]
- Fu, Z.; Xiang, J. Aptamers, the Nucleic Acid Antibodies, in Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 2793. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F. Aptamers: A New Technological Platform in Cancer Immunotherapy. Pharmaceuticals 2016, 9, 64. [Google Scholar] [CrossRef]
- Han, J.; Gao, L.; Wang, J.; Wang, J. Application and development of aptamer in cancer: From clinical diagnosis to cancer therapy. J. Cancer 2020, 11, 6902–6915. [Google Scholar] [CrossRef] [PubMed]
- Kratschmer, C.; Levy, M. Effect of chemical modifications on aptamer stability in serum. Nucleic Acid Ther. 2017, 27, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical modifications of nucleic acid aptamers for therapeutic purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.P.; Hussain, B.; Yüce, M. Current Perspectives on Aptamers as Diagnostic Tools and Therapeutic Agents. Pharmaceutics 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- Morrow, P.K.; Murthy, R.K.; Ensor, J.D.; Gordon, G.S.; Margolin, K.A.; Elias, A.D.; Urba, W.J.; Weng, D.E.; Rugo, H.S.; Hortobagyi, G.N. An open-label, phase 2 trial of RPI.4610 (Angiozyme) in the treatment of metastatic breast cancer. Cancer 2012, 118, 4098–4104. [Google Scholar] [CrossRef]
- Silverman, S.K. Deoxyribozymes: DNA catalysts for bioorganic chemistry. Org. Biomol. Chem. 2004, 2, 2701–2706. [Google Scholar] [CrossRef]
- Huo, W.; Li, X.; Wang, B.; Zhang, H.; Zhang, J.; Yang, X.; Jin, Y. Recent advances of DNAzyme-based nanotherapeutic platform in cancer gene therapy. Biophys. Rep. 2020, 123, 1–10. [Google Scholar]
- Khachigian, L.M. Deoxyribozymes as Catalytic Nanotherapeutic Agents. Cancer Res. 2019, 79, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. RNA-Cleaving DNAzymes: Old Catalysts with New Tricks for Intracellular and In Vivo Applications. Catalysts 2018, 8, 550. [Google Scholar] [CrossRef]
- Schubert, S.; Gül, D.C.; Grunert, H.-P.; Zeichhardt, H.; Erdmann, V.A.; Kurreck, J. RNA cleaving “10-23” DNAzymes with enhanced stability and activity. Nucleic Acids Res. 2003, 31, 5982–5992. [Google Scholar] [CrossRef]
- Zhou, W.; Ding, J.; Liu, J. Theranostic DNAzymes. Theranostics 2017, 7, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Thesaurus, N.C.I. C-myb Antisense Oligonucleotide G4460 (Code C1541). Available online: https://ncithesaurus.nci.nih.gov/ncitbrowser/ConceptReport.jsp?dictionary=NCI_Thesaurus&ns=ncit&code=C1541 (accessed on 22 January 2021).
- Oh, I.H.; Reddy, E.P. The myb gene family in cell growth, differentiation and apoptosis. Oncogene 1999, 18, 3017–3033. [Google Scholar] [CrossRef] [PubMed]
- Fry, E.A.; Inoue, K. c-MYB and DMTF1 in Cancer. Cancer Investig. 2019, 37, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, G.; Gewirtz, A.M.; Calabretta, B. An oligomer complementary to c-myb-encoded mRNA inhibits proliferation of human myeloid leukemia cell lines. Proc. Natl. Acad. Sci. USA 1989, 86, 3379–3383. [Google Scholar] [CrossRef] [PubMed]
- Tondelli, L.; Ricca, A.; Laus, M.; Lelli, M.; Citro, G. Highly efficient cellular uptake of c-myb antisense oligonucleotides through specifically designed polymeric nanospheres. Nucleic Acids Res. 1998, 26, 5425–5431. [Google Scholar] [CrossRef]
- Luger, S.M.; O’Brien, S.G.; Ratajczak, J.; Ratajczak, M.Z.; Mick, R.; Stadtmauer, E.A.; Nowell, P.C.; Goldman, J.M.; Gewirtz, A.M. Oligodeoxynucleotide-mediated inhibition of c-myb gene expression in autografted bone marrow: A pilot study. Blood 2002, 99, 1150–1158. [Google Scholar] [CrossRef]
- Oblimersen, A. BCL-2 antisense oligonucleotide-Genta, G 3139, GC 3139, oblimersen sodium. Drugs R. 2007, 8, 321–334. [Google Scholar]
- Harris, M.H.; Thompson, C.B. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000, 7, 1182–1191. [Google Scholar] [CrossRef]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 8. [Google Scholar] [CrossRef]
- Frenzel, A.; Grespi, F.; Chmelewskij, W.; Villunger, A. Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis 2009, 14, 584–596. [Google Scholar] [CrossRef]
