
SSAO/VAP-1 in Cerebrovascular Disorders: A Potential Therapeutic Target for Stroke and Alzheimer’s Disease




Abstract
:1. Introduction
1.1. SSAO: An Amine Oxidase

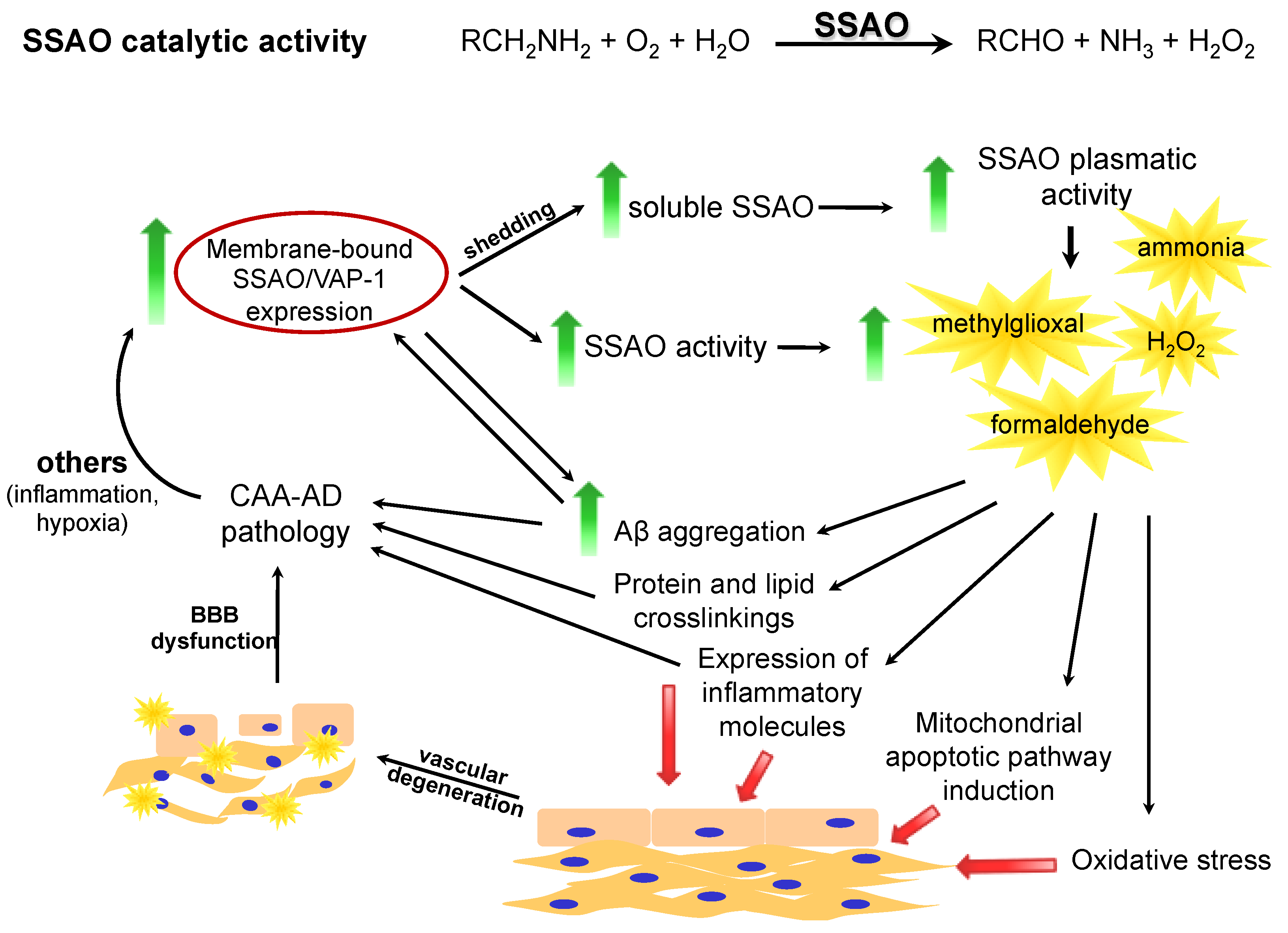{kind=link}
{kind=link}
| Tissue | Cell Type | Substrate | Function |
|---|---|---|---|
| Cerebrovascular tissue (meninges and microvessels) (human, rabbit, mouse, bovine) | Endothelial cells Smooth muscle cells | Methylamine (derived from epinephrine, adrenaline, creatine, sarcosine and choline) aminoacetone (derived from glycine and threonine) | Scavenger of endogenous dietary amines Generation of H2O2 as a signaling molecule Leukocyte trafficking under inflammation |
| Vascularized tissues (heart, kidney, lung, intestine, liver, retina and lymph nodes) and blood vessels (human, pig, rat, rabbit, bovine) | Endothelial cells Smooth muscle cells Pericytes | Phenylethylamine Dopamine Methylamine Tyramine Tryptamine | Metabolism of physiological circulating amines and xenobiotic ones Leukocyte binding and extravasation under inflammatory conditions |
| Adipose tissue (human and rat) | Adipocytes (white and brown) | Various endogenous and exogenous amines | Metabolism of endogenous amines Insulinomimetic effects through the generation of H2O2 |
| Ureter and vas deferens | Non-vascular smooth muscle cells | Dopamine | Metabolism of physiological amines and xenobiotic ones |
| Endometrium (human) | Pericytes | Methylamine | Recruiting innate immune cells |
| Skin (guinea pig) | Fibroblasts | Histamine 1–4 Methylhistamine | Metabolism of physiological amines and xenobiotic ones |
| Dental pulp (human, pig) | Odontoblasts | Serotonin Phenylethylamine Tyramine Tryptamine | Contribution to inflammatory response in dental pulp (pulpitis) |
1.2. SSAO/VAP-1: Expression and Tissular Localization
1.3. SSAO/VAP-1 Physiological Functions
1.3.1. Amine Deamination
1.3.2. Activation of Glucose Transport
1.3.3. Leukocyte Adhesion Function under Inflammation
1.4. SSAO/VAP-1 Involvement in Pathological Conditions
2. Cerebrovascular Dysfunction in Stroke and AD
2.1. The BBB and Cerebrovascular Dysfunction
2.2. Stroke
2.3. Alzheimer’s Disease and Cerebral Amyloid Angiopathy
3. SSAO/VAP-1 and Cerebrovascular Dysfunction
3.1. SSAO/VAP-1 in Stroke
3.2. SSAO/VAP-1 in Alzheimer’s Disease
| Disorder | Tissue Analyzed | Phase of the Pathology | SSAO/VAP-1 Alteration | Reference |
