
The GRKs Reactome: Role in Cell Biology and Pathology


Abstract
:1. Introduction
2. GRKs and Arrestins in GPCRs Family
2.1. GPCR Signaling Pathway; General Overview
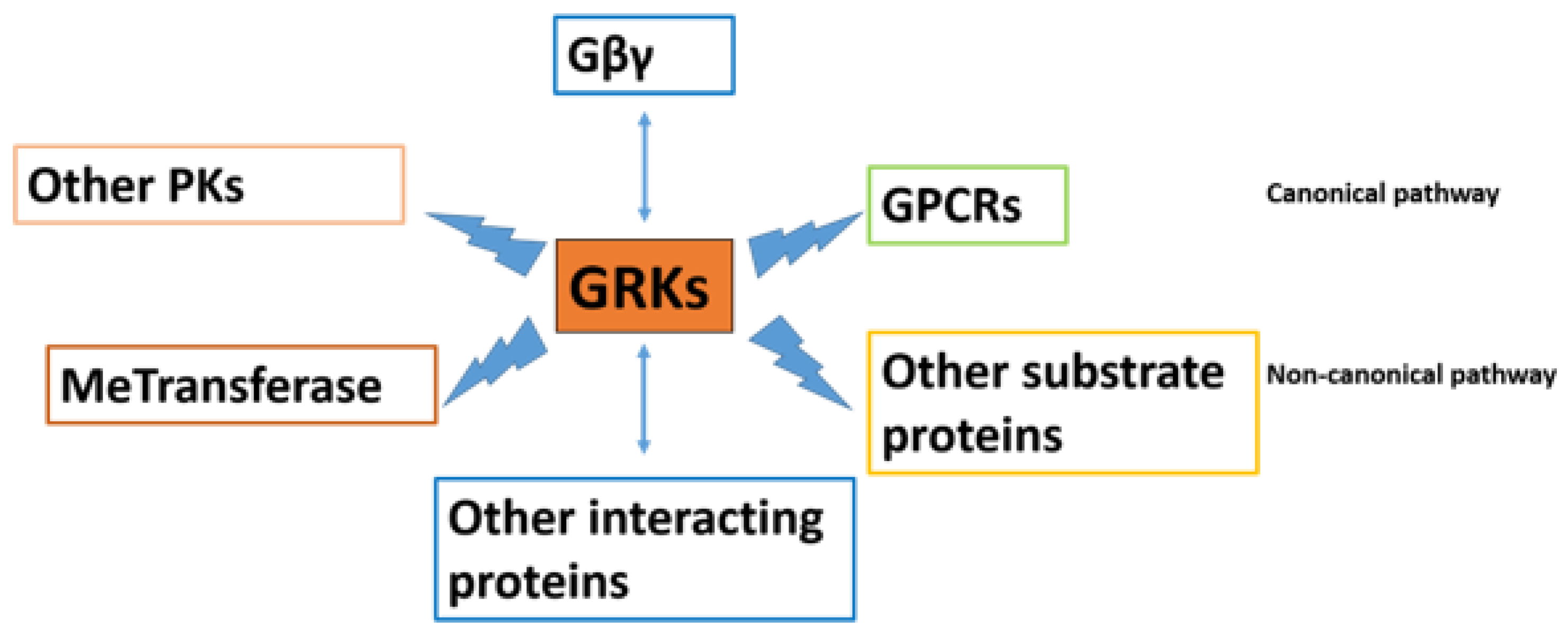
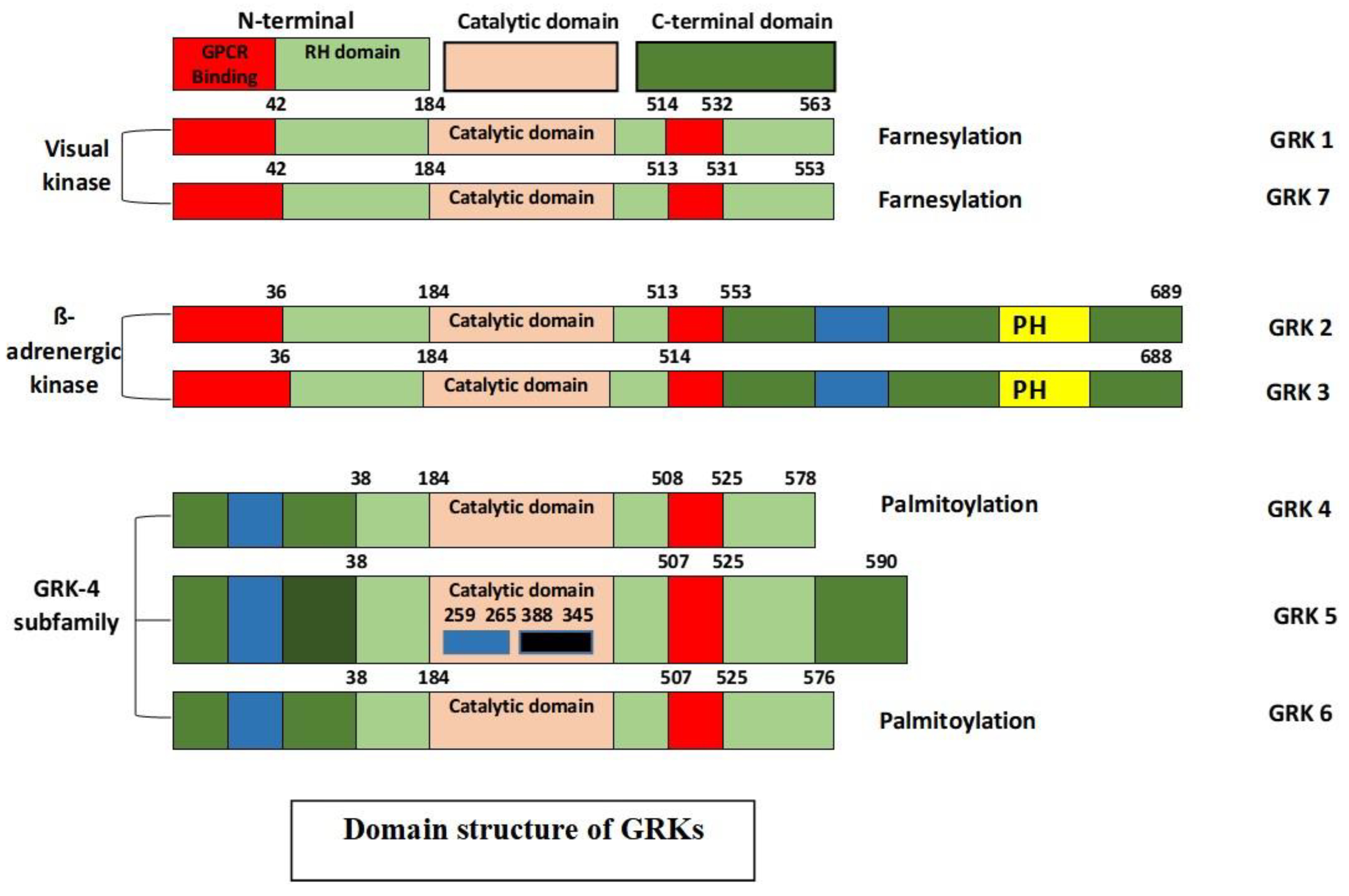
2.2. The GRK and Arrestin Family
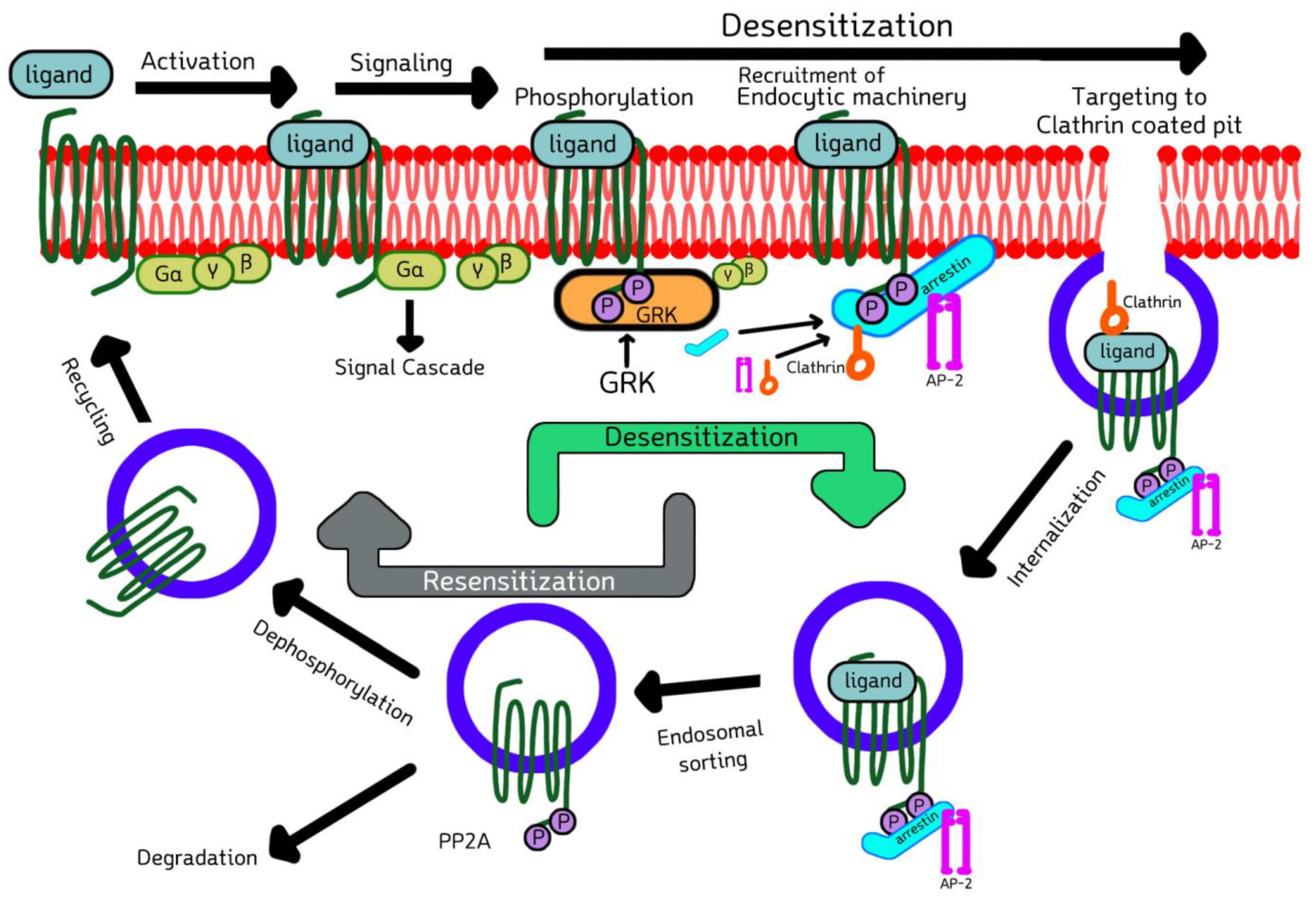
2.3. Cell Biology of GRKs and Arrestins in Signaling, Desensitization, Internalization, and Sorting of GPCRs
3. The Role of GRKs and Arrestins in Regulation of GPCRs Family
3.1. The Role of GRKs in Immune Cells and Inflammation
3.2. The Role of GRKs in Cardiovascular Diseases
3.3. The Role of GRKs in Neurodegeneration and Autoimmune Diseases
3.4. The Role of GRKs in Cancer
3.5. The Role of GRKs in Thrombosis and Hemostasis
Contribution of Platelet Beyond Thrombosis and Hemostasis
4. The Implications of GRKs in Pharmacology
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GRK | G protein-coupled receptor kinase |
| GPCR | G protein-coupled receptor |
| 7-TM | Seven-transmembrane |
| PI3K | Phosphoiositide 3-kinase |
| PIP2 | Phosphatidylinositol 4:5-bisphosphate |
| RGS | Regulator of G protein signaling domain |
| PH | Pleckstrin homology |
| AP-2 | Adaptor protein-2 |
| β2-AR | β2-adrenergic receptor |
| VEGF | Vascular endothelial growth factor |
| AD | Alzheimer’s disease |
| MS | Multiple sclerosis |
| RhoGDI | Rho GDP-dissociation inhibitor |
| PDE | Phosphodiesterase |
| TSHR | Thryoid-stimulating hormone receptor |
| IGF1-R | Insulin-like growth factor-1 receptor |
| EGR1 | Early growth factor 1 |
| NGF | Nerve growth factor |
| CXCR | Chemokine receptor |
| AT1 | Angiotensin type 1 |
| FSHR | Follicle-stimulating hormone receptor |
| vWF | Von Willebrand factor |
| TxA2 | Thromboxane A2 |
| PLC | Phospholipase C |
| DAG | Diacyl glycerol |
| PKC | Protein kinase C |
| TP | Thromboxane A2 receptor |
| GPVI | Glycoprotein VI |
| SFKs | Src family kinases |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| CRP | Collagen-related peptide |
| GF | Growth factor |