- O’Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Gregory, B.R.; Pavletic, S.; et al. Randomized Phase III Trial of Fludarabine Plus Cyclophosphamide With or Without Oblimersen Sodium (Bcl-2 antisense) in Patients With Relapsed or Refractory Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2007, 25, 1114–1120. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Rodrigueza, W.V.; Rasco, D.W.; Patnaik, A.; Papadopoulos, K.P.; Amaya, A.; Moore, T.D.; Gaylor, S.K.; Bisgaier, C.L.; Sooch, M.P.; et al. A phase 1 study of the BCL2-targeted deoxyribonucleic acid inhibitor (DNAi) PNT2258 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Harb, W.; Lakhani, N.J.; Logsdon, A.; Steigelman, M.; Smith-Green, H.; Woolliscroft, S.G.; Rodrigueza, W.V.; Sooch, M.P.; Messmann, R.A.; Al-Katib, A.M. Pharmacokinetics of the BCL2-targeted DNA interference (DNAi) nanoparticle PNT2258 in patients with recurrent or refractory non-Hodgkin lymphoma. J. Clin. Oncol. 2014, 32, 2586. [Google Scholar] [CrossRef]
- Dürig, J.; Dührsen, U.; Klein-Hitpass, L.; Worm, J.; Hansen, J.B.R.; Ørum, H.; Wissenbach, M. The novel antisense Bcl-2 inhibitor SPC2996 causes rapid leukemic cell clearance and immune activation in chronic lymphocytic leukemia. Leukemia 2011, 25, 638–647. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Amaya, M.; Strati, P.; Konopleva, M.Y. Venetoclax for AML: Changing the treatment paradigm. Blood Adv. 2019, 3, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef]
- Lahn, M.; Köhler, G.; Sundell, K.; Su, C.; Li, S.; Paterson, B.M.; Bumol, T.F. Protein kinase C alpha expression in breast and ovarian cancer. Oncology 2004, 67, 1–10. [Google Scholar] [CrossRef]
- Kazanietz, M.G. Protein Kinase C in Cancer Signaling and Therapy; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Pham, T.N.D.; Perez, W.B.E.; Zhao, H.; Mortazavi, F.; Tonetti, D.A. Protein kinase C α enhances migration of breast cancer cells through FOXC2-mediated repression of p120-catenin. BMC Cancer 2017, 17, 832. [Google Scholar] [CrossRef]
- Roychowdhury, D.; Lahn, M. Antisense therapy directed to protein kinase C-alpha (Affinitak, LY900003/ISIS 3521): Potential role in breast cancer. Semin. Oncol. 2003, 30, 30–33. [Google Scholar] [CrossRef]
- Villalona-Calero, M.A.; Ritch, P.; Figueroa, J.A.; Otterson, G.A.; Belt, R.; Dow, E.; George, S.; Leonardo, J.; McCachren, S.; Miller, G.L.; et al. A phase I/II study of LY900003, an antisense inhibitor of protein kinase C-alpha, in combination with cisplatin and gemcitabine in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 6086–6093. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Douillard, J.-Y.; Koralewski, P.; Manegold, C.; Smit, E.F.; Reyes, J.M.; Chang, G.-C.; John, W.J.; Peterson, P.M.; Obasaju, C.K.; et al. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J. Randomized phase III trial of chemotherapy and antisense oligonucleotide LY900003 (ISIS 3521) in patients with advanced NSCLC: Initial report. Am. Soc. Clin. Oncol. 2003, 22, 623. [Google Scholar]
- Haque, S.; Morris, J.C. Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccin. Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Schlingensiepen, K.-H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Schlingensiepen, K.-H.; Schlingensiepen, R.; Steinbrecher, A.; Hau, P.; Bogdahn, U.; Fischer-Blass, B.; Jachimczak, P. Targeted tumor therapy with the TGF-beta 2 antisense compound AP 12009. Cytokine Growth Factor Rev. 2006, 17, 129–139. [Google Scholar] [CrossRef]
- Vallières, L. Trabedersen, a TGFbeta2-specific antisense oligonucleotide for the treatment of malignant gliomas and other tumors overexpressing TGFbeta2. Drugs 2009, 12, 445–453. [Google Scholar]
- Hau, P.; Jachimczak, P.; Schlingensiepen, R.; Schulmeyer, F.; Jauch, T.; Steinbrecher, A.; Brawanski, A.; Proescholdt, M.; Schlaier, J.; Buchroithner, J.; et al. Inhibition of TGF-beta2 with AP 12009 in recurrent malignant gliomas: From preclinical to phase I/II studies. Oligonucleotides 2007, 17, 201–212. [Google Scholar] [CrossRef]
- Hau, P.; Jachimczak, P.; Bogdahn, U. Treatment of malignant gliomas with TGF-beta2 antisense oligonucleotides. Expert Rev. Anticancer Ther. 2009, 9, 1663–1674. [Google Scholar] [CrossRef]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro. Oncol. 2011, 13, 132–142. [Google Scholar] [CrossRef]
- The US Food and Drug Administration Trabedersen Orphan Drug Designations and Approvals. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=286709 (accessed on 23 March 2021).