|---|---|---|---|---|
| Ischemic stroke | Serum | <6 h (acute phase) | Increase | [193] |
| Plasma | 24 h after stroke | Increase vs. 1 h | [92] | |
| Plasma | 1 h after HT | Increase | [92] | |
| Serum | >24 h after stroke | No change | [93] | |
| Plasma | weeks after | Decrease | [94] | |
| Ipsilateral brain | - | Decrease | [193] | |
| Ipsilateral brain | - | Increase | [92] | |
| Hemorrhagic stroke (ICH) | Plasma | 3–4 h after ICH | Increase | [95] |
| Contralateral brain | - | Increase | [95] | |
| AD | Plasma | moderate-severe | Increase | [97] |
| Plasma | - | Increase | [98] | |
| Brain vessels | - | Increase | [96] | |
| Hippocampus | - | Increase | [98] | |
| Brain vessels | - | Increase | [204] | |
| Brain vessels | - | Increase | [212] |
4. Can SSAO/VAP-1 Be a Link between Stroke and AD?
4.1. Stroke and the Risk for AD
4.2. AD and the Risk for Stroke
4.3. SSAO/VAP-1 As a Possible Link between Stroke and AD
5. Therapeutic Approach to Stroke and AD by SSAO/VAP-1 Inhibition
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-β |
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s disease |
| ADD | AD with diabetes mellitus |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| AGEs | Advanced glycation end products |
| AOs | Amine oxidases |
| apoE | Apolipoprotein E |
| APP | Amyloid precursor protein |
| BACE-1 | Beta-secretase 1 |
| BBB | Blood–brain barrier |
| BuChE | Butyrylcholinesterase |
| CAA | Cerebral amyloid angiopathy |
| CAMs | Cell adhesion molecules |
| CNS | Central nervous system |
| DAO | Diamine oxidase |
| DM | Diabetes mellitus |
| DMBA | 7,12-dimethylbenz(alpha)anthracene |
| EAE | Experimental autoimmune encephalomyelitis |
| ECE | Endothelin-converting enzyme |
| eMCAO | Embolic MCAO |
| FAD | Flavin adenine dinucleotide |
| FDA | Food and Drug Administration |
| GLUT | Glucose transporters |
| H2O2 | Hydrogen peroxide |
| HCHWA | Hereditary cerebral hemorrhage with amyloidosis |
| hCMEC/D3 | Human cerebral microvascular endothelial cells |
| HEVs | High endothelial venules |
| HIF-1 | Hypoxia-inducible factor 1 |
| HT | Hemorrhagic transformation |
| HUVEC | Human umbilical vein endothelial cells |
| ICAM-1 | Intracellular adhesion molecule 1 |
| ICH | Intracerebral hemorrhage |
| IDE | Insulin-degrading enzyme |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| IFN- γ | Interferon- γ |
| IRF1 | Interferon regulatory factor 1 |
| JL-72 | 3-(3-hydrazinylpropyl)-1H-indole |
| JNK | c-Jun amino-terminal kinase |
| LOX | Lysyl oxidase |
| LPS | Lipopolysaccharide |
| LRP-1 | Lipoprotein receptor protein-1 |
| LTQ | Lysine tyrosyl quinone |
| MAO | Monoamine oxidase |
| MAPK | Mitogen-activated protein kinase |
| MCAO | Middle cerebral artery occlusion |
| MCI | Mild cognitive impairment |
| MMPs | Matrix metalloproteinases |
| MMSE | Mini-mental state examination |
| MS | Multiple sclerosis |
| MTDL | Multi-target directed-ligand |
| NEP | Neprilysin |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NVU | Neurovascular unit |
| O2- | Superoxide |
| OGD | Oxygen–glucose deprivation |
| OH· | Hydroxyl radical |
| PAO | Polyamine oxidase |
| PLN | Peripheral lymph node |
| PNAd | Peripheral lymph addressin |
| PrAO | Primary amine oxidase |
| PSEN | Presenilin |
| RAGE | Receptor for advanced glycation end products |
| ROS | Reactive free radicals |
| SAH | Subarachnoid hemorrhage |
| SAMP8 | Senescence accelerated mouse-prone 8 |
| SSAO | Semicarbazide-sensitive amine oxidase |
| STAT3 | Signal transducer and activator of transcription 3 |
| tMCAO | Transient MCAO |
| TNF-α | Tumor necrosis factor α |
| tPA | Tissue plasminogen activator |
| TPQ | Topa-quinone |
| VAP-1 | Vascular adhesion protein-1 |
| VCAM-1 | Vascular cell adhesion protein 1 |
| VEGF | Vascular endothelial growth factor |
References
- Gong, B.; Boor, P.J. The role of amine oxidases in xenobiotic metabolism. Expert Opin. Drug Metab. Toxicol. 2006, 2, 559–571. [Google Scholar] [CrossRef]
- Johnston, J. Some observations upon a new inhibitor of monoamine oxidase in brain tissue. Biochem. Pharmacol. 1968, 17, 1285–1297. [Google Scholar] [CrossRef]
- Knoll, J.; Magyar, K. Some puzzling pharmacological effects of monoamine oxidase inhibitors. Adv. Biochem. Psychopharmacol. 1972, 5, 393–408. [Google Scholar] [PubMed]
- Janes, S.M.; Klinman, J.P. [2] Isolation of 2,4,5-trihydroxyphenylalanine quinone (topa quinone) from copper amine oxidases. Methods in Enzymology 1995, 258, 20–34. [Google Scholar] [CrossRef]
- Jalkanen, S.; Salmi, M. NEW EMBO MEMBER’S REVIEW: Cell surface monoamine oxidases: Enzymes in search of a function. EMBO J. 2001, 20, 3893–3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinman, J.P.; Mu, D. Quinoenzymes in Biology. Annu. Rev. Biochem. 1994, 63, 299–344. [Google Scholar] [CrossRef] [PubMed]
- Lyles, G. Mammalian plasma and tissue-bound semicarba ide-sensitive amine oxidases: Biochemical, pharmacological and toxicological aspects. Int. J. Biochem. Cell Biol. 1996, 28, 259–274. [Google Scholar] [CrossRef]
- O’Sullivan, J.; Unzeta, M.; Healy, J.; O’Sullivan, M.I.; Davey, G.; Tipton, K.F. Semicarbazide-sensitive amine oxidases: En-zymes with quite a lot to do. Neurotoxicology 2004, 25, 303–315. [Google Scholar] [CrossRef]
- Lizcano, J.; Balsa, D.; Tipton, K.F.; Unzeta, M. The oxidation of dopamine by the semicarbazide-sensitive amine oxidase (SSAO) from rat vas deferens. Biochem. Pharmacol. 1991, 41, 1107–1110. [Google Scholar] [CrossRef]
- De Arriba, A.F.; Lizcano, J.; Balsa, D.; Unzeta, M. Contribution of different amine oxidases to the metabolism of dopamine in bovine retina. Biochem. Pharmacol. 1991, 42, 2355–2361. [Google Scholar] [CrossRef]
- Schayer, R.W.; Smiley, R.L.; Kaplan, E. The Metabolism of Epinephrine Containing Isotopic Carbon. II. J. Biol. Chem. 1952, 198, 545–551. [Google Scholar] [CrossRef]
- Dar, M.; Morselli, P.; Bowman, E. The enzymatic systems involved in the mammalian metabolism of methylamine. Gen. Pharmacol. Vasc. Syst. 1985, 16, 557–560. [Google Scholar] [CrossRef]
- Jones, J.D.; Brunett, P.C. Creatinine metabolism and toxicity. Kidney Int. Suppl. 1975, 294–298. [Google Scholar]
- Yu, P.; Deng, Y. Potential cytotoxic effect of chronic administration of creatine, a nutrition supplement to augment athletic performance. Med Hypotheses 2000, 54, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324. [Google Scholar] [PubMed]
- Precious, E.; Gunn, C.E.; Lyles, G.A. Deamination of methylamine by semicarbazidesensitive amine oxidase in human umbilical artery and rat aorta. Biochem. Pharmacol. 1988, 37, 707–713. [Google Scholar] [CrossRef]
- Yu, P.H.; Wright, S.; Fan, E.H.; Lun, Z.-R.; Gubisne-Harberle, D. Physiological and pathological implications of semicarbazide-sensitive amine oxidase. Biochim. et Biophys. Acta (BBA) -Bioenerg. 2003, 1647, 193–199. [Google Scholar] [CrossRef]
- Yu, P.; Zuo, D. Formaldehyde produced endogenously via deamination of methylamine. A potential risk factor for initiation of endothelial injury. Atheroscler. 1996, 120, 189–197. [Google Scholar] [CrossRef]
- Bird, M.; Nunn, P.; Lord, L. Formation of glycine and aminoacetone from l-threonine by rat liver mitochondria. Biochim. et Biophys. Acta (BBA) -Gen. Subj. 1984, 802, 229–236. [Google Scholar] [CrossRef]
- Kinemuchi, H. Selective Inhibitors of Membrane-Bound Semicarbazide-Sensitive Amine Oxidase (SSAO) Activity in Mammalian Tissues. NeuroToxicology 2004, 25, 325–335. [Google Scholar] [CrossRef]
- Esteban, G.; Bolea, I.; Sun, P.; Solé, M.; Samadi, A.; Marco-Contelles, J.; Unzeta, M. A therapeutic approach to cerebrovascular diseases based on indole substituted hydrazides and hydrazines able to interact with human vascular adhesion protein-1, monoamine oxidases (A and B), AChE and BuChE. J. Neural Transm. 2012, 120, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Dunkel, P.; Balogh, B.; Meleddu, R.; Maccioni, E.; Gyires, K.; Mátyus, P. Semicarbazide-sensitive amine oxidase/vascular adhesion protein-1: A patent survey. Expert Opin. Ther. Patents 2011, 21, 1453–1471. [Google Scholar] [CrossRef]
- Foot, J.S.; Yow, T.T.; Schilter, H.; Buson, A.; Deodhar, M.; Findlay, A.D.; Guo, L.; McDonald, I.A.; Turner, C.I.; Zhou, W.; et al. PXS-4681A, a Potent and Selective Mechanism-Based Inhibitor of SSAO/VAP-1 with Anti-Inflammatory Effects In Vivo. J. Pharmacol. Exp. Ther. 2013, 347, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Morita, M.; Tojo, T.; Nagashima, A.; Moritomo, A.; Miyake, H. Novel 1H-imidazol-2-amine derivatives as potent and orally active vascular adhesion protein-1 (VAP-1) inhibitors for diabetic macular edema treatment. Bioorganic Med. Chem. 2013, 21, 3873–3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bligt-Lindén, E.; Pihlavisto, M.; Szatmári, I.; Otwinowski, Z.; Smith, D.J.; Lázár, L.; Fülöp, F.; Salminen, T.A. Novel Pyridazinone Inhibitors for Vascular Adhesion Protein-1 (VAP-1): Old Target–New Inhibition Mode. J. Med. Chem. 2013, 56, 9837–9848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaki, S.; Koga, Y.; Nagashima, A.; Kondo, M.; Shimada, Y.; Kadono, K.; Moritomo, A.; Yoshihara, K. Synthesis and pharmacological evaluation of glycine amide derivatives as novel vascular adhesion protein-1 inhibitors without CYP3A4 and CYP2C19 inhibition. Bioorganic Med. Chem. 2017, 25, 4110–4122. [Google Scholar] [CrossRef] [PubMed]
