
Deficiency of Tristetraprolin Triggers Hyperthermia through Enhancing Hypothalamic Inflammation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
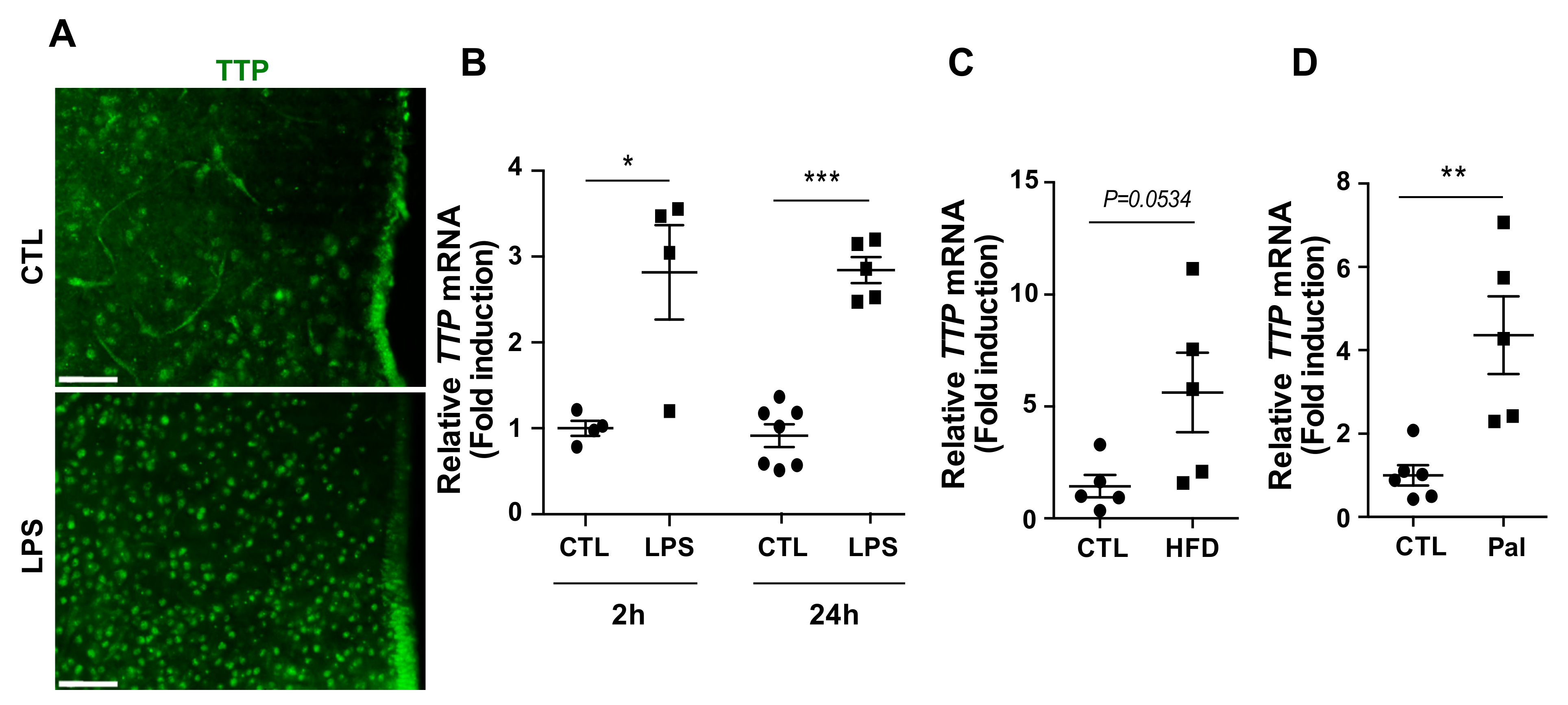
2.1. Hypothalamic TTP Responded to Inflammatory Stimuli
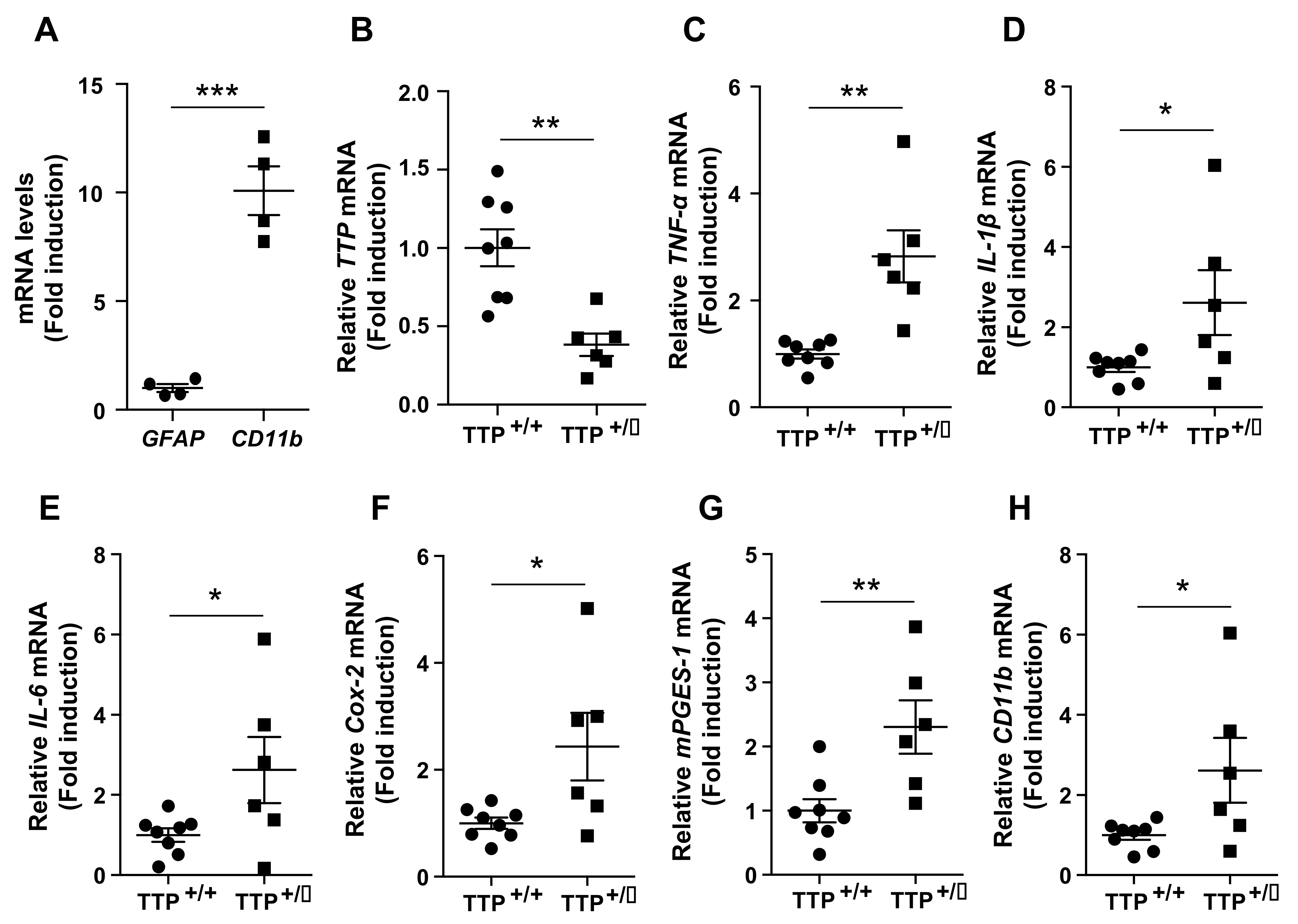
2.2. Deficiency of TTP Led to an Enhanced Inflammatory Response in the Hypothalamus
2.3. Deficiency of TTP Elicited Enhanced Inflammatory Responses in the Hypothalamic Microglia
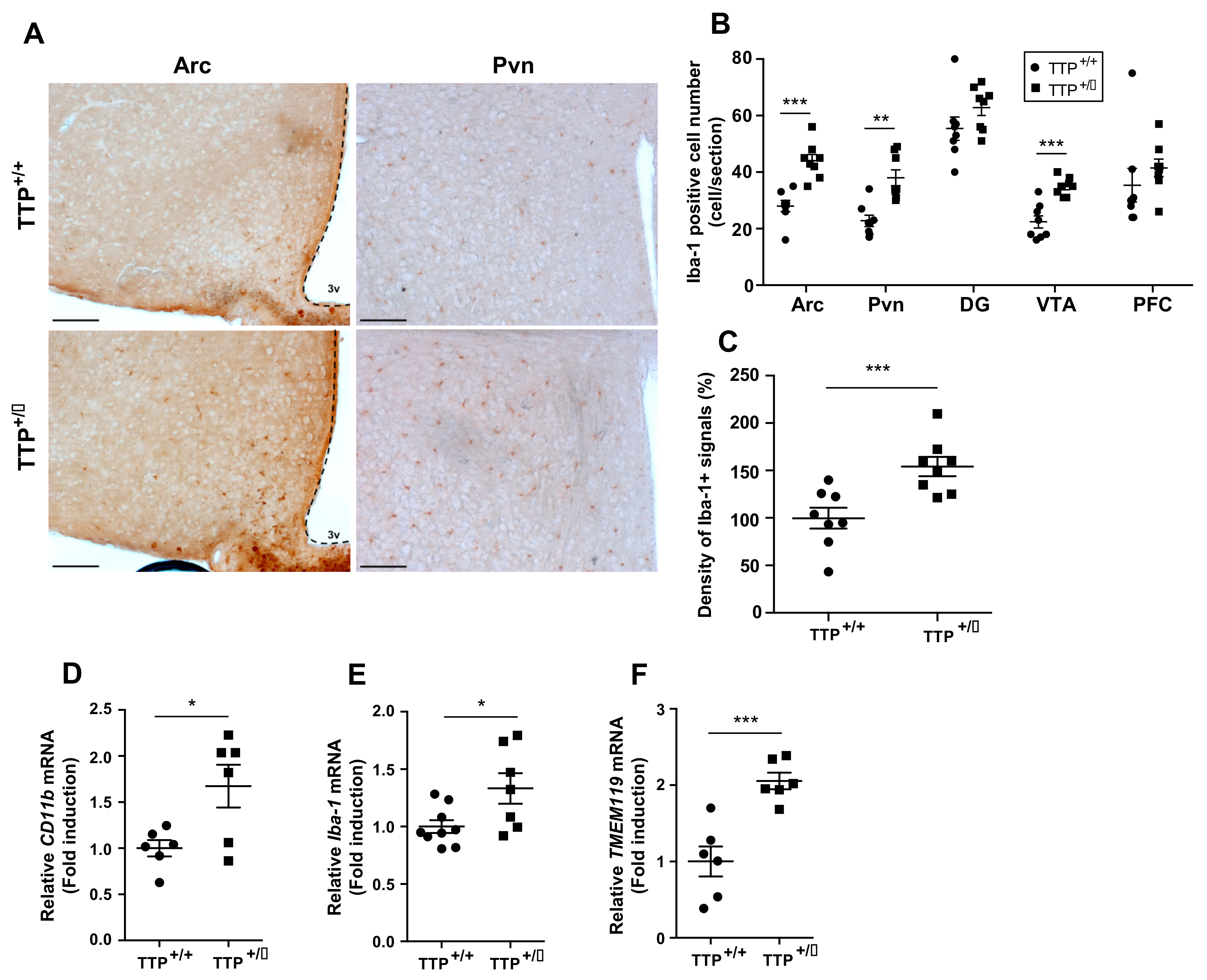
2.4. Microglial Activation Was Observed in the Hypothalamus of TTP Deficient Mice
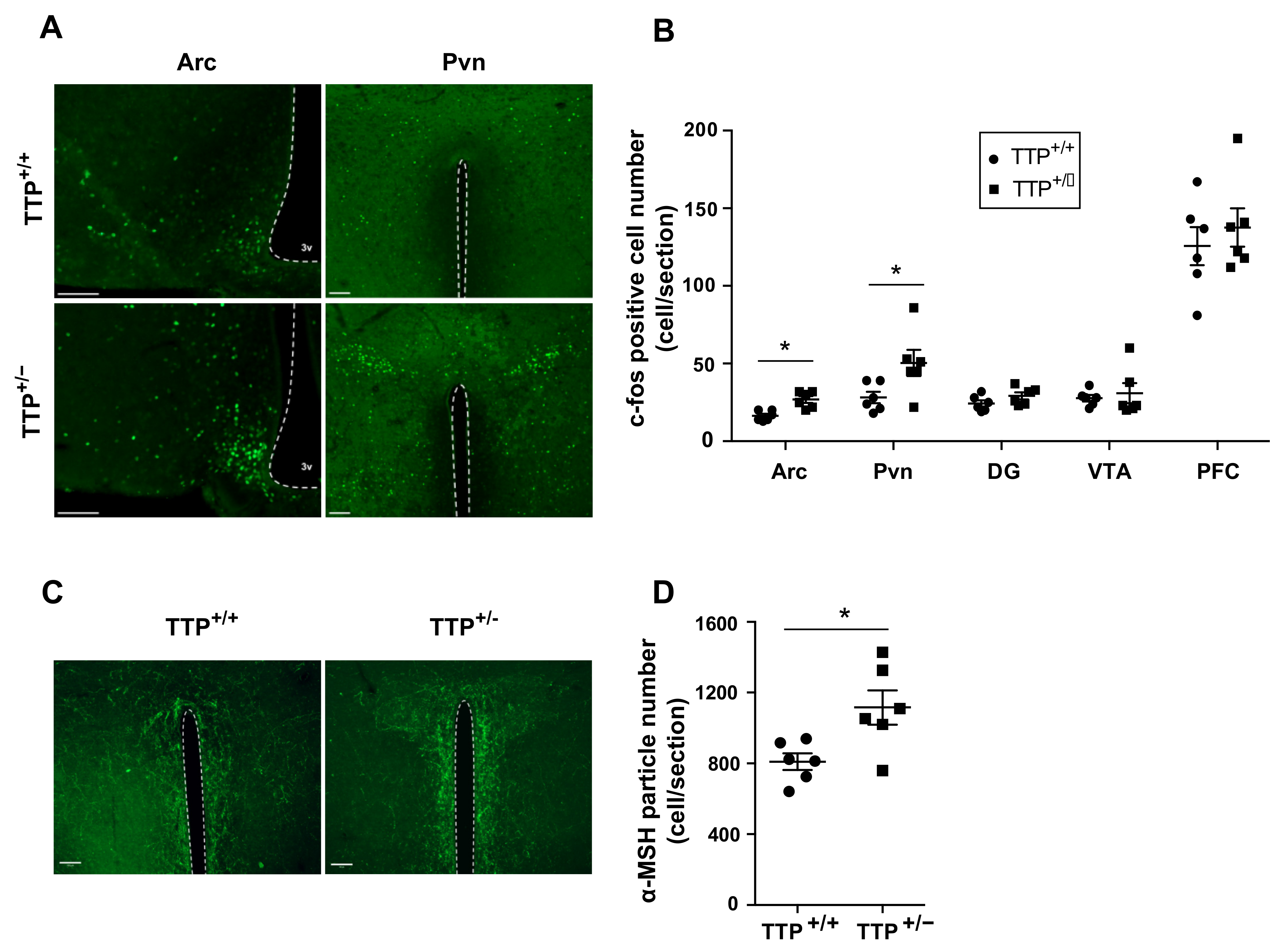
2.5. Deficiency of TTP Led to Altered Neuronal Activity in the Hypothalamic Arc
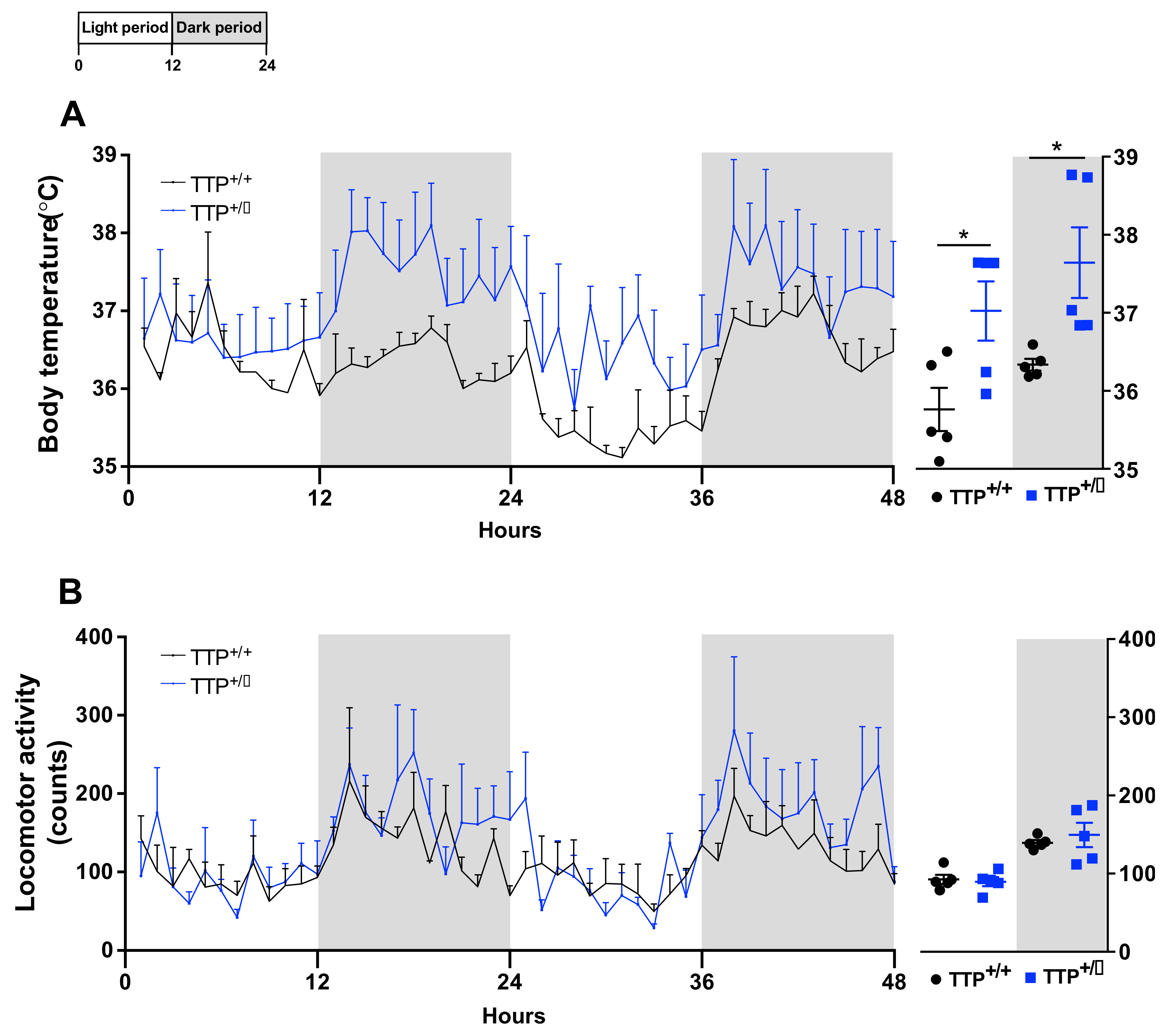
2.6. Hyperthermia Occurred in TTP-Deficient Mice
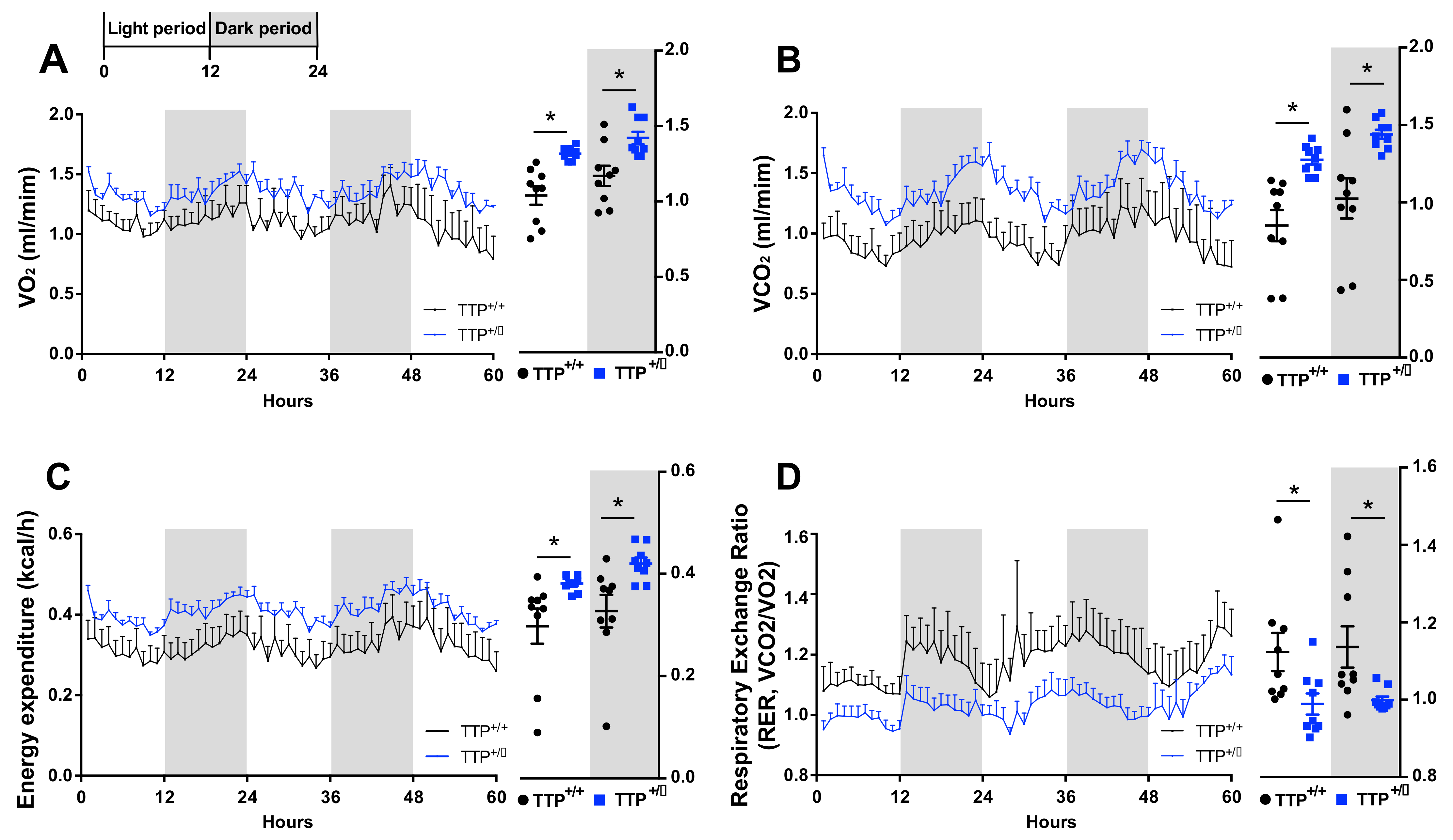
2.7. TTP-Deficient Mice Displayed Increased Energy Expenditure
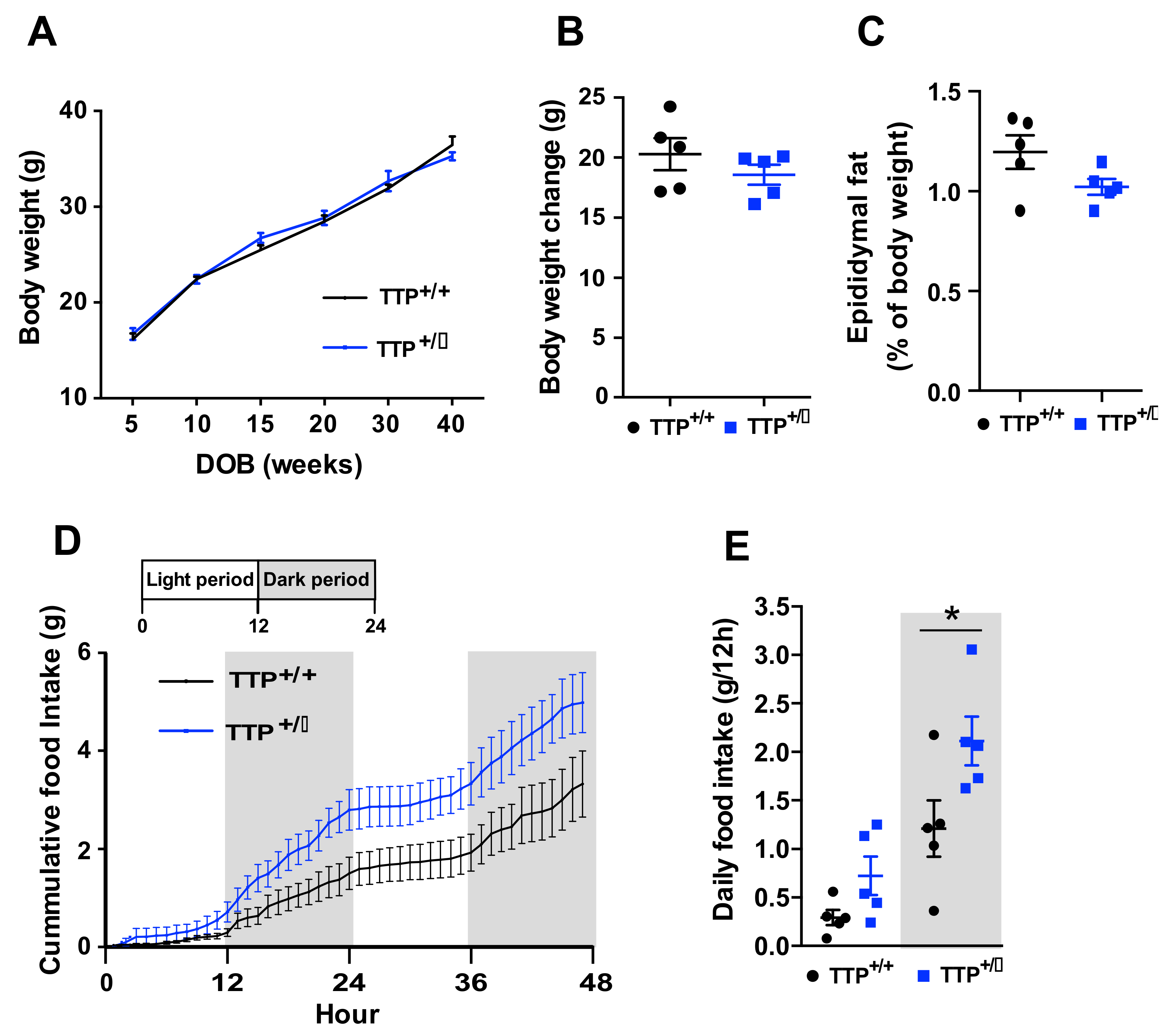
2.8. Deficiency of TTP Led to Hyperphagia without Body Weight Change
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Primary Microglia Culture