- Trougakos, I.P.; Pawelec, G.; Tzavelas, C.; Ntouroupi, T.; Gonos, E.S. Clusterin/Apolipoprotein J up-regulation after zinc exposure, replicative senescence or differentiation of human haematopoietic cells. Biogerontology 2006, 7, 375–382. [Google Scholar] [CrossRef]
- Peng, M.; Deng, J.; Zhou, S.; Tao, T.; Su, Q.; Yang, X.; Yang, X. The role of Clusterin in cancer metastasis. Cancer Manag. Res. 2019, 11, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Zhao, H.; Liang, B.; Du, Q. Clusterin confers resistance to TNF-alpha-induced apoptosis in breast cancer cells through NF-kappaB activation and Bcl-2 overexpression. J. Chemother. 2012, 24, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Hotte, S.J.; Saad, F.; Alekseev, B.; Matveev, V.; Fléchon, A.; Gravis, G.; Joly, F.; Chi, K.N.; Malik, Z.; et al. Custirsen (OGX-011) combined with cabazitaxel and prednisone versus cabazitaxel and prednisone alone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel (AFFINITY): A randomised, open-label, international, phase 3 trial. Lancet Oncol. 2017, 18, 1532–1542. [Google Scholar]
- Saad, F.; Eisenberger, M.A. Management of Castration Resistant Prostate Cancer; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Chi, K.N.; Higano, C.S.; Blumenstein, B.; Ferrero, J.-M.; Reeves, J.; Feyerabend, S.; Gravis, G.; Merseburger, A.S.; Stenzl, A.; Bergman, A.M.; et al. Custirsen in combination with docetaxel and prednisone for patients with metastatic castration-resistant prostate cancer (SYNERGY trial): A phase 3, multicentre, open-label, randomised trial. Lancet Oncol. 2017, 18, 473–485. [Google Scholar] [CrossRef]
- Abbas, R.; Larisch, S. Targeting XIAP for Promoting Cancer Cell Death-The Story of ARTS and SMAC. Cells 2020, 9, 663. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.A.S.; Zee, B.C.-Y.; Cheung, F.Y.; Kwong, P.; Chiang, C.L.; Leung, K.C.; Siu, S.W.K.; Lee, C.; Lai, M.; Kwok, C.; et al. Randomized Phase II Study of the X-linked Inhibitor of Apoptosis (XIAP) Antisense AEG35156 in Combination With Sorafenib in Patients With Advanced Hepatocellular Carcinoma (HCC). Am. J. Clin. Oncol. 2016, 39, 609–613. [Google Scholar] [CrossRef]
- Kamran, M.Z.; Patil, P.; Gude, R.P. Role of STAT3 in cancer metastasis and translational advances. Biomed Res. Int. 2013, 2013, 421821. [Google Scholar] [CrossRef]
- Pham, T.-H.; Park, H.-M.; Kim, J.; Hong, J.T.; Yoon, D.-Y. STAT3 and p53: Dual Target for Cancer Therapy. Biomedicines 2020, 8, 637. [Google Scholar] [CrossRef]
- Xu, H.; Tong, X.; Mugundu, G.; Scott, M.L.; Cook, C.; Arfvidsson, C.; Pease, E.; Zhou, D.; Lyne, P.; Al-Huniti, N. Population pharmacokinetic analysis of danvatirsen supporting flat dosing switch. J. Pharmacokinet. Pharmacodyn. 2019, 46, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Burel, S.A.; Han, S.-R.; Lee, H.-S.; Norris, D.A.; Lee, B.-S.; Machemer, T.; Park, S.-Y.; Zhou, T.; He, G.; Kim, Y.; et al. Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucleic Acid Ther. 2013, 23, 213–227. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef]
- Choi, S.-K.; Kam, H.; Kim, K.-Y.; Park, S.I.; Lee, Y.-S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Sherman, M.Y.; Ciocca, D.R. Heat Shock Proteins in Cancer; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2007; ISBN 9781402064012. [Google Scholar]
- Lelj-Garolla, B.; Kumano, M.; Beraldi, E.; Nappi, L.; Rocchi, P.; Ionescu, D.N.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Hsp27 Inhibition with OGX-427 Sensitizes Non-Small Cell Lung Cancer Cells to Erlotinib and Chemotherapy. Mol. Cancer Ther. 2015, 14, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Shipley, D.L.; Waterhouse, D.M.; Jones, S.F.; Ward, P.J.; Shih, K.C.; Hemphill, B.; McCleod, M.; Whorf, R.C.; Page, R.D.; et al. A Randomized, Double-Blinded, Phase II Trial of Carboplatin and Pemetrexed with or without Apatorsen (OGX-427) in Patients with Previously Untreated Stage IV Non-Squamous-Non-Small-Cell Lung Cancer: The SPRUCE Trial. Oncologist 2019, 24, e1409–e1416. [Google Scholar] [CrossRef]
- Yu, E.Y.; Ellard, S.L.; Hotte, S.J.; Gingerich, J.R.; Joshua, A.M.; Gleave, M.E.; Chi, K.N. A randomized phase 2 study of a HSP27 targeting antisense, apatorsen with prednisone versus prednisone alone, in patients with metastatic castration resistant prostate cancer. Investig. New Drugs 2018, 36, 278–287. [Google Scholar] [CrossRef]