| MIP | Macrophage inflammatory protein |
References
- Patel, C.B.; Noor, N.; Rockman, H.A. Functional Selectivity in Adrenergic and Angiotensin Signaling Systems. Mol. Pharmacol. 2010, 78, 983–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-Function of the G Protein–Coupled Receptor Superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular Signatures of G-Protein-Coupled Receptors. Nat. Cell Biol. 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-Transmembrane Receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef]
- Hurowitz, E.H.; Melnyk, J.M.; Chen, Y.J.; Kouros-Mehr, H.; Simon, M.I.; Shizuya, H. Genomic Characterization of the Human Heterotrimeric G Protein Alpha, Beta, and Gamma Subunit Genes. DNA Res. 2000, 7, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Stephens, L.R.; Eguinoa, A.; Erdjument-Bromage, H.; Lui, M.; Cooke, F.; Coadwell, J.; Smrcka, A.S.; Thelen, M.; Cadwallader, K.; Tempst, P.; et al. The G beta Gamma Sensitivity of a PI3K is Dependent upon a Tightly Associated Adaptor, p101. Cell 1997, 89, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G Protein Pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Bockaert, J.; Fagni, L.; Dumuis, A.; Marin, P. GPCR Interacting Proteins (GIP). Pharmacol. Ther. 2004, 103, 203–221. [Google Scholar] [CrossRef]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of Receptor Signals by Beta-Arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Ferguson, S.S. Evolving Concepts in G Protein-Coupled Receptor Endocytosis: The Role in Receptor Desensitization and Signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar] [PubMed]
- Kohout, T.A.; Lefkowitz, R.J. Regulation of G Protein-Coupled Receptor Kinases and Arrestins during Receptor Desensitization. Mol. Pharmacol. 