
Machine Learning Reduced Gene/Non-Coding RNA Features That Classify Schizophrenia Patients Accurately and Highlight Insightful Gene Clusters

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
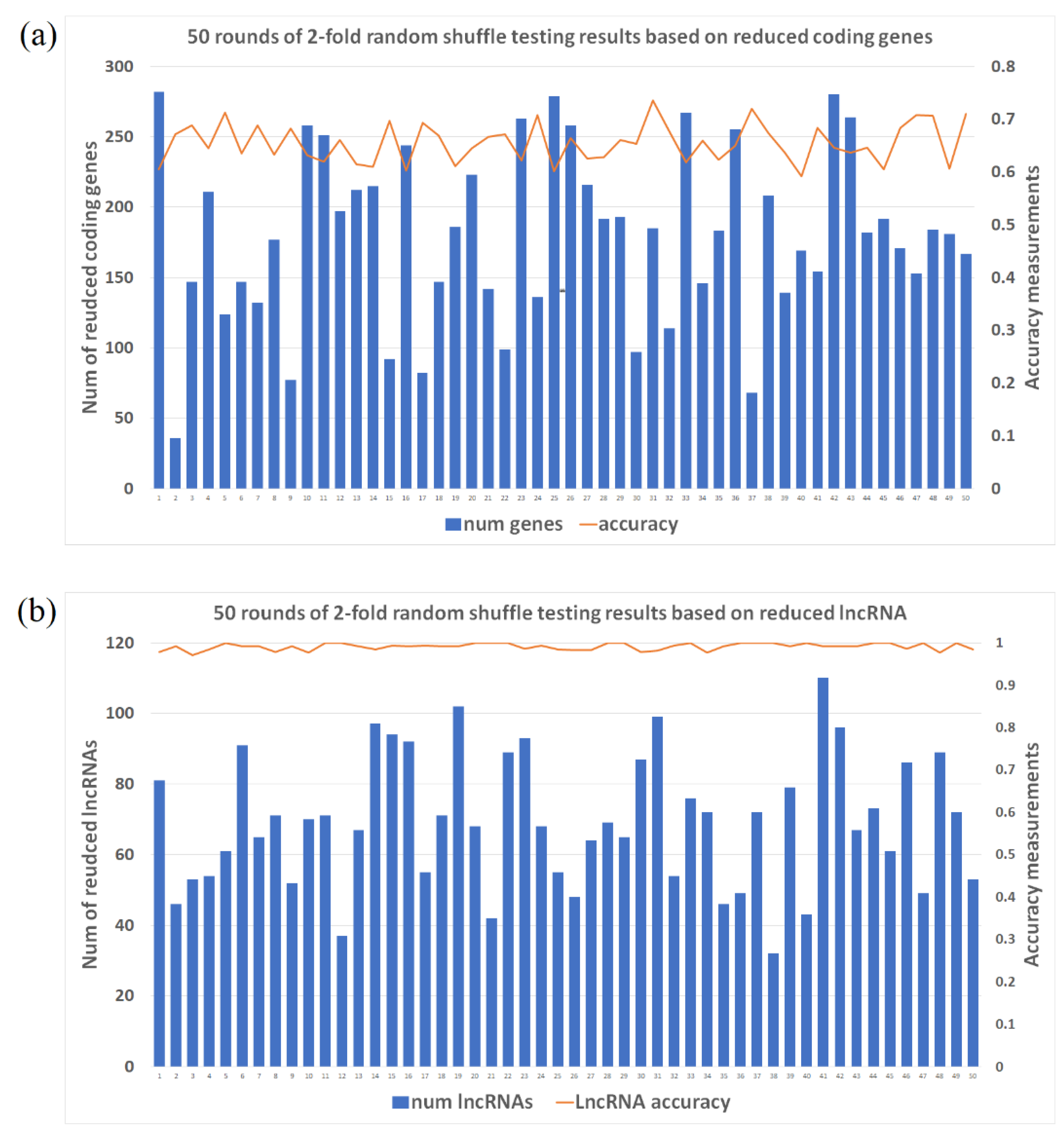
2.1. Accuracy Measure for Labeling Schizophrenia (SCZ) Patients Based on 2-Fold Shuffle Testing
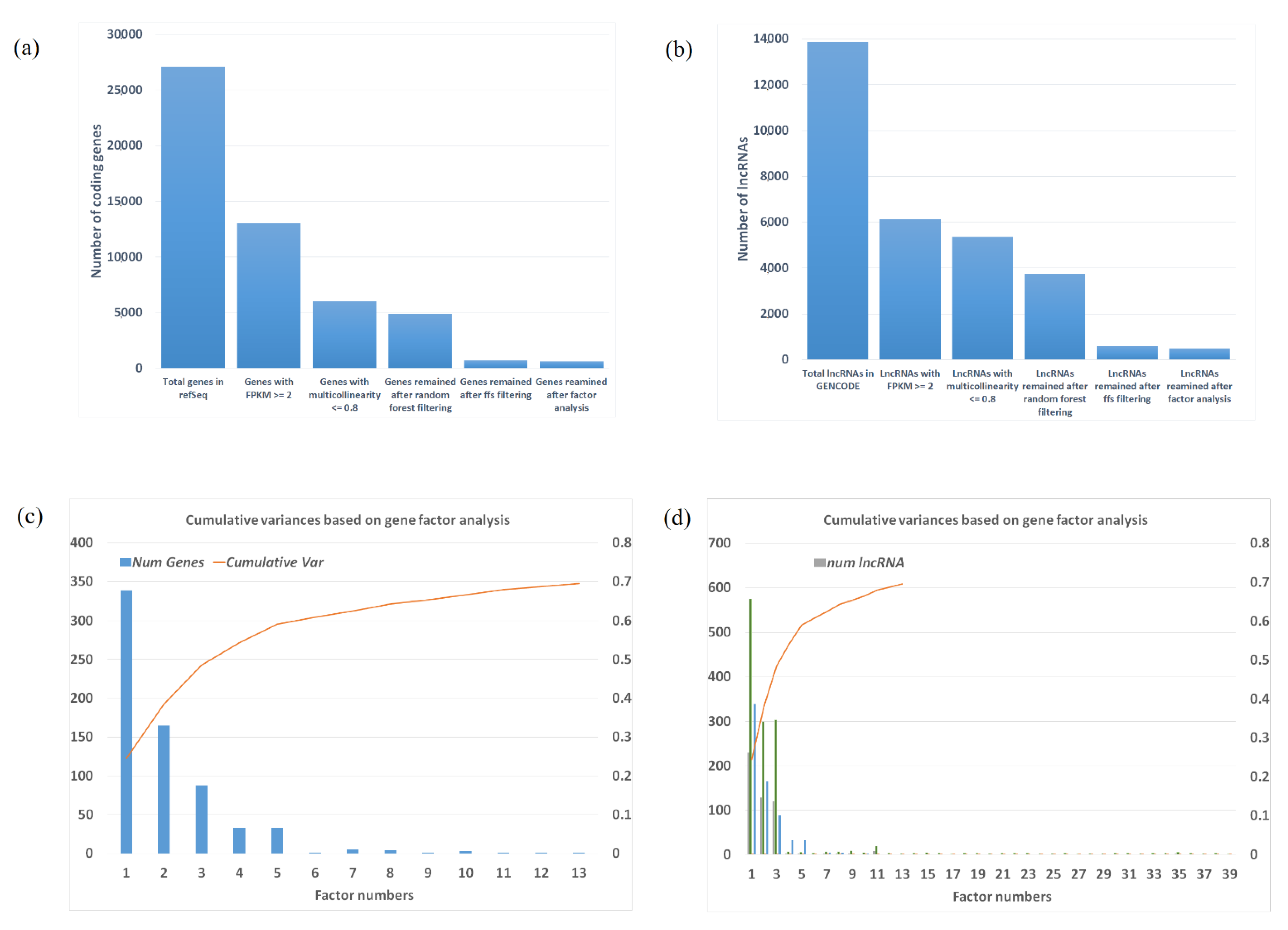
2.2. Selected Gene/lncRNA Feature Based on Machine Learning Algorithm
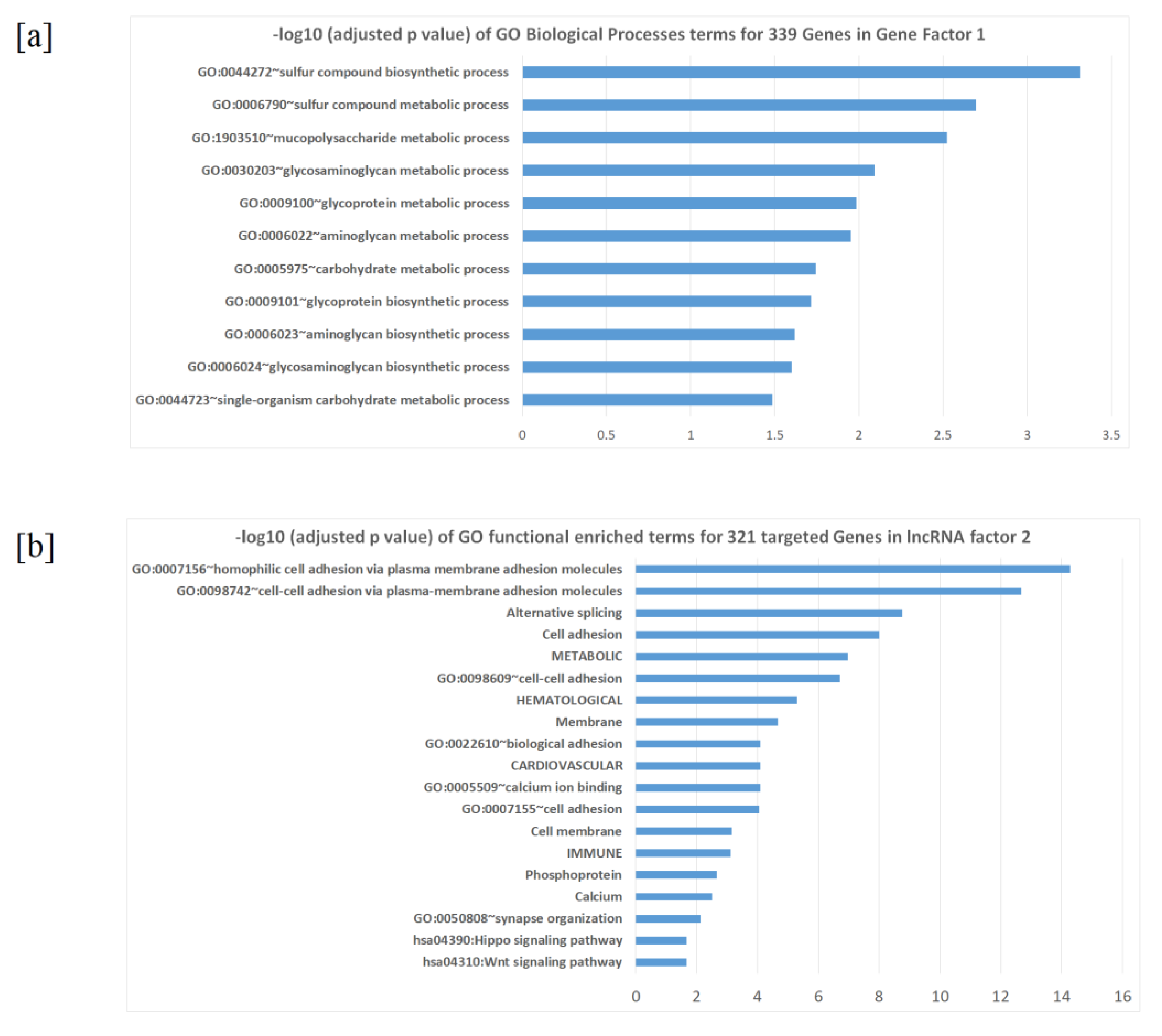
2.3. Factors and Potential Gene Modules from Factor Analysis
3. Discussion
4. Methods & Materials
4.1. RNA-Seq Data for Dorsolateral Prefrontal Cortex (DLPFC) Samples
4.2. Gene/Non-Coding RNAs Expression Matrix
4.3. Gene Reductions Using Machine Learning Algorithms
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sleiman, P.; Hakonarson, H. From schizophrenia risk locus to schizophrenia genes. Nat. Med. 2016, 22, 583–584. [Google Scholar] [CrossRef]
- Chang, X.; Lima, L.D.A.; Liu, Y.; Li, J.; Li, Q.; Sleiman, P.M.A.; Hakonarson, H. Common and Rare Genetic Risk Factors Converge in Protein Interaction Networks Underlying Schizophrenia. Front. Genet. 2018, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Vawter, M.P.; Barrett, T.; Cheadle, C.; Sokolov, B.P.; Wood III, W.H.; Donovan, D.M.; Webster, M.; Freed, W.J.; Becker, K.G. Application of cDNA microarrays to examine gene expression differences in schizophrenia. Brain Res. Bull. 2001, 55, 641–650. [Google Scholar] [CrossRef]
- Katsel, P.; Davis, K.; Gorman, J.; Haroutunian, V. Variations in differential gene expression patterns across multiple brain regions in schizophrenia. Schizophr. Res. 2005, 77, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Glaus, P.; Honkela, A.; Rattray, M. Identifying differentially expressed transcripts from RNA-seq data with biological variation. Bioinformatics 2012, 28, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Honkela, A.; Girardot, C.; Gustafson, E.H.; Liu, Y.-H.; Furlong, E.E.; Lawrence, N.D.; Rattray, M. Model-based method for transcription factor target identification with limited data. Proc. Natl. Acad. Sci. USA 2010, 107, 7793–7798. [Google Scholar] [CrossRef]