- Ko, A.H.; Murphy, P.B.; Peyton, J.D.; Shipley, D.L.; Al-Hazzouri, A.; Rodriguez, F.A.; Womack, M.S.; Xiong, H.Q.; Waterhouse, D.M.; Tempero, M.A.; et al. A Randomized, Double-Blinded, Phase II Trial of Gemcitabine and Nab-Paclitaxel Plus Apatorsen or Placebo in Patients with Metastatic Pancreatic Cancer: The RAINIER Trial. Oncologist 2017, 22, 1427–1429. [Google Scholar] [CrossRef]
- Miroshnichenko, S.K.; Patutina, O.A.; Burakova, E.A.; Chelobanov, B.P.; Fokina, A.A.; Vlassov, V.V.; Altman, S.; Zenkova, M.A.; Stetsenko, D.A. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc. Natl. Acad. Sci. USA 2019, 116, 1229–1234. [Google Scholar] [CrossRef]
- Zent, C.S.; Smith, B.J.; Ballas, Z.K.; Wooldridge, J.E.; Link, B.K.; Call, T.G.; Shanafelt, T.D.; Bowen, D.A.; Kay, N.E.; Witzig, T.E.; et al. Phase I clinical trial of CpG oligonucleotide 7909 (PF-03512676) in patients with previously treated chronic lymphocytic leukemia. Leuk. Lymphoma 2012, 53, 211–217. [Google Scholar] [CrossRef]
- Manegold, C.; van Zandwijk, N.; Szczesna, A.; Zatloukal, P.; Au, J.S.K.; Blasinska-Morawiec, M.; Serwatowski, P.; Krzakowski, M.; Jassem, J.; Tan, E.H.; et al. A phase III randomized study of gemcitabine and cisplatin with or without PF-3512676 (TLR9 agonist) as first-line treatment of advanced non-small-cell lung cancer. Ann. Oncol. 2012, 23, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, V.; Paz-Ares, L.; Boyer, M.; Rosell, R.; Middleton, G.; Eberhardt, W.E.E.; Szczesna, A.; Reiterer, P.; Saleh, M.; Arrieta, O.; et al. Randomized phase III trial of paclitaxel/carboplatin with or without PF-3512676 (Toll-like receptor 9 agonist) as first-line treatment for advanced non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Zarour, H.; Redman, B.; Trefzer, U.; O’Day, S.; van den Eertwegh, A.J.M.; Marshall, E.; Wagner, S. Randomized phase 2/3 trial of CpG oligodeoxynucleotide PF-3512676 alone or with dacarbazine for patients with unresectable stage III and IV melanoma. Cancer 2009, 115, 3944–3954. [Google Scholar] [CrossRef]
- Ruzsa, A.; Sen, M.; Evans, M.; Lee, L.W.; Hideghety, K.; Rottey, S.; Klimak, P.; Holeckova, P.; Fayette, J.; Csoszi, T.; et al. Phase 2, open-label, 1:1 randomized controlled trial exploring the efficacy of EMD 1201081 in combination with cetuximab in second-line cetuximab-naïve patients with recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN). Investig. New Drugs 2014, 32, 1278–1284. [Google Scholar] [CrossRef]
- Friedberg, J.W.; Kim, H.; McCauley, M.; Hessel, E.M.; Sims, P.; Fisher, D.C.; Nadler, L.M.; Coffman, R.L.; Freedman, A.S. Combination immunotherapy with a CpG oligonucleotide (1018 ISS) and rituximab in patients with non-Hodgkin lymphoma: Increased interferon-alpha/beta-inducible gene expression, without significant toxicity. Blood 2005, 105, 489–495. [Google Scholar] [CrossRef]
- Ribas, A.; Medina, T.; Kummar, S.; Amin, A.; Kalbasi, A.; Drabick, J.J.; Barve, M.; Daniels, G.A.; Wong, D.J.; Schmidt, E.V.; et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov. 2018, 8, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, E.; Neveu, B.; Kostantin, E.; Tsongalis, G.J.; De Guire, V. How close are miRNAs from clinical practice? A perspective on the diagnostic and therapeutic market. Ejifcc 2019, 30, 114–127. [Google Scholar]
- Foss, F.M.; Querfeld, C.; Porcu, P.; Kim, Y.H.; Pacheco, T.; Halwani, A.S.; DeSimone, J.; William, B.M.; Seto, A.G.; Ruckman, J.; et al. Phase 1 trial evaluating MRG-106, a synthetic inhibitor of microRNA-155, in patients with cutaneous t-cell lymphoma (CTCL). J. Clin. Oncol. 2017, 35, 7564. [Google Scholar] [CrossRef]
- Reid, G.; Pel, M.E.; Kirschner, M.B.; Cheng, Y.Y.; Mugridge, N.; Weiss, J.; Williams, M.; Wright, C.; Edelman, J.J.B.; Vallely, M.P.; et al. Restoring expression of miR-16: A novel approach to therapy for malignant pleural mesothelioma. Ann. Oncol. 2013, 24, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Varghese, A.M.; Ang, C.; Dimaio, C.J.; Javle, M.M.; Gutierrez, M.; Yarom, N.; Stemmer, S.M.; Golan, T.; Geva, R.; Semenisty, V.; et al. A phase II study of siG12D-LODER in combination with chemotherapy in patients with locally advanced pancreatic cancer (PROTACT). J. Clin. Orthod. 2020, 38, 4672. [Google Scholar] [CrossRef]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Oncol. 2015, 23, 11006. [Google Scholar] [CrossRef]