4.3. Quantitative Real-Time PCR
4.4. Immunohistochemistry
4.5. Measurement of Body Temperature and Locomotor Activity
4.6. Measurement of VO2, VCO2, Energy Expenditure and Food Intake
4.7. Measurement of Cytokine Levels
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Williams, G.; Bing, C.; Cai, X.J.; Harrold, J.A.; King, P.J.; Liu, X.H. The hypothalamus and the control of energy homeostasis: Different circuits, different purposes. Physiol. Behav. 2001, 74, 683–701. [Google Scholar] [CrossRef]
- Schneeberger, M.; Gomis, R.; Claret, M. Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J. Endocrinol. 2014, 220, T25–T46. [Google Scholar] [CrossRef]
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Models Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Xu, A.W.; Koliwad, S.K. Hypothalamic inflammation in the control of metabolic function. Annu. Rev. Physiol. 2015, 77, 131–160. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Teeling, J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunology 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Kim, J.G.; Park, J.W.; Koch, M.; Horvath, T.L.; Lee, B.J. Hypothalamic TLR2 triggers sickness behavior via a microglia-neuronal axis. Sci. Rep. 2016, 6, 29424. [Google Scholar] [CrossRef]
- Díaz-Muñoz, M.D.; Turner, M. Uncovering the Role of RNA-Binding Proteins in Gene Expression in the Immune System. Front. Immunol. 2018, 9, 1094. [Google Scholar] [CrossRef]
- Newman, R.; McHugh, J.; Turner, M. RNA binding proteins as regulators of immune cell biology. Clin. Exp. Immunol. 2016, 183, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.S.; Carballo, E.; Strum, J.R.; Kennington, E.A.; Phillips, R.S.; Blackshear, P.J. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 1999, 19, 4311–4323. [Google Scholar] [CrossRef]
- Tiedje, C.; Diaz-Muñoz, M.D.; Trulley, P.; Ahlfors, H.; Laaß, K.; Blackshear, P.J.; Turner, M.; Gaestel, M. The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res. 2016, 44, 7418–7440. [Google Scholar] [CrossRef]
- Ross, E.A.; Naylor, A.J.; O’Neil, J.D.; Crowley, T.; Ridley, M.L.; Crowe, J.; Smallie, T.; Tang, T.J.; Turner, J.D.; Norling, L.V.; et al. Treatment of inflammatory arthritis via targeting of tristetraprolin, a