- Salminen, T.A.; Smith, D.J.; Jalkanen, S.; Johnson, M.S. Structural model of the catalytic domain of an enzyme with cell adhe-sion activity: Human vascular adhesion protein-1 (HVAP-1) D4 domain is an amine oxidase. Protein engineering 1998, 11, 1195–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, E.; Nilsson, J.; Ogg, D.; Kleywegt, G.J. Structure of human semicarbazide-sensitive amine oxidase/vascular adhesion protein-1. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Salmi, M.; Jalkanen, S. A 90-kilodalton endothelial cell molecule mediating lymphocyte binding in humans. Sci. 1992, 257, 1407–1409. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Salmi, M.; Bono, P.; Hellman, J.; Leu, T.; Jalkanen, S. Cloning of Vascular Adhesion Protein 1 Reveals a Novel Multifunctional Adhesion Molecule. J. Exp. Med. 1998, 188, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Lewinsohn, R. Mammalian monoamine-oxidizing enzymes, with special reference to benzylamine oxidase in human tissues. Braz. J. Med Biol. Res. 1984, 17, 223–256. [Google Scholar]
- Lizcano, J.M.; Balsa, D.; Tipton, K.F.; Unzeta, M. Amine oxidase activities in bovine lung. Amine Oxidases Their Impact Neurobiol. 1990, 32, 341–344. [Google Scholar] [CrossRef]
- Lyles, G.A.; Singh, I. Vascular smooth muscle cells: A major source of the semicarbazide-sensitive amine oxidase of the rat aorta. J. Pharm. Pharmacol. 1985, 37, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Salmi, M.; Jalkanen, S. Different forms of human vascular adhesion protein-1 (VAP-1) in blood vesselsin vivo and in cultured endothelial cells: Implications for lymphocyte-endothelial cell adhesion models. Eur. J. Immunol. 1995, 25, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Ramonet, D.; Rodriguez, M.J.; Saura, J.; Lizcano, J.M.; Romera, M.; Unzeta, M.; Finch, C.; Billett, E.; Mahy, N. Localization of monoamine oxidase A and B and semicarbazide-sensitive amine oxidase in human peripheral tissues. Inflammopharmacology 2003, 11, 111–117. [Google Scholar] [CrossRef]
- Andrés, N.; Lizcano, J.M.; Rodríguez, M.J.; Romera, M.; Unzeta, M.; Mahy, N. Tissue activity and cellular localization of human semicarbazide-sensitive amine oxidase. J. Histochem. Cytochem. 2001, 49, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, M.; MacDougall, M.B.; Unzeta, M.; Lizcano, J.-M.; Tipton, K.F. Semicarbazide-sensitive amine oxidases in pig dental pulp. Biochim. et Biophys. Acta (BBA) -Bioenerg. 2003, 1647, 333–336. [Google Scholar] [CrossRef]
- O’Sullivan, M.; Tipton, K.F.; E McDevitt, W. Immunolocalization of semicarbazide-sensitive amine oxidase in human dental pulp and its activity towards serotonin. Arch. Oral Biol. 2002, 47, 399–406. [Google Scholar] [CrossRef]
- Vavilova, T.; Ostrovskaya, I.; Axenova, L.; Buneeva, O.; Medvedev, A. Monoamine oxidase and semicarbazide sensitive amine oxidase activities in normal and inflamed human dental pulp. Med Sci. Monit. 2009, 15, 289–292. [Google Scholar]
- Lizcano, J.M.; Tipton, K.F.; Unzeta, M. Purification and characterization of membrane-bound semicarbazide-sensitive amine oxidase (SSAO) from bovine lung. Biochem. J. 1998, 331, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, V.; Lizcano, J.M.; Visa, J.; Unzeta, M. Semicarbazide-sensitive amine oxidase (SSAO) from human and bovine cerebrovascular tissues: Biochemical and immunohistological characterization. Neurochem. Int. 1998, 33, 415–423. [Google Scholar] [CrossRef]
- Morin, N.; Lizcano, J.M.; Fontana, E.; Marti, L.; Smih, F.; Rouet, P.; Prevot, D.; Zorzano, A.; Unzeta, M.; Carpene, C. Semi-carbazide-sensitive amine oxidase substrates stimulate glucose transport and inhibit lipolysis in human adipocytes. J Pharma-col Exp. Ther. 2001, 297, 563–572. [Google Scholar]
- Salmi, M.; Kalimo, K.; Jalkanen, S. Induction and function of vascular adhesion protein-1 at sites of inflammation. J. Exp. Med. 1993, 178, 2255–2260. [Google Scholar] [CrossRef] [Green Version]
- Salmi, M.; Yegutkin, G.G.; Lehvonen, R.; Koskinen, K.; Salminen, T.; Jalkanen, S. A Cell Surface Amine Oxidase Directly Controls Lymphocyte Migration. Immunity 2001, 14, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Zuo, D.-M.; Yu, P.H. Semicarbazide-sensitive amine oxidase and monoamine oxidase in rat brain microvessels, meninges, retina and eye sciera. Brain Res. Bull. 1994, 33, 307–311. [Google Scholar] [CrossRef]
- Castillo, V.; Lizcano, J.M.; Unzeta, M. Presence of SSAO in human and bovine meninges and microvessels. Neurobiology 1999, 7, 263–272. [Google Scholar] [PubMed]
- Smeraldi, C.; Castillo, V.; Lizcano, J.M.; Unzeta, M. Some properties of semicarbazide-sensitive amine oxidase (SSAO) from human cerebrovascular tissues. Inflamm. Res. 2001, 50, 144–145. [Google Scholar]
- Jaakkola, K.; Kaunismäki, K.; Tohka, S.; Yegutkin, G.; Vänttinen, E.; Havia, T.; Pelliniemi, L.J.; Virolainen, M.; Jalkanen, S.; Salmi, M. Human Vascular Adhesion Protein-1 in Smooth Muscle Cells. Am. J. Pathol. 1999, 155, 1953–1965. [Google Scholar] [CrossRef] [Green Version]
- Souto, R.P.; Vallega, G.; Wharton, J.; Vinten, J.; Tranum-Jensen, J.; Pilch, P.F. Immunopurification and Characterization of Rat Adipocyte Caveolae Suggest Their Dissociation from Insulin Signaling. J. Biol. Chem. 2003, 278, 18321–18329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.A.; London, E. Structure and Function of Sphingolipid- and Cholesterol-rich Membrane Rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [Green Version]
- Boomsma, F.; Bhaggoe, U.M.; Van Der Houwen, A.M.; Meiracker, A.H.V.D. Plasma semicarbazide-sensitive amine oxidase in human (patho)physiology. Biochim. et Biophys. Acta (BBA) -Bioenerg. 2003, 1647, 48–54. [Google Scholar] [CrossRef]
- Tipton, K.F.; Strolin Benedetti, M. Amine oxidases and the metabolism of xenobiotics. Enzym. Syst. Metab. Drugs Other Xenobiotics 2001, 95. [Google Scholar] [CrossRef]
- Kunsch, C.; Medford, R.M. Oxidative Stress as a Regulator of Gene Expression in the Vasculature. Circ. Res. 1999, 85, 753–766. [Google Scholar] [CrossRef]
- Finkel, T. Oxygen radicals and signaling. Curr. Opin. Cell Biol. 1998, 10, 248–253. [Google Scholar] [CrossRef]
- Fischer, Y.; Thomas, J.; Sevilla, L.; Muñoz, P.; Becker, C.; Holman, G.; Kozka, I.J.; Palacín, M.; Testar, X.; Kammermeier, H.; et al. Insulin-induced Recruitment of Glucose Transporter 4 (GLUT4) and GLUT1 in Isolated Rat Cardiac Myocytes. J. Biol. Chem. 1997, 272, 7085–7092. [Google Scholar] [CrossRef] [Green Version]
- Enrique-Tarancón, G.; Marti, L.; Morin, N.; Lizcano, J.; Unzeta, M.; Sevilla, L.; Camps, M.; Palacín, M.; Testar, X.; Carpéné, C.; et al. Role of Semicarbazide-sensitive Amine Oxidase on Glucose Transport and GLUT4 Recruitment to the Cell Surface in Adipose Cells. J. Biol. Chem. 1998, 273, 8025–8032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorzano, A. Semicarbazide-sensitive amine oxidase activity exerts insulin-like effects on glucose metabolism and insulin-signaling pathways in adipose cells. Biochim. et Biophys. Acta (BBA) -Proteins Proteom. 2003, 1647, 3–9. [Google Scholar] [CrossRef]
- Salmi, M.; Jalkanen, S. VAP-1: An adhesin and an enzyme. Trends Immunol. 2001, 22, 211–216. [Google Scholar] [CrossRef]
- Butcher, E.C.; Picker, L.J. Lymphocyte Homing and Homeostasis. Science 1996, 272, 60–67. [Google Scholar] [CrossRef]
- Salmi, M.; Jalkanen, S. How Do Lymphocytes Know Where to Go: Current Concepts and Enigmas of Lymphocyte Homing. Adv. Immunol. 1997, 64, 139–218. [Google Scholar] [CrossRef]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Salmi, M.; Jalkanen, S. Cell-surface enzymes in control of leukocyte trafficking. Nat. Rev. Immunol. 2005, 5, 760–771. [Google Scholar] [CrossRef]
- Lalor, P.F.; Sun, P.J.; Weston, C.J.; Wakelam, M.J.O.; Adams, D.H.; Martin-Santos, A. Activation of vascular adhesion protein-1 on liver endothelium results in an NF-κB–dependent increase in lymphocyte adhesion. Hepatology 2007, 45, 465–474. [Google Scholar] [CrossRef]
- Arvilommi, A.-M.; Salmi, M.; Jalkanen, S. Organ-selective regulation of vascular adhesion protein-1 expression in man. Eur. J. Immunol. 1997, 27, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Tanaka, T.; Kawakami, T.; Takano, H.; Sugahara, M.; Saito, H.; Higashijima, Y.; Yamaguchi, J.; Inagi, R.; Nangaku, M. Vascular adhesion protein-1 enhances neutrophil infiltration by generation of hydrogen peroxide in renal ischemia/reperfusion injury. Kidney Int. 2017, 92, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-L.; Salter-Cid, L.; Linnik, M.D.; Wang, E.Y.; Paisansathan, C.; Pelligrino, D.A. Vascular Adhesion Protein-1 Plays an Important Role in Postischemic Inflammation and Neuropathology in Diabetic, Estrogen-Treated Ovariectomized Female Rats Subjected to Transient Forebrain Ischemia. J. Pharmacol. Exp. Ther. 2005, 317, 19–29. [Google Scholar] [CrossRef]
- Xu, H.; Testai, F.D.; Valyi-Nagy, T.; Pavuluri, M.N.; Zhai, F.; Nanegrungsunk, D.; Paisansathan, C.; Pelligrino, D.A. VAP-1 blockade prevents subarachnoid hemorrhage-associated cerebrovascular dilating dysfunction via repression of a neutrophil recruitment-related mechanism. Brain Res. 2015, 1603, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Manaenko, A.; Khatibi, N.H.; Chen, W.; Zhang, J.H.; Tang, J. Vascular Adhesion Protein-1 Inhibition Provides Antiinflammatory Protection after an Intracerebral Hemorrhagic Stroke in Mice. Br. J. Pharmacol. 2010, 31, 881–893. [Google Scholar] [CrossRef] [Green Version]