- Dannull, J.; Haley, N.R.; Archer, G.; Nair, S.; Boczkowski, D.; Harper, M.; De Rosa, N.; Pickett, N.; Mosca, P.J.; Burchette, J.; et al. Melanoma immunotherapy using mature DCs expressing the constitutive proteasome. J. Clin. Investig. 2013, 123, 3135–3145. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Bambury, R.M.; Van Allen, E.M.; Drabkin, H.A.; Lara, P.N., Jr.; Harzstark, A.L.; Wagle, N.; Figlin, R.A.; Smith, G.W.; Garraway, L.A.; et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Investig. New Drugs 2014, 32, 178–187. [Google Scholar] [CrossRef]
- Steurer, M.; Montillo, M.; Scarfò, L.; Mauro, F.R.; Andel, J.; Wildner, S.; Trentin, L.; Janssens, A.; Burgstaller, S.; Frömming, A.; et al. Olaptesed pegol (NOX-A12) with bendamustine and rituximab: A phase IIa study in patients with relapsed/refractory chronic lymphocytic leukemia. Haematologica 2019, 104, 2053–2060. [Google Scholar] [CrossRef]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef]
- Liao, W.-H.; Yang, L.-F.; Liu, X.-Y.; Zhou, G.-F.; Jiang, W.-Z.; Hou, B.-L.; Sun, L.-Q.; Cao, Y.; Wang, X.-Y. DCE-MRI assessment of the effect of Epstein-Barr virus-encoded latent membrane protein-1 targeted DNAzyme on tumor vasculature in patients with nasopharyngeal carcinomas. BMC Cancer 2014, 14, 835. [Google Scholar] [CrossRef]
- Cho, E.-A.; Moloney, F.J.; Cai, H.; Au-Yeung, A.; China, C.; Scolyer, R.A.; Yosufi, B.; Raftery, M.J.; Deng, J.Z.; Morton, S.W.; et al. Safety and tolerability of an intratumorally injected DNAzyme, Dz13, in patients with nodular basal-cell carcinoma: A phase 1 first-in-human trial (DISCOVER). Lancet 2013, 381, 1835–1843. [Google Scholar] [CrossRef]
- Dou, Y.; Jiang, X.; Xie, H.; He, J.; Xiao, S. The Jun N-terminal kinases signaling pathway plays a “seesaw” role in ovarian carcinoma: A molecular aspect. J. Ovarian Res. 2019, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Fokina, A.A.; Stetsenko, D.A.; François, J.-C. DNA enzymes as potential therapeutics: Towards clinical application of 10-23 DNAzymes. Expert Opin. Biol. Ther. 2015, 15, 689–711. [Google Scholar] [CrossRef]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the “undruggable” cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Krichevsky, A.M.; Uhlmann, E.J. Oligonucleotide Therapeutics as a New Class of Drugs for Malignant Brain Tumors: Targeting mRNAs, Regulatory RNAs, Mutations, Combinations, and Beyond. Neurotherapeutics 2019, 16, 319–347. [Google Scholar] [CrossRef] [PubMed]
- Milella, M.; Estrov, Z.; Kornblau, S.M.; Carter, B.Z.; Konopleva, M.; Tari, A.; Schober, W.D.; Harris, D.; Leysath, C.E.; Lopez-Berestein, G.; et al. Synergistic induction of apoptosis by simultaneous disruption of the Bcl-2 and MEK/MAPK pathways in acute myelogenous leukemia. Blood 2002, 99, 3461–3464. [Google Scholar] [CrossRef]
- Elez, R.; Piiper, A.; Kronenberger, B.; Kock, M.; Brendel, M.; Hermann, E.; Pliquett, U.; Neumann, E.; Zeuzem, S. Tumor regression by combination antisense therapy against Plk1 and Bcl-2. Oncogene 2003, 22, 69–80. [Google Scholar] [CrossRef]
- Sharma, V.K.; Watts, J.K. Oligonucleotide therapeutics: Chemistry, delivery and clinical progress. Future Med. Chem. 2015, 7, 2221–2242. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef]
- The US FDA Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-cancer-drugs-and-biologics (accessed on 23 March 2021).
- Gandara, D.; Narayan, S.; Lara, P.N., Jr.; Goldberg, Z.; Davies, A.; Lau, D.H.M.; Mack, P.; Gumerlock, P.; Vijayakumar, S. Integration of novel therapeutics into combined modality therapy of locally advanced non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 5057s–5062s. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Xia, S.; Yu, R.Z.; Viney, N.J.; Wang, Y.; Tsimikas, S.; Geary, R.S. Integrated Assessment of the Clinical Performance of GalNAc-Conjugated 2′-O-Methoxyethyl Chimeric Antisense Oligonucleotides: I. Human Volunteer Experience. Nucleic Acid Ther. 2019, 29, 16–32. [Google Scholar] [CrossRef]
- Richard, M.J.; John, H.C. Designing Multi-Target Drugs; Royal Society of Chemistry: London, UK, 2012; ISBN 9781849734912. [Google Scholar]