master regulator of pro-inflammatory gene expression. Ann. Rheum. Dis. 2017, 76, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Sanduja, S.; Blanco, F.F.; Young, L.E.; Kaza, V.; Dixon, D.A. The role of tristetraprolin in cancer and inflammation. Front. Biosci. (Landmark Ed.) 2012, 17, 174–188. [Google Scholar] [CrossRef]
- Schaljo, B.; Kratochvill, F.; Gratz, N.; Sadzak, I.; Sauer, I.; Hammer, M.; Vogl, C.; Strobl, B.; Müller, M.; Blackshear, P.J.; et al. Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J. Immunol. 2009, 183, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Huang, Z.; Sun, X.; Zhu, X.; Zhou, L.; Li, M.; Cheng, B.; Liu, X.; He, C. Microglia Polarization with M1/M2 Phenotype Changes in rd1 Mouse Model of Retinal Degeneration. Front. Neuroanat. 2017, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Fung, Y.K.; Liu, X.; Pahan, K. Up-regulation of microglial CD11b expression by nitric oxide. J. Biol. Chem. 2006, 281, 14971–14980. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Li, W.; Rodriguez, M.L. Role of microglia in CNS inflammation. FEBS Lett. 2011, 585, 3798–3805. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197.e3. [Google Scholar] [CrossRef]