2003, 63, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Santulli, G.; Iaccarino, G. Adrenergic Signaling in Heart Failure and Cardiovascular Aging. Maturitas 2016, 93, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorriento, D.; Ciccarelli, M.; Cipolletta, E.; Trimarco, B.; Iaccarino, G. “Freeze, Don’t Move”: How to Arrest a Suspect in Heart Failure–a Review on Available GRK2 Inhibitors. Front. Cardiovasc. Med. 2016, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Beautrait, A.; Michalski, K.R.; Lopez, T.S.; Mannix, K.M.; McDonald, D.J.; Cutter, A.R.; Medina, C.B.; Hebert, A.M.; Francis, C.J.; Bouvier, M.; et al. Mapping the Putative G Protein-Coupled Receptor (GPCR) Docking Site on GPCR Kinase 2: Insights from Intact Cell Phosphor-Ylation and Recruitment Assays. J. Biol. Chem. 2014, 289, 25262–25275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallese, M.; Mariggiò, S.; Collodel, G.; Moretti, E.; Piomboni, P.; Baccetti, B.; De Blasi, A. G Protein-Coupled Receptor Kinase GRK4: Molecular Analysis of the Four Isoforms and Ultrastructural Localization in Spermatozoa and Germinal Cells. J. Biol. Chem. 1997, 272, 10188–10195. [Google Scholar] [CrossRef] [Green Version]
- Virlon, B.; Firsov, D.; Cheval, L.; Reiter, E.; Troispoux, C.; Guillou, F.; Elalouf, J.-M. Rat G Protein-Coupled Receptor Kinase GRK4: Identification, Functional Expression, and Differential Tissue Distribution of Two Splice Variants. Endocrinology 1998, 139, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Willets, J.M.; Challiss, R.; Nahorski, S.R. Non-visual GRKs: Are We Seeing the Whole Picture? Trends Pharmacol. Sci. 2003, 24, 626–633. [Google Scholar] [CrossRef] [Green Version]
- DebBurman, S.K.; Ptasienski, J.; Benovic, J.L.; Hosey, M.M. G Protein-Coupled Receptor Kinase GRK2 Is a Phospholip-ID-Dependent Enzyme That Can Be Conditionally Activated by G Protein Betagamma Subunits. J. Biol. Chem. 1996, 271, 22552–22562. [Google Scholar] [CrossRef] [Green Version]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G Protein-Coupled Receptor Kinases. Annu. Rev. Biochem. 1998, 67, 653–692. [Google Scholar] [CrossRef] [Green Version]
- Penela, P.; Ribas, C.; Mayor, F., Jr. Mechanisms of Regulation of the Expression and Function of G Protein-Coupled Receptor Kinases. Cell Signal. 2003, 15, 973–981. [Google Scholar] [CrossRef]