- Xie, L.; Bourne, P.E. Developing multi-target therapeutics to fine-tune the evolutionary dynamics of the cancer ecosystem. Front. Pharmacol. 2015, 6, 209. [Google Scholar] [CrossRef]
- Adjiri, A. DNA Mutations May Not Be the Cause of Cancer. Oncol. Ther. 2017, 5, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboezem, E.N.; Duvall, C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev. 2018, 130, 73–89. [Google Scholar] [CrossRef]
- Bijsterbosch, M.K.; Rump, E.T.; De Vrueh, R.L.; Dorland, R.; van Veghel, R.; Tivel, K.L.; Biessen, E.A.; van Berkel, T.J.; Manoharan, M. Modulation of plasma protein binding and in vivo liver cell uptake of phosphorothioate oligodeoxynucleotides by cholesterol conjugation. Nucleic Acids Res. 2000, 28, 2717–2725. [Google Scholar] [CrossRef]
- Kilanowska, A.; Studzińska, S. In vivo and in vitro studies of antisense oligonucleotides–a review. RSC Adv. 2020, 10, 34501–34516. [Google Scholar] [CrossRef]
- Sparreboom, A.; Loos, W.J. Protein binding of anticancer drugs. In Handbook of Anticancer Pharmacokinetics and Pharmacodynamics; Springer: Berlin/Heidelberg, Germany, 2004; pp. 169–188. [Google Scholar]
- Schneider, E.K.; Huang, J.X.; Carbone, V.; Baker, M.; Azad, M.A.K.; Cooper, M.A.; Li, J.; Velkov, T. Drug-drug plasma protein binding interactions of ivacaftor. J. Mol. Recognit. 2015, 28, 339–348. [Google Scholar] [CrossRef]
- Lee, J.H.; Nan, A. Combination drug delivery approaches in metastatic breast cancer. J. Drug Deliv. 2012, 2012, 915375. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug Names | Market Names | Companies | FDA Approved | Indications | Drug Modality | Mechanisms | Targets |
|---|---|---|---|---|---|---|---|
| fomivirsen | Vitravene | Ionis Pharmaceuticals, Novartis | 1998 | Cytomegalovirus (CMV) retinitis | ASO | Translation block | CMV protein IE2 |
| pegaptanib | Macugen | OSI Pharmaceuticals | 2004 | Neovascular age-related macular degeneration | Aptamer | Binding and blocking | Heparin-binding domain of VEGF-165 |
| mipomersen | Kynamro | Kastle Therapeutics, Ionis Pharmaceuticals, Genzyme | 2013 | Homozygous familial hypercholesterolemia | ASO | RNase H degradation | Apolipoprotein B100 |
| eteplirsen | Exondys 51 | Sarepta Therapeutics | 2016 | Duchenne muscular dystrophy | ASO | Splicing modulation | Exon 51 of DMD |
| nusinersen | Spinraza | Ionis Pharmaceuticals, Biogen | 2016 | Spinal muscular atrophy | ASO | Splicing modulation | Exon 7 of SMN2 |
| defibrotide | Defitelio | Jazz Pharmaceuticals | 2016 | Veno-occlusive disease in liver | Aptamer | Binding and activating | Adenosine A1/A2 receptor |
| inotersen | Tegsedi | Akcea Therapeutics | 2018 | Polyneuropathy caused by hereditary transthyretin-mediated (hATTR) amyloidosis | ASO | RNase H degradation | Transthyretin |
| milasen | Not applicable | Boston Children’s Hospital | 2018 | Mila Makovec’s CLN7 gene associated with Batten disease | ASO | Splicing modulation | CLN7 |
| patisiran | Onpattro | Alnylam | 2018 | Polyneuropathy caused by hATTR amyloidosis | siRNA | RNAi | Transthyretin |
| golodirsen | Vyondys 53 | Sarepta Therapeutics | 2019 | Duchenne muscular dystrophy | ASO | Splicing modulation | Exon 53 of DMD |
| givosiran | Givlaari | Alnylam | 2019 | Acute hepatic porphyria (AHP) | siRNA | RNAi | 5-aminolevulinic acid synthase |
| volanesorsen | Waylivra | Akcea Therapeutics | EMA approved in 2019 1 | Familial chylomicronemia syndrome (FCS) | ASO | RNase H degradation | Apolipoprotein C3 |
| viltolarsen | Viltepso | NS Pharma | 2020 | Duchenne muscular dystrophy | ASO | Splicing modulation | Exon 53 of DMD |
| casimersen | Amondys 45 | Sarepta Therapeutics | 2021 | Duchenne muscular dystrophy | ASO | Splicing modulation | Exon 45 of DMD |
| Oligonucleotide Therapeutics | Target | Drug Modality | Cancer Types | Clinical Trials |
|---|---|---|---|---|
| DNAzyme targeting EBV-LMP1 (DZ1) | EBV-LMP1 | DNAzyme | Nasopharyngeal Cancer | NCT01449942 |
| 68Ga-Sgc8 | PTK7 | aptamer | Colorectal Cancer | NCT03385148 |
| EYE001 (Anti-VEGF Pegylated Aptamer) | VEGF | aptamer | Retinal Cancer | NCT00056199 |
| NOX-A12 | CXCL12 | aptamer | Pancreatic Cancer|Colorectal Cancer|Myeloma|Leukemia | NCT01521533|NCT01521533|NCT03168139 |
| AS1411 | NCL | aptamer | Acute Myeloid Leukemia | NCT01034410|NCT00881244|NCT00740441|NCT00512083 |