- Lee, C.H.; Kim, H.J.; Lee, Y.S.; Kang, G.M.; Lim, H.S.; Lee, S.H.; Song, D.K.; Kwon, O.; Hwang, I.; Son, M.; et al. Hypothalamic Macrophage Inducible Nitric Oxide Synthase Mediates Obesity-Associated Hypothalamic Inflammation. Cell Rep. 2018, 25, 934–946.e5. [Google Scholar] [CrossRef]
- Valdearcos, M.; Myers, M.G., Jr.; Koliwad, S.K. Hypothalamic microglia as potential regulators of metabolic physiology. Nat. Metab. 2019, 1, 314–320. [Google Scholar] [CrossRef]
- Reis, W.L.; Yi, C.X.; Gao, Y.; Tschöp, M.H.; Stern, J.E. Brain innate immunity regulates hypothalamic arcuate neuronal activity and feeding behavior. Endocrinology 2015, 156, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.H.; Nam-Goong, I.S.; Lee, J.; Yang, S.; Kim, J.G. Visfatin Triggers Anorexia and Body Weight Loss through Regulating the Inflammatory Response in the Hypothalamic Microglia. Mediat. Inflamm. 2017, 2017, 1958947. [Google Scholar] [CrossRef]
- Jin, S.; Kim, K.K.; Park, B.S.; Kim, D.H.; Jeong, B.; Kang, D.; Lee, T.H.; Park, J.W.; Kim, J.G.; Lee, B.J. Function of astrocyte MyD88 in high-fat-diet-induced hypothalamic inflammation. J. Neuroinflammation 2020, 17, 195. [Google Scholar] [CrossRef]
- Rajbhandari, P.; Thomas, B.J.; Feng, A.-C.; Hong, C.; Wang, J.; Vergnes, L.; Sallam, T.; Wang, B.; Sandhu, J.; Seldin, M.M.; et al. IL-10 Signaling Remodels Adipose Chromatin Architecture to Limit Thermogenesis and Energy Expenditure. Cell 2018, 172, 218–233.e17. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- García-Mauriño, S.M.; Rivero-Rodríguez, F.; Velázquez-Cruz, A.; Hernández-Vellisca, M.; Díaz-Quintana, A.; De la Rosa, M.A.; Díaz-Moreno, I. RNA Binding Protein Regulation and Cross-Talk in the Control of AU-rich mRNA Fate. Front. Mol. Biosci. 2017, 4, 71. [Google Scholar] [CrossRef]
- Abizaid, A.; Gao, Q.; Horvath, T.L. Thoughts for food: Brain mechanisms and peripheral energy balance. Neuron 2006, 51, 691–702. [Google Scholar] [CrossRef]