- Merinen, M.; Irjala, H.; Salmi, M.; Jaakkola, I.; Hänninen, A.; Jalkanen, S. Vascular Adhesion Protein-1 Is Involved in Both Acute and Chronic Inflammation in the Mouse. Am. J. Pathol. 2005, 166, 793–800. [Google Scholar] [CrossRef] [Green Version]
- Becchi, S.; Buson, A.; Foot, J.; Jarolimek, W.; Balleine, B.W. Inhibition of semicarbazide-sensitive amine oxidase/vascular adhesion protein-1 reduces lipopolysaccharide-induced neuroinflammation. Br. J. Pharmacol. 2017, 174, 2302–2317. [Google Scholar] [CrossRef] [Green Version]
- Schilter, H.C.; Collison, A.; Russo, R.C.; Foot, J.S.; Yow, T.T.; Vieira, A.T.; Tavares, L.D.; Mattes, J.; Teixeira, M.M.; Jarolimek, W. Effects of an anti-inflammatory VAP-1/SSAO inhibitor, PXS-4728A, on pulmonary neutrophil migration. Respir. Res. 2015, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Tuncer, C.; Oo, Y.H.; Murphy, N.; Adams, D.H.; Lalor, P.F. The regulation of T-cell recruitment to the human liver during acute liver failure. Liver Int. 2013, 33, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Bonder, C.S.; Norman, M.U.; Swain, M.G.; Zbytnuik, L.D.; Yamanouchi, J.; Santamaria, P.; Ajuebor, M.; Salmi, M.; Jalkanen, S.; Kubes, P. Rules of Recruitment for Th1 and Th2 Lymphocytes in Inflamed Liver: A Role for Alpha-4 Integrin and Vascular Adhesion Protein-1. Immunity 2005, 23, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspinall, A.I.; Curbishley, S.M.; Lalor, P.F.; Weston, C.J.; Blahova, M.; Liaskou, E.; Adams, R.M.; Holt, A.P.; Adams, D.H. CX3CR1 and vascular adhesion protein-1-dependent recruitment of CD16+ monocytes across human liver sinusoidal endothelium. Hepatology 2010, 51, 2030–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, S.; Weston, C.J.; Oo, Y.H.; Westerlund, N.; Stamataki, Z.; Youster, J.; Hubscher, S.G.; Salmi, M.; Jalkanen, S.; Lalor, P.F.; et al. Common Lymphatic Endothelial and Vascular Endothelial Receptor-1 Mediates the Transmigration of Regulatory T Cells across Human Hepatic Sinusoidal Endothelium. J. Immunol. 2011, 186, 4147–4155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelius, T.; Salmi, M.; Krogerus, L.; Loginov, R.; Schoultz, M.; Karikoski, M.; Miiluniemi, M.; Soots, A.; Höckerstedt, K.; Jalkanen, S.; et al. Inhibition of Semicarbazide-Sensitive Amine Oxidases Decreases Lymphocyte Infiltration in the Early Phases of Rat Liver Allograft Rejection. Int. J. Immunopathol. Pharmacol. 2008, 21, 911–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelius, T.; Salaspuro, V.; Salmi, M.; Krogerus, L.; Höckerstedt, K.; Jalkanen, S.; Lautenschlager, I. Blockade of Vascular Adhesion Protein-1 Inhibits Lymphocyte Infiltration in Rat Liver Allograft Rejection. Am. J. Pathol. 2004, 165, 1993–2001. [Google Scholar] [CrossRef] [Green Version]
- Marttila-Ichihara, F.; Castermans, K.; Auvinen, K.; Egbrink, M.G.A.O.; Jalkanen, S.; Griffioen, A.W.; Salmi, M. Small-Molecule Inhibitors of Vascular Adhesion Protein-1 Reduce the Accumulation of Myeloid Cells into Tumors and Attenuate Tumor Growth in Mice. J. Immunol. 2010, 184, 3164–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, S.; Noda, K.; Zandi, S.; Sun, D.; Taher, M.; Schering, A.; Xie, F.; Mashima, Y.; Hafezi-Moghadam, A. VAP-1–Mediated M2 Macrophage Infiltration Underlies IL-1β– but Not VEGF-A–Induced Lymph- and Angiogenesis. Am. J. Pathol. 2011, 178, 1913–1921. [Google Scholar] [CrossRef]
- Noda, K.; Nakao, S.; Zandi, S.; Engelstädter, V.; Mashima, Y.; Hafezi-Moghadam, A. Vascular adhesion protein-1 regulates leukocyte transmigration rate in the retina during diabetes. Exp. Eye Res. 2009, 89, 774–781. [Google Scholar] [CrossRef] [Green Version]
- Noda, K.; Miyahara, S.; Nakazawa, T.; Almulki, L.; Nakao, S.; Hisatomi, T.; She, H.; Thomas, K.L.; Garland, R.C.; Miller, J.W.; et al. Inhibition of vascular adhesion protein-1 suppresses endotoxin-induced uveitis. FASEB J. 2007, 22, 1094–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmi, M.; Tohka, S.; Jalkanen, S. Human vascular adhesion protein-1 (VAP-1) plays a critical role in lymphocyte-endothelial cell adhesion cascade under shear. Circ. Res. 2000, 86, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, K.; Vainio, P.J.; Smith, D.J.; Pihlavisto, M.; Ylä-Herttuala, S.; Jalkanen, S.; Salmi, M. Granulocyte transmigration through the endothelium is regulated by the oxidase activity of vascular adhesion protein-1 (VAP-1). Blood 2004, 103, 3388–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bour, S.; Caspar-Bauguil, S.; Iffiú-Soltész, Z.; Nibbelink, M.; Cousin, B.; Miiluniemi, M.; Salmi, M.; Stolen, C.; Jalkanen, S.; Casteilla, L.; et al. Semicarbazide-Sensitive Amine Oxidase/Vascular Adhesion Protein-1 Deficiency Reduces Leukocyte Infiltration into Adipose Tissue and Favors Fat Deposition. Am. J. Pathol. 2009, 174, 1075–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyus, P.; Dajka-Halasz, B.; Foldi, A.; Haider, N.; Barlocco, D.; Magyar, K. Semicarbazide-sensitive amine oxidase: Current status and perspectives. Curr. Med. Chem. 2004, 11, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Solé, M.; Hernandez-Guillamon, M.; Boada, M.; Unzeta, M. p53 phosphorylation is involved in vascular cell death induced by the catalytic activity of membrane-bound SSAO/VAP-1. Biochim. et Biophys. Acta (BBA) -Bioenerg. 2008, 1783, 1085–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, A.; Desai, K.; Kazachmov, M.; Yu, P.; Wu, L. Methylglyoxal production in vascular smooth muscle cells from different metabolic precursors. Metabolism 2008, 57, 1211–1220. [Google Scholar] [CrossRef]
- Mathys, K.C.; Ponnampalam, S.N.; Padival, S.; Nagaraj, R.H. Semicarbazide-sensitive amine oxidase in aortic smooth muscle cells mediates synthesis of a methylglyoxal-AGE: Implications for vascular complications in diabetes. Biochem. Biophys. Res. Commun. 2002, 297, 863–869. [Google Scholar] [CrossRef]
- Kurkijärvi, R.; Adams, D.H.; Leino, R.; Möttönen, T.; Jalkanen, S.; Salmi, M. Circulating form of human vascular adhesion protein-1 (VAP-1): Increased serum levels in inflammatory liver diseases. J. Immunol. 1998, 161, 1549–1557. [Google Scholar]
- Kurkijärvi, R.; Yegutkin, G.G.; Gunson, B.K.; Jalkanen, S.; Salmi, M.; Adams, D.H. Circulating soluble vascular adhesion protein 1 accounts for the increased serum monoamine oxidase activity in chronic liver disease. Gastroenterology 2000, 119, 1096–1103. [Google Scholar] [CrossRef]
- Boomsma, F.; van Veldhuisen, D.J.; de Kam, P.J.; Man in’t Veld, A.J.; Mosterd, A.; Lie, K.I.; Schalekamp, M.A. Plasma semi-carbazide-sensitive amine oxidase is elevated in patients with congestive heart failure. Cardiovasc. Res. 1997, 33, 387–391. [Google Scholar] [CrossRef]
- Hernandez-Guillamon, M.; Garcia-Bonilla, L.; Solé, M.; Sosti, V.; Parés, M.; Campos, M.; Ortega-Aznar, A.; Domínguez, C.; Rubiera, M.; Ribó, M.; et al. Plasma VAP-1/SSAO Activity Predicts Intracranial Hemorrhages and Adverse Neurological Outcome After Tissue Plasminogen Activator Treatment in Stroke. Stroke 2010, 41, 1528–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garpenstrand, H.; Ekblom, J.; Von Arbin, M.; Oreland, L.; Murray, V. Plasma Semicarbazide-Sensitive Amine Oxidase in Stroke. Eur. Neurol. 1999, 41, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, F. Plasma Benzylamine Oxidase Activity in Cerebrovascular Disease. Eur. Neurol. 1990, 30, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Guillamon, M.; Solé, M.; Delgado, P.; García-Bonilla, L.; Giralt, D.; Boada, C.; Penalba, A.; Garcia, S.; Flores, A.; Ribó, M.; et al. VAP-1/SSAO Plasma Activity and Brain Expression in Human Hemorrhagic Stroke. Cerebrovasc. Dis. 2012, 33, 55–63. [Google Scholar] [CrossRef]
- Ferrer, I.; Lizcano, J.; Hernández, M.; Unzeta, M. Overexpression of semicarbazide sensitive amine oxidase in the cerebral blood vessels in patients with Alzheimer’s disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Neurosci. Lett. 2002, 321, 21–24. [Google Scholar] [CrossRef]
- Hernandez, M.D.M.; Esteban, M.; Szabo, P.; Boada, M.; Unzeta, M. Human plasma semicarbazide sensitive amine oxidase (SSAO), β-amyloid protein and aging. Neurosci. Lett. 2005, 384, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Wang, W.; Luo, W.; Lv, J.; Li, H.; Luo, H.; Jia, J.; He, R. Urine Formaldehyde Predicts Cognitive Impairment in Post-Stroke Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 55, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Brands, A.M.; Kessels, R.P.; De Haan, E.H.; Kappelle, L.; Biessels, G.J. Cerebral dysfunction in type 1 diabetes: Effects of insulin, vascular risk factors and blood-glucose levels. Eur. J. Pharmacol. 2004, 490, 159–168. [Google Scholar] [CrossRef]
- Carro, E.; Torres-Aleman, I. The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur. J. Pharmacol. 2004, 490, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haan, M.N. Therapy Insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Garpenstrand, H.; Ekblom, J.; Backlund, L.B.; Oreland, L.; Rosenqvist, U. Elevated plasma semicarbazide-sensitive amine oxidase (SSAO) activity in Type 2 diabetes mellitus complicated by retinopathy. Diabet. Med. 1999, 16, 514–521. [Google Scholar] [CrossRef]