| KRAS G12D siRNA | KRASG12D | siRNA | Pancreatic Cancer | NCT03608631 |
| EphA2-targeting DOPC-encapsulated siRNA | EPHA2 | siRNA | Solid Tumors | NCT01591356 |
| APN401 | CBLB | siRNA | Brain Cancer|Melanoma|Pancreatic Cancer|Renal Cell Cancer | NCT03087591|NCT02166255 |
| Proteasome siRNA and tumor antigen RNA-transfected dendritic cells | LMP2, LMP7, MECL1 | siRNA | Melanoma | NCT00672542 |
| TKM-080301 | PLK1 | siRNA | Cancer with hepatic metastases| Liver Cancer|Hepatocellular Cancer| Adrenocortical Cancer | NCT01437007|NCT02191878|NCT01262235 |
| Atu027 | PNK3 | siRNA | Solid Tumors|Pancreatic Cancer | NCT00938574|NCT01808638 |
| DCR-MYC | MYC | siRNA | Solid Tumors|Hepatocellular Cancer | NCT02110563|NCT02314052 |
| CALAA-01 | M2 subunit of ribonucleotide reductase (R2) | siRNA | Solid Tumors | NCT00689065 |
| siG12D LODER | KRASG12D | siRNA | Pancreatic Cancer | NCT01676259|NCT01188785 |
| ARO-HIF2 | HIF2A | siRNA | Clear Cell Renal Cell Carcinoma | NCT04169711 |
| SV40 vectors carrying siRNA | Unknown | siRNA | Chronic Myeloid Leukemia | NCT00257647 |
| MRX34 | 30 unique oncogenes, including but not limited to MET, MYC, PDGFRA, CDK4/6 and BCL2 | miRNA | Liver Cancer|Lung Cancer |Lymphoma |Melanoma|Multiple Myeloma|Renal Cell Cancer| | NCT01829971|NCT02862145 |
| INT-1B3 | JNK1 | miRNA | Solid Tumor | NCT04675996 |
| TargomiRs | Multiple oncogenes, including BCL2, MCL1, CCND1, and WNT3A | miRNA | Malignant Pleural Mesothelioma|Non-Small Cell Lung Cancer | NCT02369198 |
| Cobomarsen (MRG-106) | MIR155 | miRNA | Cutaneous T-Cell Lymphoma|Lymphoma|Leukemia | NCT03837457|NCT03713320|NCT02580552 |
| 1018 ISS | TLR9 | ASO | Non-Hodgkin’s Lymphoma|Colorectal Cancer | NCT00251394|NCT00403052 |
| AEG35156 | XIAP | ASO | Hepatocellular Cancer|Pancreatic Cancer|Breast Cancer|Non-Small Cell Lung Cancer| Leukemia|Lymphoma | NCT00357747|NCT00363974|NCT00372736|NCT00385775|NCT00557596|NCT00558545|NCT00558922|NCT00768339|NCT00882869|NCT01018069 |
| Apatorsen (OGX-427) | HSP27 | ASO | Urologic Cancer|Bladder Cancer|Prostate Cancer|Urothelial Cancer|Non-Small Cell Lung Cancer | NCT00487786|NCT01454089|NCT01681433|NCT01780545|NCT01829113 |
| ARRx (AZD5312) | AR | ASO | Prostate Cancer | NCT02144051|NCT03300505 |
| AZD4785 | KRAS | ASO | Non-Small Cell Lung Cancer | NCT03101839 |
| AZD8701 | FOXP3 | ASO | Advanced Cancer | NCT04504669 |
| AZD9150 | STAT3 | ASO | Bladder Cancer|Lymphoma|Malignancies | NCT02546661|NCT02549651|NCT03394144|NCT03527147|NCT03819465 |
| BP1001 | GRB2 | ASO | Ph1 Positive Leukemia|Acute Myeloid Leukemia|Chronic Myelogenous Leukemia | NCT01159028|NCT02781883|NCT02923986|NCT04196257 |
| Cenersen (EL625) | TP53 | ASO | Acute Myelogenous Leukemia|Lymphoma | NCT00074737|NCT00636155|NCT00967512|NCT02243124 |
| CpG 7909 (PF03512676) | TLR9 | ASO | Melanoma|Breast Cancer|Renal Cancer|Lymphoma|Non-Small Cell Lung Cancer|Esophageal Cancer|Prostate Cancer | NCT00031278|NCT00040950|NCT00043368|NCT00043394|NCT00043407|NCT00043420|NCT00070629|NCT00070642|NCT00085189|NCT00112242|NCT00145145|NCT00185965|NCT00199836|NCT00226993|NCT00233506|NCT00292045|NCT00299728|NCT00369291|NCT00438880|NCT00471471|NCT00490529|NCT00669292|NCT00819806|NCT00824733|NCT00880581|NCT01266603|NCT01588015 |
| CpG ODN (GNKG168) | TLR9 | ASO | Leukemia | NCT01035216|NCT01743807 |
| CpG Oligonucleotide 1 | TLR9 | ASO | Breast Cancer | NCT00640861 |
| CpG-ODN 1 | TLR9 | ASO | Glioblastoma | NCT00190424 |
| Custirsen (OGX-011) | ApoJ | ASO | Prostate Cancer|Breast Cancer|Non-Small Cell Lung Cancer | NCT00054106|NCT00138658|NCT00138918|NCT00258375|NCT00258388|NCT00327340|NCT00471432|NCT01083615|NCT01188187|NCT01497470|NCT01578655|NCT01630733 |
| Danvatirsen (AZD9150, ISIS STAT3Rx) | STAT3 | ASO | Advanced Cancers | NCT01563302|NCT01839604|NCT02417753|NCT02417753|NCT02499328|NCT02983578|NCT03334617 |
| EGFR Antisense DNA | EGFR | ASO | Head and Neck Squamous Cell Cancer|Gastric Cancer|Ovarian Cancer|Prostate Cancer | NCT00009841|NCT00023634|NCT00903461|NCT01592721|NCT03433027 |
| EZN-2968 (RO7070179,SPC2968) | HIF1A | ASO | Hepatocellular Cancer|Lymphoma | NCT00466583|NCT01120288|NCT02564614|NCT00466583 |
| G4460 | CMYB | ASO | Leukemia| Hematologic Malignancies | NCT00002592| NCT00780052 |
| IGF-1R/AS ODN | IGF1 | ASO | Glioma | NCT01550523|NCT02507583 |