- Thaler, J.P.; Choi, S.J.; Schwartz, M.W.; Wisse, B.E. Hypothalamic inflammation and energy homeostasis: Resolving the paradox. Front. Neuroendocrinol. 2010, 31, 79–84. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Michaelis, K.A.; Marks, D.L. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin. Cell Dev. Biol. 2016, 54, 42–52. [Google Scholar] [CrossRef]
- Deierborg, T. Preparation of primary microglia cultures from postnatal mouse and rat brains. Methods Mol. Biol. 2013, 1041, 25–31. [Google Scholar] [PubMed]
- Masuda, K.; Marasa, B.; Martindale, J.L.; Halushka, M.K.; Gorospe, M. Tissue- and age-dependent expression of RNA-binding proteins that influence mRNA turnover and translation. Aging 2009, 1, 681–698. [Google Scholar] [CrossRef]
- Magno, L.A.V.; Tenza-Ferrer, H.; Collodetti, M.; Aguiar, M.F.G.; Rodrigues, A.P.C.; da Silva, R.S.; Silva, J.D.P.; Nicolau, N.F.; Rosa, D.V.F.; Birbrair, A.; et al. Optogenetic Stimulation of the M2 Cortex Reverts Motor Dysfunction in a Mouse Model of Parkinson’s Disease. J. Neurosci. 2019, 39, 3234–3248. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, D.Y.; Song, N.; Yang, H.R.; Tu, T.H.; Park, B.S.; Kang, H.; Park, J.W.; Lee, B.J.; Yang, S.; Kim, J.G. Deficiency of Tristetraprolin Triggers Hyperthermia through Enhancing Hypothalamic Inflammation. Int. J. Mol. Sci. 2021, 22, 3328. https://doi.org/10.3390/ijms22073328
Jeong DY, Song N, Yang HR, Tu TH, Park BS, Kang H, Park JW, Lee BJ, Yang S, Kim JG. Deficiency of Tristetraprolin Triggers Hyperthermia through Enhancing Hypothalamic Inflammation. International Journal of Molecular Sciences. 2021; 22(7):3328. https://doi.org/10.3390/ijms22073328
Chicago/Turabian StyleJeong, Da Yeon, Nuri Song, Hye Rim Yang, Thai Hien Tu, Byong Seo Park, Hara Kang, Jeong Woo Park, Byung Ju Lee, Sunggu Yang, and Jae Geun Kim. 2021. "Deficiency of Tristetraprolin Triggers Hyperthermia through Enhancing Hypothalamic Inflammation" International Journal of Molecular Sciences 22, no. 7: 3328. https://doi.org/10.3390/ijms22073328
APA StyleJeong, D. Y., Song, N., Yang, H. R., Tu, T. H., Park, B. S., Kang, H., Park, J. W., Lee, B. J., Yang, S., & Kim, J. G. (2021). Deficiency of Tristetraprolin Triggers Hyperthermia through Enhancing Hypothalamic Inflammation. International Journal of Molecular Sciences, 22(7), 3328. https://doi.org/10.3390/ijms22073328