- Irjala, H.; Salmi, M.; Alanen, K.; Grénman, R.; Jalkanen, S. Vascular Adhesion Protein 1 Mediates Binding of Immunotherapeutic Effector Cells to Tumor Endothelium. J. Immunol. 2001, 166, 6937–6943. [Google Scholar] [CrossRef] [PubMed]
- Abella, A.; Garcia-Vicente, S.; Viguerie, N.; Ros-Baro, A.; Camps, M.; Palacín, M.; Zorzano, A.; Marti, L. Adipocytes release a soluble form of VAP-1/SSAO by a metalloprotease-dependent process and in a regulated manner. Diabetologia 2004, 47, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Stolen, C.M.; Yegutkin, G.G.; Kurkijärvi, R.; Bono, P.; Alitalo, K.; Jalkanen, S. Origins of Serum Semicarbazide-Sensitive Amine Oxidase. Circ. Res. 2004, 95, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boomsma, F.; Meiracker, A.H.V.D.; Winkel, S.; Aanstoot, H.J.; Batstra, M.R.; Veld, A.J.M.I. ’T; Bruining, G.J. Circulating semicarbazide-sensitive amine oxidase is raised both in Type I (insulin-dependent), in Type II (non-insulin-dependent) diabetes mellitus and even in childhood Type I diabetes at first clinical diagnosis. Diabetologia 1999, 42, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mészáros, Z.; Karádi, I.; Csányi, A.; Szombathy, T.; Romics, L.; Magyar, K. Determination of human serum semicarbazide-sensitive amine oxidase activity: A possible clinical marker of atherosclerosis. Eur. J. Drug Metab. Pharmacokinet. 1999, 24, 299–302. [Google Scholar] [CrossRef]
- Grönvall-Nordquist, J.L.; Bäcklund, L.B.; Garpenstrand, H.; Ekblom, J.; Landin, B.; Yu, P.H.; Oreland, L.; Rosenqvist, U. Follow-up of plasma semicarbazide-sensitive amine oxidase activity and retinopathy in Type 2 diabetes mellitus. J. Diabetes its Complicat. 2001, 15, 250–256. [Google Scholar] [CrossRef]
- Karádi, I.; Mészáros, Z.; Csányi, A.; Szombathy, T.; Hosszúfalusi, N.; Romics, L.; Magyar, K. Serum semicarbazide-sensitive amine oxidase (SSAO) activity is an independent marker of carotid atherosclerosis. Clin. Chim. Acta 2002, 323, 139–146. [Google Scholar] [CrossRef]
- Weiss, H.G.; Klocker, J.; Labeck, B.; Nehoda, H.; Aigner, F.; Klingler, A.; Ebenbichler, C.; Föger, B.; Lechleitner, M.; Patsch, J.R.; et al. Plasma amine oxidase: A postulated cardiovascular risk factor in nondiabetic obese patients. Metabolism 2003, 52, 688–692. [Google Scholar] [CrossRef]
- Yu, P.H.; Zuo, D.-M. Oxidative Deamination of Methylamine by Semicarbazide-Sensitive Amine Oxidase Leads to Cytotoxic Damage in Endothelial Cells: Possible Consequences for Diabetes. Diabetes 1993, 42, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Vainio, P.J. Targeting Vascular Adhesion Protein-1 to Treat Autoimmune and Inflammatory Diseases. Ann. New York Acad. Sci. 2007, 1110, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Lewinsohn, R. Human serum amine oxidase. Enzyme activity in severely burnt patients and in patients with cancer. Clin. Chim. Acta 1977, 81, 247–256. [Google Scholar] [CrossRef]
- Lizcano, J.; Escrich, E.; Ribalta, T.; Muntané, J.; Unzeta, M. Amine oxidase activities in rat breast cancer induced experimentally with 7,12-dimethylbenz(α)anthracene. Biochem. Pharmacol. 1991, 42, 263–269. [Google Scholar] [CrossRef]
- Chang, S.-J.; Tu, H.-P.; Lai, Y.-C.C.; Luo, C.-W.; Nejo, T.; Tanaka, S.; Chai, C.-Y.; Kwan, A.-L. Increased Vascular Adhesion Protein 1 (VAP-1) Levels Are Associated with Alternative M2 Macrophage Activation and Poor Prognosis for Human Gliomas. Diagnostics 2020, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Kostoro, J.; Chang, S.-J.; Lai, Y.-C.C.; Wu, C.-C.; Chai, C.-Y.; Kwan, A.-L. Overexpression of vascular adhesion protein-1 is associated with poor prognosis of astrocytomas. APMIS 2016, 124, 462–468. [Google Scholar] [CrossRef]
- Airas, L.; Mikkola, J.; Vainio, J.M.; Elovaara, I.; Smith, D.J. Elevated serum soluble vascular adhesion protein-1 (VAP-1) in patients with active relapsing remitting multiple sclerosis. J. Neuroimmunol. 2006, 177, 132–135. [Google Scholar] [CrossRef]
- Elo, P.; Tadayon, S.; Liljenbäck, H.; Teuho, J.; Käkelä, M.; Koskensalo, K.; Saunavaara, V.; Virta, J.; Veres, T.Z.; Kiviniemi, A. Vascular adhesion protein-1 is actively involved in the development of inflammatory lesions in rat models of multiple sclero-sis. J. Neuroinflammation 2018, 15, 1–17. [Google Scholar] [CrossRef]
- Pannecoeck, R.; Serruys, D.; Benmeridja, L.; Delanghe, J.R.; Van Geel, N.; Speeckaert, R.; Speeckaert, M.M. Vascular adhesion protein-1: Role in human pathology and application as a biomarker. Crit. Rev. Clin. Lab. Sci. 2015, 52, 284–300. [Google Scholar] [CrossRef]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood–brain barrier: An overview. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaria, R.N. Cerebral vessels in ageing and Alzheimer’s disease. Pharmacol. Ther. 1996, 72, 193–214. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Zhang, K.; Li, P.; Zhu, L.; Xu, J.; Yang, B.; Hu, X.; Lu, Z.; Chen, J. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Res. Rev. 2017, 34, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takechi, R.; Lam, V.; Brook, E.; Giles, C.; Fimognari, N.; Mooranian, A.; Al-Salami, H.; Coulson, S.H.; Nesbit, M.; Mamo, J.C.L. Blood-Brain Barrier Dysfunction Precedes Cognitive Decline and Neurodegeneration in Diabetic Insulin Resistant Mouse Model: An Implication for Causal Link. Front. Aging Neurosci. 2017, 9, 399. [Google Scholar] [CrossRef]
- Toth, P.; Tarantini, S.; Csiszar, A.; Ungvari, Z. Functional vascular contributions to cognitive impairment and dementia: Mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am. J. Physiol. Circ. Physiol. 2017, 312, H1–H20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Zacchigna, S.; Lambrechts, D.; Carmeliet, P. Neurovascular signalling defects in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, D.A.; Jin, K. From angiogenesis to neuropathology. Nat. Cell Biol. 2005, 438, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.W.; Filosa, J.A. Stroke and the neurovascular unit: Glial cells, sex differences, and hypertension. Am. J. Physiol. Physiol. 2019, 316, C325–C339. [Google Scholar] [CrossRef] [PubMed]
- Michalicova, A.; A Banks, W.; Legath, J.; Kovac, A. Tauopathies-focus on changes at the neurovascular unit. Curr. Alz-heimer Res. 2017, 14, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Rom, S.; Ramirez, S.H.; Persidsky, Y. Emerging Roles of Pericytes in the Regulation of the Neurovascular Unit in Health and Disease. J. Neuroimmune Pharmacol. 2014, 9, 591–605. [Google Scholar] [CrossRef] [Green Version]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 2011, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Thundyil, J.; Tang, S.-C.; Sobey, C.G.; Taylor, S.M.; Arumugam, T.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003, 4, 399–414. [Google Scholar] [CrossRef]
- E Lakhan, S.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Fiskum, G.; Murphy, A.N.; Beal, M.F. Mitochondria in Neurodegeneration: Acute Ischemia and Chronic Neurodegenerative Diseases. Br. J. Pharmacol. 1999, 19, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.H. Reactive Oxygen Radicals in Signaling and Damage in the Ischemic Brain. Br. J. Pharmacol. 2001, 21, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.V.; Woodruff, T.M.; Lathia, J.D.; Selvaraj, P.K.; Mattson, M.P.; Taylor, S.M. Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience 2009, 158, 1074–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, S.-M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147, S232–S240. [Google Scholar] [CrossRef] [Green Version]
- Swanson, R.A.; Ying, W.; Kauppinen, T.M. Astrocyte Influences on Ischemic Neuronal Death. Curr. Mol. Med. 2004, 4, 193–205. [Google Scholar] [CrossRef]
- Schroeter, M.; Jander, S.; Witte, O.W.; Stoll, G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J. Neuroimmunol. 1994, 55, 195–203. [Google Scholar] [CrossRef]
- Zhang, R.; Chopp, M.; Zhang, Z.; Jiang, N.; Powers, C. The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Res. 1998, 785, 207–214. [Google Scholar] [CrossRef]
- Lindsberg, P.J.; Carpe’ n, O.; Paetau, A.; Karjalainen-Lindsberg, M.-L.; Kaste, M. Endothelial ICAM-1 expression associated with inflammatory cell response in human ischemic stroke. Circulation 1996, 94, 939–945. [Google Scholar] [CrossRef]
- Rallidis, L.S.; Zolindaki, M.G.; Vikelis, M.; Kaliva, K.; Papadopoulos, C.; Kremastinos, D.T. Elevated soluble intercellular ad-hesion molecule-1 levels are associated with poor short-term prognosis in middle-aged patients with acute ischaemic stroke. Int. J. Cardiol. 2009, 132, 216–220. [Google Scholar] [CrossRef]
- Kivipelto, M.; Helkala, E.-L.; Laakso, M.P.; Hänninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. BMJ 2001, 322, 1447–1451. [Google Scholar] [CrossRef] [Green Version]
- Mayeux, R. Epidemiology Ofneurodegeneration. Annu. Rev. Neurosci. 2003, 26, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Poirier, J.; Bertrand, P.; Kogan, S.; Gauthier, S.; Davignon, J.; Bouthillier, D. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 1993, 342, 697–699. [Google Scholar] [CrossRef]