- Lodowski, D.T.; Tesmer, V.M.; Benovic, J.L.; Tesmer, J.J. The Structure of G Protein-coupled Receptor Kinase (GRK)-6 Defines a Second Lineage of GRKs. J. Biol. Chem. 2006, 281, 16785–16793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-Arrestins and Cell Signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [Green Version]
- Krupnick, J.G.; Benovic, J.L. The Role of Receptor Kinases and Arrestins in G Protein–Coupled Receptor Regulation. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 289–319. [Google Scholar] [CrossRef]
- Premont, R.T.; Gainetdinov, R.R. Physiological Roles of G Protein–Coupled Receptor Kinases and Arrestins. Annu. Rev. Physiol. 2007, 69, 511–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, S.K.; Lefkowitz, R.J. Multifaceted Roles of Beta-Arrestins in the Regulation of Seven-Membrane-Spanning Receptor Trafficking and Signalling. Biochem. J. 2003, 375, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Gurevich, V.V. Arrestins: Ubiquitous Regulators of Cellular Signaling Pathways. Genome Biol. 2006, 7, 236. [Google Scholar] [CrossRef] [Green Version]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced Morphine Analgesia in Mice Lacking Beta-Arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Conner, D.A.; Mathier, M.A.; Mortensen, R.M.; Christe, M.; Vatner, S.F.; Seidman, C.E.; Seidman, J.G. Beta-arrestin1 Knockout Mice Appear Normal but Demonstrate Altered Cardiac Responses to Beta-Adrenergic Stimulation. Circ. Res. 1997, 81, 1021–1026. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Hara, M.R.; Davenport, C.L.; Kim, J.; Lefkowitz, R.J. Arrestin Development: Emerging Roles for Beta-Arrestins in Developmental Signaling Pathways. Dev. Cell 2009, 17, 443–458. [Google Scholar] [CrossRef] [Green Version]
- Sorkin, A.; von Zastrow, M. Endocytosis and Signalling: Intertwining Molecular Networks. Nature Reviews. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar]