- Liu, Y.; Chang, X.; Hahn, C.-G.; Gur, R.E.; Sleiman, P.A.M.; Hakonarson, H. Non-coding RNA dysregulation in the amygdala region of schizophrenia patients contributes to the pathogenesis of the disease. Transl. Psychiatry 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Tian, T.; Wei, Z.; Chang, X.; Liu, Y.; Gur, R.E.; Sleiman, P.M.; Hakonarson, H. The Long Noncoding RNA Landscape in Amygdala Tissues from Schizophrenia Patients. EBioMedicine 2018, 34, 171–181. [Google Scholar] [CrossRef]
- Emerelo, V.; Edurand, D.; Lescallette, A.R.; Vrana, K.E.; Hong, L.E.; Faghihi, M.A.; Ebellon, A. Associating schizophrenia, long non-coding RNAs and neurostructural dynamics. Front. Mol. Neurosci. 2015, 8, 57. [Google Scholar] [CrossRef]
- Gibbons, A.; Udawela, M.; Dean, B. Non-Coding RNA as Novel Players in the Pathophysiology of Schizophrenia. Non-Coding RNA 2018, 4, 11. [Google Scholar] [CrossRef]
- Hoffman, G.E.; Bendl, J.; Voloudakis, G.; Montgomery, K.S.; Sloofman, L.; Wang, Y.-C.; Shah, H.R.; Hauberg, M.E.; Johnson, J.S.; Girdhar, K.; et al. CommonMind Consortium provides transcriptomic and epigenomic data for Schizophrenia and Bipolar Disorder. Sci. Data 2019, 6, 1–14. [Google Scholar] [CrossRef]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2010, 39, D691–D697. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.S.; Kavanagh, D.H.; Perumal, T.M.; Ruderfer, D.M.; Oh, E.C.; Topol, A.; Shah, H.R.; et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nat. Cell Biol. 2014, 511, 421–427. [Google Scholar] [CrossRef]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.R.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Non-Coding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Goff, L.A.; Groff, A.F.; Sauvageau, M.; Trayes-Gibson, Z.; Sanchez-Gomez, D.B.; Morse, M.; Martin, R.D.; Elcavage, L.E.; Liapis, S.C.; Gonzalez-Celeiro, M.; et al. Spatiotemporal expression and transcriptional perturbations by long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2015, 112, 6855–6862. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.; Engreitz, J.M.; Konermann, S.; Abudayyeh, O.O.; Verdine, V.K.; Aguet, F.; Gootenberg, J.S.; Sanjana, N.E.; Wright, J.B.; Fulco, C.P.; et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nat. Cell Biol. 2017, 548, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Kong, Y.; Yu, T. A Deep Neural Network Model using Random Forest to Extract Feature Representation for Gene Expression Data Classification. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Van Ijzendoorn, D.G.P.; Szuhai, K.; Bruijn, I.H.B.-D.; Kostine, M.; Kuijjer, M.L.; Bovée, J.V.M.G. Machine learning analysis of gene expression data reveals novel diagnostic and prognostic biomarkers and identifies therapeutic targets for soft tissue sarcomas. PLoS Comput. Biol. 2019, 15, e1006826. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Chang, H.Y. Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol. 2010, 7, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Gejman, P.V.; Sanders, A.R.; Duan, J. The Role of Genetics in the Etiology of Schizophrenia. Psychiatr. Clin. N. Am. 2010, 33, 35–66. [Google Scholar] [CrossRef]
- Alachkar, A.; Wang, L.; Yoshimura, R.; Hamzeh, A.R.; Wang, Z.; Sanathara, N.; Lee, S.M.; Xu, X.; Abbott, G.W.; Civelli, O. Prenatal one-carbon metabolism dysregulation programs schizophrenia-like deficits. Mol. Psychiatry 2017, 23, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.D.; Coulson-Thomas, V.J.; Foscarin, S.; Kwok, J.C.; Fawcett, J.W. “GAG-ing with the neuron”: The role of glycosaminoglycan patterning in the central nervous system. Exp. Neurol. 2015, 274, 100–114. [Google Scholar] [CrossRef]
- Emaeda, N. Proteoglycans and neuronal migration in the cerebral cortex during development and disease. Front. Neurosci. 2015, 9, 98. [Google Scholar] [CrossRef]
- Berretta, S.; Pantazopoulos, H.; Markota, M.; Brown, C.; Batzianouli, E.T. Losing the sugar coating: Potential impact of perineuronal net abnormalities on interneurons in schizophrenia. Schizophr. Res. 2015, 167, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Hoosain, F.G.; Choonara, Y.E.; Tomar, L.K.; Kumar, P.; Tyagi, C.; du Toit, L.C.; Pillay, V. Bypassing P-Glycoprotein Drug Efflux Mechanisms: Possible Applications in Pharmacoresistant Schizophrenia Therapy. BioMed Res. Int. 2015, 2015, 1–21. [Google Scholar] [CrossRef]