- Solis, E., Jr.; Hascup, K.N.; Hascup, E.R. Alzheimer’s disease: The link between amyloid-β and neurovascular dysfunction. J. Alzheimer’s Dis. 2020, 76, 1179–1198. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease: Genes, Proteins, and Therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Vassar, R. BACE1: The β-Secretase Enzyme in Alzheimer’s Disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Hussain, I.; Powell, D.; Howlett, D.R.; Tew, D.G.; Meek, T.D.; Chapman, C.; Gloger, I.S.; Murphy, K.E.; Southan, C.D.; Ryan, D.M.; et al. Identification of a Novel Aspartic Protease (Asp 2) as β-Secretase. Mol. Cell. Neurosci. 1999, 14, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J. Puri-fication and cloning of amyloid precursor protein β-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carson, J.A.; Turner, A.J. β-Amyloid catabolism: Roles for neprilysin (NEP) and other metallopeptidases? J. Neurochem. 2002, 81, 1–8. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Tanzi, R.; Moir, R.; Wagner, S. Clearance of Alzheimer’s Aβ Peptide. Neuron 2004, 43, 605–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/Amyloid β-Peptide Interaction Mediates Differential Brain Efflux of Aβ Isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Van Nostrand, W.E.; Melchor, J.P.; Romanov, G.; Zeigler, K.; Davis, J. Pathogenic Effects of Cerebral Amyloid Angiopathy Mutations in the Amyloid β-Protein Precursor. Ann. N. Y. Acad. Sci. 2002, 977, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Rostagno, A.; Ghiso, J.; Frangione, B. Sporadic and familial cerebral amyloid angiopathies. Brain Pathol. 2006, 12, 343–357. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; Van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease — one peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Leurgans, S.E.; Wang, Z.; Wilson, R.S.; Bennett, D.A.; Schneider, J.A. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 2010, 69, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Wattendorff, A.R.; Frangione, B.; Luyendijk, W.; Bots, G.T. Hereditary cerebral haemorrhage with amyloidosis, Dutch type (HCHWA-D): Clinicopathological studies. J. Neurol. Neurosurg. Psychiatry 1995, 58, 699–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grammas, P. A damaged microcirculation contributes to neuronal cell death in Alzheimer’s disease☆. Neurobiol. Aging 2000, 21, 199–205. [Google Scholar] [CrossRef]
- Snyder, H.M.; Corriveau, R.A.; Craft, S.; Faber, J.E.; Greenberg, S.M.; Knopman, D.; Lamb, B.T.; Montine, T.J.; Nedergaard, M.; Schaffer, C.B.; et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 710–717. [Google Scholar] [CrossRef] [Green Version]
- Iturria-Medina, Y.; Initiative, T.A.D.N.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, C.E.; Hays, C.C.; Zlatar, Z.Z. Cerebral Blood Flow Measured by Arterial Spin Labeling MRI as a Preclinical Marker of Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 42, S411–S419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Xing, A.; Xu, C.; Cai, Q.; Liu, H.; Li, L. Cerebrovascular Hypoperfusion Induces Spatial Memory Impairment, Synaptic Changes, and Amyloid-β Oligomerization in Rats. J. Alzheimer’s Dis. 2010, 21, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; Van Osch, M.J.P.; Van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2017, 282, 615. [Google Scholar] [CrossRef] [PubMed]
- Aliev, G.; Priyadarshini, M.; P Reddy, V.; Grieg, N.H.; Kaminsky, Y.; Cacabelos, R.; Md Ashraf, G.; Jabir, N.R.; Kamal, M.A.; Nikolenko, V. Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease. Curr. Med. Chem. 2014, 21, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Suo, Z.; Tan, J.; Placzek, A.; Crawford, F.; Fang, C.; Mullan, M. Alzheimer’s β-amyloid peptides induce inflammatory cascade in human vascular cells: The roles of cytokines and CD40. Brain Res. 1998, 807, 110–117. [Google Scholar] [CrossRef]
- Thirumangalakudi, L.; Samany, P.G.; Owoso, A.; Wiskar, B.; Grammas, P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 10, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, G.; Cavalieri, M.; Galvani, M.; Passaro, A.; Munari, M.; Bosi, C.; Zurlo, A.; Fellin, R. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J. Neurol. Sci. 2008, 272, 164–170. [Google Scholar] [CrossRef]
- MacKenzie, I.R.; Hao, C.; Muñoz, D.G. Role of microglia in senile plaque formation. Neurobiol. Aging 1995, 16, 797–804. [Google Scholar] [CrossRef]
- Emmerling, M.R.; Watson, M.; Raby, C.A.; Spiegel, K. The role of complement in Alzheimer’s disease pathology. Biochim. et Biophys. Acta (BBA) -Mol. Basis Dis. 2000, 1502, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Michaud, J.-P.; Hallé, M.; Lampron, A.; Thériault, P.; Préfontaine, P.; Filali, M.; Tribout-Jover, P.; Lanteigne, A.-M.; Jodoin, R.; Cluff, C.; et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc. Natl. Acad. Sci. USA 2013, 110, 1941–1946. [Google Scholar] [CrossRef] [Green Version]
- Lue, L.-F.; Brachova, L.; Civin, W.H.; Rogers, J. Inflammation, Aβ deposition, and neurofibrillary tangle formation as corre-lates of Alzheimer’s disease neurodegeneration. J. Neuropathol. Exp. Neurol. 1996, 55, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Airas, L.; Lindsberg, P.J.; Karjalainen-Lindsberg, M.-L.; Mononen, I.; Kotisaari, K.; Smith, D.J.; Jalkanen, S. Vascular adhesion protein-1 in human ischaemic stroke. Neuropathol. Appl. Neurobiol. 2008, 34, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Ziliotto, N.; Zivadinov, R.; Jakimovski, D.; Bergsland, N.; Ramasamy, D.P.; Weinstock-Guttman, B.; Ramanathan, M.; Marchetti, G.; Bernardi, F. Are Plasma Levels of Vascular Adhesion Protein-1 Associated Both with Cerebral Microbleeds in Multiple Sclerosis and Intracerebral Haemorrhages in Stroke? Thromb. Haemost. 2018, 119, 175–178. [Google Scholar] [CrossRef]
- Kiss, J.; Jalkanen, S.; Fülöp, F.; Savunen, T.; Salmi, M. Ischemia-reperfusion injury is attenuated in VAP-1-deficient mice and by VAP-1 inhibitors. Eur. J. Immunol. 2008, 38, 3041–3049. [Google Scholar] [CrossRef]
- Yang, W.; Li, H.; Luo, H.; Luo, W. Inhibition of semicarbazide-sensitive amine oxidase attenuates myocardial ischemia–reperfusion injury in an in vivo rat model. Life Sci. 2011, 88, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, S.; Karikoski, M.; Mercier, N.; Koskinen, K.; Henttinen, T.; Elima, K.; Salmivirta, K.; Salmi, M. The oxidase activity of vascular adhesion protein-1 (VAP-1) induces endothelial E- and P-selectins and leukocyte binding. Blood 2007, 110, 1864–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Hernandez-Guillamón, M.; Campos-Martorell, M.; Simats, A.; Montaner, J.; Unzeta, M.; Solé, M. Simvastatin blocks soluble SSAO/VAP-1 release in experimental models of cerebral ischemia: Possible benefits for stroke-induced inflammation control. Biochim. et Biophys. Acta (BBA) -Mol. Basis Dis. 2018, 1864, 542–553. [Google Scholar] [CrossRef]
- Sun, P.; Solé, M.; Unzeta, M. Involvement of SSAO/VAP-1 in Oxygen-Glucose Deprivation-Mediated Damage Using the Endothelial hSSAO/VAP-1-Expressing Cells as an Experimental Model of Cerebral Ischemia. Cerebrovasc. Dis. 2014, 37, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.; Solé, M.; Boada, M.; Unzeta, M. Soluble Semicarbazide Sensitive Amine Oxidase (SSAO) catalysis induces apoptosis in vascular smooth muscle cells. Biochim. et Biophys. Acta (BBA) -Bioenerg. 2006, 1763, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, P. Involvement of cerebrovascular semicarbazide-sensitive amine oxidase in the pathogenesis of Alzheimer’s disease and vascular dementia. Med. Hypotheses 2001, 57, 175–179. [Google Scholar] [CrossRef]
- Valente, T.; Gella, A.; Fernàndez-Busquets, X.; Unzeta, M.; Durany, N. Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer’s disease and diabetes mellitus. Neurobiol. Dis. 2010, 37, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, T.; Gella, A.; Solé, M.; Durany, N.; Unzeta, M. Immunohistochemical study of semicarbazide-sensitive amine oxidase/vascular adhesion protein-1 in the hippocampal vasculature: Pathological synergy of Alzheimer’s disease and diabetes mellitus. J. Neurosci. Res. 2012, 90, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Broce, I.J.; Tan, C.H.; Fan, C.C.; Jansen, I.; Savage, J.E.; Witoelar, A.; Wen, N.; Hess, C.P.; Dillon, W.P.; Glastonbury, C.M.; et al. Dissecting the genetic relationship between cardiovascular risk factors and Alzheimer’s disease. Acta Neuropathol. 2019, 137, 209–226. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-H.; Yu, T.-Y.; Tsai, F.-C.; Weston, C.J.; Lin, M.-S.; Hung, C.-S.; Kao, H.-L.; Li, Y.-I.; Solé, M.; Unzeta, M.; et al. Inhibition of semicarbazide-sensitive amine oxidase reduces atherosclerosis in apolipoprotein E-deficient mice. Transl. Res. 2018, 197, 12–31. [Google Scholar] [CrossRef]
- Boor, P.J.; Trent, M.B.; Lyles, G.A.; Tao, M.; Ansari, G. Methylamine metabolism to formaldehyde by vascular semicarbazide-sensitive amine oxidase. Toxicology 1992, 73, 251–258. [Google Scholar] [CrossRef]
- Gubisne-Haberle, D.; Hill, W.; Kazachkov, M.; Richardson, J.S.; Yu, P.H. Protein Cross-Linkage Induced by Formaldehyde Derived from Semicarbazide-Sensitive Amine Oxidase-Mediated Deamination of Methylamine. J. Pharmacol. Exp. Ther. 2004, 310, 1125–1132. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Maley, J.; Yu, P.H. Potential implications of endogenous aldehydes in ?-amyloid misfolding, oligomerization and fibrillogenesis. J. Neurochem. 2006, 99, 1413–1424. [Google Scholar] [CrossRef]
- Chen, K.; Kazachkov, M.; Yu, P.H. Effect of aldehydes derived from oxidative deamination and oxidative stress on β-amyloid aggregation; pathological implications to Alzheimer’s disease. J. Neural Transm. 2007, 114, 835–839. [Google Scholar] [CrossRef]