- Goodman, O.B.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. β-Arrestin Acts as a Clathrin Adaptor in Endocytosis of the β2-Adrenergic Receptor. Nat. Cell Biol. 1996, 383, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Laporte, S.A.; Oakley, R.H.; Zhang, J.; Holt, J.A.; Ferguson, S.S.; Caron, M.G.; Barak, L.S. The β2-adrenergic Recep-Tor/βArrestin Complex Recruits the Clathrin Adaptor AP-2 during Endocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3712–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilden, U.; Hall, S.W.; Kuhn, H. Phosphodiesterase Activation by Photoexcited Rhodopsin Is Quenched When Rhodopsin Is Phosphorylated and Binds the Intrinsic 48-kDa Protein of Rod Outer Segments. Proc. Natl. Acad. Sci. USA 1986, 83, 1174–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasser, R.H.; Sibley, D.R.; Lefkowitz, R.J. A Novel Catecholamine-Activated Adenosine Cyclic 3’, 5’-Phosphate Independent Pathway for. Beta-Adrenergic Receptor Phos-Phorylation in Wild-Type and Mutant s49 Lymphoma Cells: Mechanism of Homologous Desensitization of Adenylate Cyclase. Biochemistry 1986, 25, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-K.; Burns, M.E.; Spencer, M.; Niemi, G.A.; Chen, J.; Hurley, J.B.; Baylor, D.A.; Simon, M.I. Abnormal Photore-Sponses and Light-Induced Apoptosis in Rods Lacking Rhodopsin Kinase. Proc. Natl. Acad. Sci. USA 1999, 96, 3718–3722. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Dodd, R.L.; Makino, C.L.; Simon, M.I.; Baylor, D.A.; Chen, J. Prolonged Photoresponses in Transgenic Mouse Rods Lacking Arrestin. Nat. Cell Biol. 1997, 389, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Wilden, U. Duration and Amplitude of the Light-Induced cGMP Hydrolysis in Vertebrate Photoreceptors Are Regulated by Mul-Tiple Phos-Phorylation of Rhodopsin and by Arrestin Binding. Biochemistry 1995, 34, 1446–1454. [Google Scholar] [CrossRef]
- Krupnick, J.G.; Gurevich, V.V.; Benovic, J.L. Mechanism of Quenching of Phototransduction: Binding Competition between Arrestin and Transducin for Phosphorhodopsin. J. Biol. Chem. 1997, 272, 18125–18131. [Google Scholar] [CrossRef] [Green Version]