| IGV-001 containing autologous GBM cells treated with antisense oligonucleotide (IMV-001) | IGF1R | ASO | Glioblastoma | NCT04485949 |
| IMO-2055 (EMD 1201081) | TLR9 | ASO | Renal Cell Cancer|Colorectal Cancer|Non-Small Cell Lung Cancer|Head and Neck Cancer | NCT00729053|NCT01040832|NCT00633529|NCT00719199|NCT01360827 |
| ION251 | IRF4 | ASO | Myeloma | NCT04398485 |
| ION537 | YAP1 | ASO | Advanced Solid Tumors | NCT04659096 |
| ISIS 183750(ISIS-EIF4ERx, LY2275796) | EIF4E | ASO | Castrate-Resistant Prostate Cancer|Non-Small Cell Lung Cancer|Colorectal Cancer | NCT00903708|NCT01234025|NCT01234038|NCT01675128 |
| ISIS 2503 | HRAS | ASO | Colorectal Cancer|Pancreatic Cancer | NCT00004193|NCT00005594|NCT00006467 |
| ISIS 5132 | CRAF | ASO | Ovarian Cancer | NCT00003892 |
| L-Bcl-2 antisense oligonucleotide | BCL2 | ASO | Advanced Lymphoid Malignancies | NCT04072458 |
| LErafAON | CRAF | ASO | Cancers | NCT00024648|NCT00024661|NCT00100672 |
| Lucanix | TGFB2 | ASO | Non-small Cell Lung Cancer | NCT01058785|NCT01279798 |
| LY2181308 | BIRC5 | ASO | Non-small Cell Lung Cancer | NCT01107444 |
| LY900003 (ISIS 3521, Affinitak) | PKCA | ASO | Melanoma|Lung Cancer|Non-Small Cell Lung Cancer|Breast Cancer | NCT00003989|NCT00017407|NCT00034268|NCT00042679|NCT00042679|NCT00003236 |
| MTL-CEBPA | CEBPA | ASO | Hepatocellular Cancer | NCT02716012|NCT04105335|NCT04710641 |
| Oblimersen (G3139) | BCL2 | ASO | Cancers | NCT00003103|NCT00004862|NCT00004870|NCT00005032|NCT00016263|NCT00017251|NCT00017589|NCT00017602|NCT00021749|NCT00024440|NCT00030641|NCT00039117|NCT00039481|NCT00042978|NCT00047229|NCT00049192|NCT00049374|NCT00054548|NCT00054639|NCT00055822|NCT00059813|NCT00060112|NCT00062244|NCT00063934|NCT00064259|NCT00070083|NCT00070343|NCT00078234|NCT00079131|NCT00080847|NCT00085124|NCT00085228|NCT00086944|NCT00091078|NCT00301795|NCT00409383|NCT00517218|NCT00518895|NCT00542893|NCT00543205|NCT00543231|NCT00636545|NCT00736450|NCT01200342 |
| OGX-427 | HSP27 | ASO | Cancers | NCT00487786|NCT00959868|NCT01120470|NCT01844817|NCT02423590 |
| PNT2258 | BCL2 | ASO | Prostate Cancer|Lymphoma|Melanoma | NCT01191775|NCT01733238|NCT02226965 |
| SPC2996 | BCL2 | ASO | Chronic Lymphocytic Leukemia | NCT00285103 |
| TGFβ2 Antisense-GMCSF Gene Modified Autologous Tumor Cell (TAG) Vaccine | TGFB2 | ASO | Advanced Cancer | NCT00684294 |
| SD-101 | TLR9 | ASO | Cancers | NCT01042379|NCT01745354|NCT02254772|NCT02266147|NCT02521870|NCT02731742|NCT02927964|NCT03007732|NCT03322384|NCT03410901|NCT04050085|NCT03831295 |
| Trabedersen (AP 12009, OT-101) | TGFB2 | ASO | Glioblastoma|Anaplastic Astrocytoma|Pancreatic Cancer|Melanoma|Colorectal Cancer | NCT00431561|NCT00761280|NCT00844064 |
| VEGF-Antisense Oligonucleotide | VEGF | ASO | Mesothelioma | NCT00668499 |
| ASO Modification | Modification Type | Nuclease Resistance | RNAse H Cleavage | Target Affinity | Toxicity |
|---|---|---|---|---|---|
| Phosphorothioate (PS) | Phosphate | + | Yes | - | ++ |
| Phosphoramidate (NP) | Phosphate | + | No | + | + |
| Methyl-phosphonate (MP) | Phosphate | + | No | - | + |
| 2′-O-methyl (2′-OMe) | Ribose | ++ | No | ++ | + Reduced immune activation; less toxic than PS |
| 2′-O-methoxyethyl (2′-MOE) | Ribose | ++ | No | ++ | + Reduced immune activation; Less than 2′-OMe; less toxic than PS |
| Phosphorodiamidate morpholino (PMO) | Ribose phosphate | ++ | No | ++ | Safer |
| Locked nucleic acid (LNA) | Ribose | ++ | No | +++ | +++ |
| Constrained ethyl (cEt) | Ribose | +++ | No | +++ | ++ |
| Peptide nucleic acid (PNA) | Ribose phosphate | ++ | No | ++ | ++ No immune activation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. https://doi.org/10.3390/ijms22073295
Xiong H, Veedu RN, Diermeier SD. Recent Advances in Oligonucleotide Therapeutics in Oncology. International Journal of Molecular Sciences. 2021; 22(7):3295. https://doi.org/10.3390/ijms22073295
Chicago/Turabian StyleXiong, Haoyu, Rakesh N. Veedu, and Sarah D. Diermeier. 2021. "Recent Advances in Oligonucleotide Therapeutics in Oncology" International Journal of Molecular Sciences 22, no. 7: 3295. https://doi.org/10.3390/ijms22073295
APA StyleXiong, H., Veedu, R. N., & Diermeier, S. D. (2021). Recent Advances in Oligonucleotide Therapeutics in Oncology. International Journal of Molecular Sciences, 22(7), 3295. https://doi.org/10.3390/ijms22073295