- Berridge, M.J. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 2013, 7, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Stepan, J.; Anderzhanova, E.; Gassen, N.C. Hippo Signaling: Emerging Pathway in Stress-Related Psychiatric Disorders? Front. Psychiatry 2018, 9, 715. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Lin, Y.; Ghazanfar, S.; Wang, K.Y.X.; Gagnon-Bartsch, J.A.; Lo, K.K.; Su, X.; Han, Z.-G.; Ormerod, J.T.; Speed, T.P.; Yang, P.; et al. scMerge leverages factor analysis, stable expression, and pseudoreplication to merge multiple single-cell RNA-seq datasets. Proc. Natl. Acad. Sci. USA 2019, 116, 9775–9784. [Google Scholar] [CrossRef]
- Iacob, E.; Light, A.R.; Donaldson, G.W.; Okifuji, A.; Hughen, R.W.; White, A.T.; Light, K.C. Gene Expression Factor Analysis to Differentiate Pathways Linked to Fibromyalgia, Chronic Fatigue Syndrome, and Depression in a Diverse Patient Sample. Arthritis Rheum. 2015, 68, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yao, Y.-G.; Luo, X.-J. SZDB: A Database for Schizophrenia Genetic Research. Schizophr. Bull. 2016, 43, 459–471. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Gene | GWAS | Linkage | CNV | Integrative | Diff Methy | Diff Exp | Exome | Brain Exp | GO | Score |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ASPHD1 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 1 (26.68) | 0 | 5 |
| 1 | AK4 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 (13.55) | 1 | 4 |
| 1 | APH1A | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 (43.24) | 1 | 4 |
| 1 | FPGS | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 (14.38) | 1 | 4 |
| 1 | FSCN1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 (48.13) | 0 | 4 |
| 1 | INO80E | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 1 (20.22) | 0 | 4 |
| 1 | P2RX6 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 (4.99) | 1 | 4 |
| 1 | PCCB | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 (26.64) | 0 | 4 |
| 1 | PRODH | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 (24.04) | 0 | 4 |
| 1 | SCN1B | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 (25.12) | 1 | 4 |