- Mohan, M.L.; Vasudevan, N.T.; Gupta, M.K.; Martelli, E.E.; Naga Prasad, S.V. G-Protein Coupled Receptor Resensitiza-Tion-Appreciating the Balancing Act of Receptor Function. Curr. Mol. Pharmacol. 2012, 5, 350–361. [Google Scholar] [CrossRef]
- Pals-Rylaarsdam, R.; Xu, Y.; Witt-Enderby, P.; Benovic, J.L.; Hosey, M.M. Desensitization and Internalization of the m2 Muscarinic Acetylcholine Receptor Are Directed by Independent Mechanisms. J. Biol. Chem. 1995, 270, 29004–29011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakata, H.; Kameyama, K.; Haga, K.; Haga, T. Location of Agonist-Dependent-Phosphorylation Sites in the Third Intracellular Loop of Muscarinic Acetylcholine Receptors (m2 subtype). JBIC J. Biol. Inorg. Chem. 1994, 220, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Jojima, E.; Saito, H.; Haga, T. Role of the Third Intracellular Loop in the Subtype-Specific Internalization and Recy-Cling of Muscarinic M2 and M4 Receptors. Biomed. Res. 2014, 35, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, M.; Zhang, W.; Tian, Y.; Xu, C.; Xu, T.; Liu, J.; Zhang, R. Unraveling a Molecular Determinant for Clathrin-Independent Internalization of the M2 Muscarinic Acetylcholine Receptor. Sci. Rep. 2015, 5, 11408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billard, M.J.; Fitzhugh, D.J.; Parker, J.S.; Brozowski, J.M.; McGinnis, M.W.; Timoshchenko, R.G.; Serafin, D.S.; Lininger, R.; Klauber-Demore, N.; Sahagian, G.; et al. G Protein Coupled Receptor Kinase 3 Regulates Breast Cancer Migration, Invasion, and Metastasis. PLoS ONE 2016, 11, e0152856. [Google Scholar] [CrossRef] [PubMed]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct Phosphorylation Sites on the β(2)-Adrenergic Receptor Establish a Barcode That Encodes Differential Functions of β-Arrestin. Sci. Sig. 2011, 4, ra51. [Google Scholar] [CrossRef] [Green Version]