- Qiang, M.; Xiao, R.; Su, T.; Wu, B.-B.; Tong, Z.-Q.; Liu, Y.; He, R.-Q. A novel mechanism for endogenous formaldehyde ele-vation in SAMP8 mouse. J. Alzheimer’s Dis. 2014, 40, 1039–1053. [Google Scholar] [CrossRef]
- Unzeta, M.; Solé, M.; Boada, M.; Hernández, M. Semicarbazide-sensitive amine oxidase (SSAO) and its possible contribution to vascular damage in Alzheimer’s disease. J. Neural Transm. 2007, 114, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.J.; Richardson, J.S.; Yu, P.H. The contribution of cerebral vascular semicarbazide-sensitive amine oxidase to cerebral amyloid angiopathy in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2008, 34, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Rossi, B.; Santos-Lima, B.; Terrabuio, E.; Zenaro, E.; Constantin, G. Common Peripheral Immunity Mechanisms in Multiple Sclerosis and Alzheimer’s Disease. Front. Immunol. 2021, 12, 639369. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Cha, M.-Y.; Hyun, Y.-M.; Cho, H.; Hamza, B.; Kim, D.K.; Han, S.-H.; Choi, H.; Kim, K.H.; Moon, M.; et al. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 2014, 35, 1286–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease–like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Hernández, J.C.C.; Bracko, O.; Kersbergen, C.J.; Muse, V.; Haft-Javaherian, M.; Berg, M.; Park, L.; Vinarcsik, L.K.; Ivasyk, I.; Rivera, D.A.; et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 2019, 22, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Pietronigro, E.; Zenaro, E.; Della Bianca, V.; Dusi, S.; Terrabuio, E.; Iannoto, G.; Slanzi, A.; Ghasemi, S.; Nagarajan, R.; Piacentino, G.; et al. Blockade of α4 integrins reduces leukocyte–endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer’s disease. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Solé, M.; Hernández, M.; Boada, M.; Unzeta, M. Characterization of A7r5 cell line transfected in a stable form by hSSAO/VAP-1 gene (A7r5 hSSAO/VAP-1 cell line). J. Neural Transm. 2007, 114, 763–767. [Google Scholar] [CrossRef]
- Solé, M.; Miñano-Molina, A.J.; Unzeta, M. A cross-talk between Aβ and endothelial SSAO/VAP-1 accelerates vascular dam-age and Aβ aggregation related to CAA-AD. Neurobiol. Aging 2015, 36, 762–775. [Google Scholar] [CrossRef]
- Sun, P.; Esteban, G.; Inokuchi, T.; Marco-Contelles, J.; Weksler, B.B.; A Romero, I.; O Couraud, P.; Unzeta, M.; Solé, M. Protective effect of the multitarget compound DPH-4 on human SSAO/VAP-1-expressing hCMEC/D3 cells under oxygen-glucose deprivation conditions: Anin vitroexperimental model of cerebral ischaemia. Br. J. Pharmacol. 2015, 172, 5390–5402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solé, M.; Unzeta, M. Vascular cell lines expressing SSAO/VAP-1: A new experimental tool to study its involvement in vascular diseases. Biol. Cell 2011, 103, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010, 120, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Solé, M.; Esteban-Lopez, M.; Taltavull, B.; Fábregas, C.; Fadó, R.; Casals, N.; Rodríguez-Álvarez, J.; Miñano-Molina, A.J.; Unzeta, M. Blood-brain barrier dysfunction underlying Alzheimer’s disease is induced by an SSAO/VAP-1-dependent cere-brovascular activation with enhanced Aβ deposition. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 2189–2202. [Google Scholar] [CrossRef] [PubMed]
- Weksler, B.B.; Subileau, E.A.; Perrière, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef] [PubMed]
- Honig, L.S.; Kukull, W.; Mayeux, R. Atherosclerosis and AD: Analysis of data from the US National Alzheimer’s Coordinating Center. Neurology 2005, 64, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Goulay, R.; Romo, L.M.; Hol, E.M.; Dijkhuizen, R.M. From Stroke to Dementia: A Comprehensive Review Exposing Tight Interactions Between Stroke and Amyloid-β Formation. Transl. Stroke Res. 2020, 11, 601–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Yu, J.-T.; Wang, H.-F.; Meng, X.-F.; Tan, C.-C.; Wang, J.; Wang, C.; Tan, L. Association between stroke and Alz-heimer’s disease: Systematic review and meta-analysis. J. Alzheimer’s Dis. 2015, 43, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Reddy, P.H. Stroke, Vascular Dementia, and Alzheimer’s Disease: Molecular Links. J. Alzheimer’s Dis. 2016, 54, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Yang, S.; Stubley, L.; Day, A.; Simpkins, J. Hypoperfusion induces overexpression of β-amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res. 2000, 853, 1–4. [Google Scholar] [CrossRef]
- Nihashi, T.; Inao, S.; Kawai, T.; Sugimoto, T.; Niwa, M.; Hata, N.; Hayashi, S.; Yoshida, J.; Kajita, Y.; Kabeya, R. Expression and Distribution of Beta Amyloid Precursor Protein and Beta Amyloid Peptide in Reactive Astrocytes After Transient Middle Cerebral Artery Occlusion. Acta Neurochir. 2001, 143, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Rash, K.; Clemens, J.A. Amyloid precursor protein accumulates in regions of neurodegeneration following focal cerebral ischemia in the rat. Brain Res. 1992, 593, 128–135. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Bhatti, S.U.; Palatinsky, E.A.; Pennington, D.H.; Shelton, E.R.; Chan, H.W.; Perry, G.; Lust, W.D. Accumulation of the β amyloid precursor protein at sites of ischemic injury in rat brain. NeuroReport 1993, 4, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Badan, I.; Dinca, I.; Buchhold, B.; Suofu, Y.; Walker, L.C.; Gratz, M.; Platt, D.H.; Kessler, C.; Popawagner, A. Accelerated accumulation of N- and C-terminal betaAPP fragments and delayed recovery of microtubule-associated protein 1B expression following stroke in aged rats. Eur. J. Neurosci. 2004, 19, 2270–2280. [Google Scholar] [CrossRef]
- Van Groen, T.; Puurunen, K.; Mäki, H.-M.; Sivenius, J.; Jolkkonen, J. Transformation of Diffuse β-Amyloid Precursor Protein and β-Amyloid Deposits to Plaques in the Thalamus After Transient Occlusion of the Middle Cerebral Artery in Rats. Stroke 2005, 36, 1551–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkinen, S.; Van Groen, T.; Clarke, J.; Thornell, A.; Corbett, D.; Hiltunen, M.; Soininen, H.; Jolkkonen, J. Coaccumulation of Calcium and β-Amyloid in the Thalamus after Transient Middle Cerebral Artery Occlusion in Rats. Br. J. Pharmacol. 2007, 28, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, M.; Mäkinen, P.; Peräniemi, S.; Sivenius, J.; Van Groen, T.; Soininen, H.; Jolkkonen, J. Focal cerebral ischemia in rats alters APP processing and expression of Aβ peptide degrading enzymes in the thalamus. Neurobiol. Dis. 2009, 35, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.-F.; Xu, H.; Zhang, Y.-W. Hypoxia-inducible Factor 1α (HIF-1α)-mediated Hypoxia Increases BACE1 Expression and β-Amyloid Generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef] [Green Version]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1α. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef]
- Nalivaevaa, N.N.; Fisk, L.; Kochkina, E.G.; Plesneva, S.A.; Zhuravin, I.A.; Babusikova, E.; Dobrota, D.; Turner, A.J. Effect of Hypoxia/Ischemia and Hypoxic Preconditioning/Reperfusion on Expression of Some Amyloid-Degrading Enzymes. Ann. N. Y. Acad. Sci. 2004, 1035, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Fisk, L.; Nalivaeva, N.N.; Boyle, J.P.; Peers, C.S.; Turner, A.J. Effects of Hypoxia and Oxidative Stress on Expression of Neprilysin in Human Neuroblastoma Cells and Rat Cortical Neurones and Astrocytes. Neurochem. Res. 2007, 32, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Le, W. Pathological role of hypoxia in Alzheimer’s disease. Exp. Neurol. 2010, 223, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Pichiule, P.; Chavez, J.C.; Schmidt, A.M.; Vannucci, S.J. Hypoxia-inducible Factor-1 Mediates Neuronal Expression of the Receptor for Advanced Glycation End Products following Hypoxia/Ischemia. J. Biol. Chem. 2007, 282, 36330–36340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.-J.; Xu, J.; LaHousse, S.A.; Caggiano, N.L.; De La Monte, S.M. Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: Potential strategies for neuroprotection1. J. Alzheimer’s Dis. 2003, 5, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Yang, S.; Liu, R.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Transient Cerebral Ischemia Induces Aberrant Neuronal Cell Cycle Re-entry and Alzheimer’s Disease-like Tauopathy in Female Rats. J. Biol. Chem. 2004, 279, 22684–22692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnecom, C.; Lev, M.H.; Wendell, L.; Smith, E.E.; Rosand, J.; Frosch, M.P.; Greenberg, S.M. Course of cerebral amyloid angiopathy-related inflammation. Neurology 2007, 68, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics 2008, 5, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtunik-Kulesza, K.A.; Oniszczuk, A.; Oniszczuk, T.; Waksmundzka-Hajnos, M. The influence of common free radicals and antioxidants on development of Alzheimer’s Disease. Biomed. Pharmacother. 2016, 78, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.-L.; Guo, Z.-N.; Yang, Y. Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxidative Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease—Lessons from pathology. BMC Med. 2014, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Han, B.H.; Zhou, M.-L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [Green Version]