| 1 | SEMA7A | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 (13.64) | 1 | 4 |
| 2 | BCCIP | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 (26.92) | 1 | 5 |
| 2 | HNRNPU | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 (108.84) | 0 | 4 |
| 2 | HSP90AA1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 (411.41) | 1 | 4 |
| 2 | NRG1 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 (4.47) | 1 | 4 |
| 2 | PDE4B | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1 (38.88) | 1 | 4 |
| 3 | TIMP2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 (107.98) | 1 | 4 |
| 7 | BCL6 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 (29.50) | 1 | 5 |
| 9 | RERE | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 (25.51) | 1 | 4 |
| Factor | lncRNA | Target Gene | GWAS | Linkage | CNV | Integrative | Diff Methy | Diff Exp | Exome | Brain Exp | GO | Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ENSG00000247735.2 | ASPHD1 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 1 (26.68) | 0 | 5 |
| 1 | ENSG00000232912.1 | RERE | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 (25.51) | 1 | 4 |
| 1 | ENSG00000235770.1 | FN1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 (38.33) | 1 | 4 |
| 1 | ENSG00000235831.2 | ITPR1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 (25.59) | 1 | 4 |
| 1 | ENSG00000239569.2 | SRPK2 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 (75.17) | 1 | 4 |
| 1 | ENSG00000243762.1 | RANBP1 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 1 (30.73) | 0 | 4 |
| 1 | ENSG00000247735.2 | SEZ6L2 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 (38.77) | 1 | 4 |
| 1 | ENSG00000257126.1 | FOXG1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 (22.05) | 1 | 4 |
| 1 | ENSG00000261220.2 | ST3GAL1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 (8.93) | 1 | 4 |
| 1 | ENSG00000271849.1 | PJA2 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 (144.54) | 1 | 4 |
| 2 | ENSG00000224563.1 | BCL6 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 (29.50) | 1 | 5 |
| 2 | ENSG00000226978.1 | MAGI2 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 (24.46) | 1 | 4 |
| 2 | ENSG00000236031.1 | AKT3 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 (35.93) | 1 | 4 |
| 2 | ENSG00000248816.1 | TENM3 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 (8.06) | 1 | 4 |
| 2 | ENSG00000272367.1 | RASA1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 (20.45) | 1 | 4 |
| 3 | ENSG00000272989.1 | DLG1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 (52.38) | 1 | 5 |
| 3 | ENSG00000239569.2 | SRPK2 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 (75.17) | 1 | 4 |
| 3 | ENSG00000273164.1 | PRODH | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 (24.04) | 0 | 4 |
| 3 | ENSG00000273164.1 | DGCR2 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 1 (30.80) | 0 | 4 |
| 37 | ENSG00000248816.1 | TENM3 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 (8.06) | 1 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Qu, H.-Q.; Chang, X.; Tian, L.; Qu, J.; Glessner, J.; Sleiman, P.M.A.; Hakonarson, H. Machine Learning Reduced Gene/Non-Coding RNA Features That Classify Schizophrenia Patients Accurately and Highlight Insightful Gene Clusters. Int. J. Mol. Sci. 2021, 22, 3364. https://doi.org/10.3390/ijms22073364
Liu Y, Qu H-Q, Chang X, Tian L, Qu J, Glessner J, Sleiman PMA, Hakonarson H. Machine Learning Reduced Gene/Non-Coding RNA Features That Classify Schizophrenia Patients Accurately and Highlight Insightful Gene Clusters. International Journal of Molecular Sciences. 2021; 22(7):3364. https://doi.org/10.3390/ijms22073364
Chicago/Turabian StyleLiu, Yichuan, Hui-Qi Qu, Xiao Chang, Lifeng Tian, Jingchun Qu, Joseph Glessner, Patrick M. A. Sleiman, and Hakon Hakonarson. 2021. "Machine Learning Reduced Gene/Non-Coding RNA Features That Classify Schizophrenia Patients Accurately and Highlight Insightful Gene Clusters" International Journal of Molecular Sciences 22, no. 7: 3364. https://doi.org/10.3390/ijms22073364
APA StyleLiu, Y., Qu, H. -Q., Chang, X., Tian, L., Qu, J., Glessner, J., Sleiman, P. M. A., & Hakonarson, H. (2021). Machine Learning Reduced Gene/Non-Coding RNA Features That Classify Schizophrenia Patients Accurately and Highlight Insightful Gene Clusters. International Journal of Molecular Sciences, 22(7), 3364. https://doi.org/10.3390/ijms22073364