- Freeze, W.M.; Bacskai, B.J.; Frosch, M.P.; Jacobs, H.I.; Backes, W.H.; Greenberg, S.M.; Van Veluw, S.J. Blood-Brain Barrier Leakage and Microvascular Lesions in Cerebral Amyloid Angiopathy. Stroke 2019, 50, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinters, H.V.; Gilbert, J.J. Cerebral amyloid angiopathy: Incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 1983, 14, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attems, J.; Jellinger, K.; Thal, D.; Van Nostrand, W. Review: Sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 2010, 37, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Marco, S.; Skaper, S.D. Amyloid β-peptide1–42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci. Lett. 2006, 401, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.; Hoozemans, J.J.; Van Der Vies, S.M.; Rozemuller, A.J.; Van Horssen, J.; De Vries, H.E. Amyloid Beta Induces Oxidative Stress-Mediated Blood–Brain Barrier Changes in Capillary Amyloid Angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef]
- Zipfel, G.J.; Han, H.; Ford, A.L.; Lee, J.-M. Cerebral Amyloid Angiopathy: Progressive Disruption of the Neurovascular Unit. Stroke 2008, 40, S16–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, L.; Uekawa, K.; Garcia-Bonilla, L.; Koizumi, K.; Murphy, M.; Pistik, R.; Younkin, L.; Younkin, S.; Zhou, P.; Carlson, G.; et al. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Aβ Peptides. Circ. Res. 2017, 121, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Shen, Y.; Stins, M.; Du Yan, S.; Schmidt, A.M.; Stern, D.; Kim, K.-S.; Zlokovic, B.; Kalra, V.K. β-Amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am. J. Physiol. Physiol. 2000, 279, C1772–C1781. [Google Scholar] [CrossRef]
- Hartz, A.M.; Bauer, B.; Soldner, E.L.; Wolf, A.; Boy, S.; Backhaus, R.; Mihaljevic, I.; Bogdahn, U.; Klünemann, H.H.; Schuierer, G.; et al. Amyloid-β Contributes to Blood–Brain Barrier Leakage in Transgenic Human Amyloid Precursor Protein Mice and in Humans with Cerebral Amyloid Angiopathy. Stroke 2012, 43, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrano, A.; Hoozemans, J.J.; Van Der Vies, S.M.; Van Horssen, J.; De Vries, H.E.; Rozemuller, A.J. Neuroinflammation and Blood-Brain Barrier Changes in Capillary Amyloid Angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Kooij, G.; Reijerkerk, A.; Van Doorn, R.; Gringhuis, S.I.; Van Der Pol, S.; Weksler, B.B.; Romero, I.A.; Couraud, P.; Piontek, J.; et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007, 21, 3666–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiso, J.; Fossati, S.; Rostagno, A. Amyloidosis Associated with Cerebral Amyloid Angiopathy: Cell Signaling Pathways Elicited in Cerebral Endothelial Cells. J. Alzheimer’s Dis. 2014, 42, S167–S176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gireud-Goss, M.; Mack, A.F.; McCullough, L.D.; Urayama, A. Cerebral Amyloid Angiopathy and Blood-Brain Barrier Dys-function. Neuroscientist 2020. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V.; Buccafusco, J.J.; Wilson, C. Cognitive dysfunction in neuropsychiatric disorders: Selected serotonin receptor subtypes as therapeutic targets. Behav. Brain Res. 2008, 195, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Esteban, G.; Ojima, M.; Bautista-Aguilera, O.M.; Inokuchi, T.; Moraleda, I.; Iriepa, I.; Samadi, A.; Youdim, M.B.; Romero, A.; et al. Donepezil + propargylamine + 8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 80, 543–561. [Google Scholar] [CrossRef] [Green Version]
- Davies, P. Selective Loss of Central Cholinergic Neurons in Alzheimer’s Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Geula, C.; Mesulam, M. Cholinergic systems in Alzheimer’s disease. In: Terry RD, Katzman R, Bick KL, Sisodia SS, editors. Alzheimer Dis. 1999, 2, 269–292. [Google Scholar]
- Buccafusco, J.J.; Terry, A.V., Jr. Multiple central nervous system targets for eliciting beneficial effects on memory and cogni-tion. J. Pharmacol. Exp. Ther. 2000, 295, 438–446. [Google Scholar]
- Youdim, M.B.H.; Buccafusco, J.J. CNS Targets for multi-functional drugs in the treatment of Alzheimer?s and Parkinson?s diseases. J. Neural Transm. 2005, 112, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Eunzeta, M.; Eesteban, G.; Ebolea, I.; Fogel, W.A.; Ramsay, R.; Youdim, M.B.H.; Tipton, K.F.; Emarco-Contelles, J. Multi-Target Directed Donepezil-Like Ligands for Alzheimer’s Disease. Front. Neurosci. 2016, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Bernabé, M.; Fernández-Alvarez, E.; Lora-Tamayo, M.; Nieto, O. [Potential psychotropic drugs. V. Monoamine oxidase inhibitors. Preparation and study of some indolyl-3 alkyl-hydrazines]. Bull. de la Soc. Chim. de Fr. 1971, 5, 1882–1887. [Google Scholar]
- Monge, A.; Palop, J.A.; Goñí, T.; Martínez, A.; Fernandez-Alvarez, E. About the synthesis of [1,2]diazepinoindole derivatives from ethyl 2-(1-methylindole)acetate, 2-indole and 3-indoleacetohydrazones. J. Heterocycl. Chem. 1985, 22, 1445–1451. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, J.; Chen, X.; Li, J.; Yang, Y.; Weng, J.; Deng, C.; Yenari, M.A. Statins: Multiple neuroprotective mechanisms in neurodegenerative diseases. Exp Neurol 2011, 230, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhuang, Q.K.; Yang, J.N.; Zhang, Y.Y. Statins excert neuroprotection on cerebral ischemia independent of their li-pid-lowering action: The potential molecular mechanisms. Eur. Rev. Med. Pharmacol Sci. 2014, 18, 1113–1126. [Google Scholar]
- García-Bonilla, L.; Campos, M.; Giralt, D.; Salat, D.; Chacón, P.; Hernández-Guillamon, M.; Rosell, A.; Montaner, J. Evidence for the efficacy of statins in animal stroke models: A meta-analysis. J. Neurochem. 2012, 122, 233–243. [Google Scholar] [CrossRef] [PubMed]


| Inflammatory Stimulus | Organ/Tissue | Type of Leukocytes Bound by SSAO/VAP-1 | Type of Leukocytes not Bound by SSAO/VAP-1 | Reference |
|---|---|---|---|---|
| Ischemia/reperfusion | Kidney | Neutrophils | Macrophages/T-lymphocytes | [65] |
| Postischemic inflammation | Brain | Neutrophils | - | [66] |
| Subarachnoid hemorrhage | Brain | Neutrophils | - | [67] |
| Intracerebral hemorrhage | Brain | Neutrophils | - | [68] |
| Peritonitis | Peritoneum | Granulocytes | - | |
| Air pouch inflammation | Subcutaneous | Monocytes/lymphocytes | - | [69] |
| LPS | Brain | Neutrophils | - | [70] |
| LPS, Klebsiella pneumoniae | Lungs | Polymorphonuclear cells, neutrophils | - | [71] |
| Acute liver failure | Liver | Leukocytes | Monocytes | [72] |
| ConA hepatitis | Liver | CD4+ Th2 cells | - | [73] |
| Hepatic chronic inflammation and fibrosis | Liver | CD16+ monocytes | - | [74] |
| Liver inflammation | Liver | CD4+ T cell | - | [75] |
| Liver allograft rejection | Liver | CD4+ and CD8+ lymphocytes | - | [76,77] |
| Tumors (adhesion function) | Skin | CD45+, CD3+, CD8+ | CD4+, T-reg cells, Type2 macrophages, GR-1+CD11b+ | [78] |
| Tumors (enzymatic function) | Skin | CD45+, CD8+, CD11b+, granulocytes, | CD4+, type2 macrophages | [78] |
| Cytokine-induced angiogenesis | Eyes | CD11b+ cells, granulocytes | - | [79] |
| Diabetic retinopathy | Eyes | Leukocytes | - | [80] |
| Uveitis | Eyes | CD45+ | - | [81] |
| In vitro | Endothelial cells | Lymphocytes, T-killer cells | Neutrophils, monocytes | [82] |
| In vitro | Endothelial cells | Polymorphonuclear leukocytes | - | [83] |
| AOC3 knockout | Adipose tissue | CD45+, T cells, macrophages, natural killer | - | [84] |
| Physiological Function | Pathological Effect Upon SSAO/VAP-1 Overexpression | Involvement in Pathologies |
|---|---|---|
| Oxidative deamination of primary amines of endogenous and xenobiotic origin Molecular signaling through H2O2 generation | Toxicity of metabolic products (formaldehyde, methylglyoxal, H2O2) | Stroke AD |
| Protein cross-linking and Aβ aggregation | Diabetes | |
| Oxidative stress | Atherosclerosis | |
| AGEs generation | Congestive heart failure | |
| Inflammation | Fibrotic liver disease | |
| Pathological angiogenesis | Cancer Age-related macular degeneration | |
| Leukocyte trafficking under inflammatory conditions | Excessive inflammatory response | MS |
| Insulinomimetic action by recruitment of GLUT4 receptors to the cell membrane | Unknown | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unzeta, M.; Hernàndez-Guillamon, M.; Sun, P.; Solé, M. SSAO/VAP-1 in Cerebrovascular Disorders: A Potential Therapeutic Target for Stroke and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 3365. https://doi.org/10.3390/ijms22073365
Unzeta M, Hernàndez-Guillamon M, Sun P, Solé M. SSAO/VAP-1 in Cerebrovascular Disorders: A Potential Therapeutic Target for Stroke and Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(7):3365. https://doi.org/10.3390/ijms22073365
Chicago/Turabian StyleUnzeta, Mercedes, Mar Hernàndez-Guillamon, Ping Sun, and Montse Solé. 2021. "SSAO/VAP-1 in Cerebrovascular Disorders: A Potential Therapeutic Target for Stroke and Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 7: 3365. https://doi.org/10.3390/ijms22073365
APA StyleUnzeta, M., Hernàndez-Guillamon, M., Sun, P., & Solé, M. (2021). SSAO/VAP-1 in Cerebrovascular Disorders: A Potential Therapeutic Target for Stroke and Alzheimer’s Disease. International Journal of Molecular Sciences, 22(7), 3365. https://doi.org/10.3390/ijms22073365