- Reiter, E.; Marion, S.; Robert, F.; Troispoux, C.; Boulay, F.; Guillou, F.; Crepieux, P. Kinase-Inactive G-Protein-Coupled Receptor Kinases Are Able to Attenuate Follicle-Stimulating Hormone-Induced Signaling. Biochem. Biophys. Res. Commun. 2001, 282, 71–78. [Google Scholar] [CrossRef]
- Kim, O.J.; Gardner, B.R.; Williams, D.B.; Marinec, P.S.; Cabrera, D.M.; Peters, J.D.; Mak, C.C.; Kim, K.-M.; Sibley, D.R. The Role of Phosphorylation in D1 Dopamine Receptor Desensitization: Evidence for a Novel Mechanism of Arrestin Association. J. Biol. Chem. 2004, 279, 7999–8010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, M.J.; Lee, K.A.; Niemi, G.A.; Craven, K.B.; Garwin, G.G.; Saari, J.C.; Hurley, J.B. Multiple Phosphorylation of Rhodopsin and the in Vivo Chemistry Underlying Rod Photoreceptor Dark Adaptation. Neuron 2001, 31, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Onorato, J.J.; Palczewski, K.; Regan, J.W.; Caron, M.G.; Lefkowitz, R.J.; Benovic, J.L. Role of Acidic Amino Acids in Peptide Substrates of the Beta-Adrenergic Receptor Kinase and Rhodopsin Kinase. Biochemistry 1991, 30, 5118–5125. [Google Scholar] [CrossRef]
- Kim, J.; Ahn, S.; Ren, X.R.; Whalen, E.J.; Reiter, E.; Wei, H.; Lefkowitz, R.J. Functional Antagonism of Different G Protein-Coupled Receptor Kinases for Beta-Arrestin-Mediated Angiotensin II Receptor Signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 1442–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.R.; Reiter, E.; Ahn, S.; Kim, J.; Chen, W.; Lefkowitz, R.J. Different G Protein-Coupled Receptor Kinases Govern G Protein and Beta-Arrestin-Mediated Signaling of V2 Vasopressin Receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homan, K.T.; Tesmer, J.J.G. Structural Insights into G Protein-Coupled Receptor Kinase Function. Curr. Opin. Cell Biol. 2014, 27, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotton, M.; Claing, A. G Protein-Coupled Receptors Stimulation and the Control of Cell Migration. Cell Signal 2009, 21, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Insall, R. The Interaction between Pseudopods and Extracellular Signalling during Chemotaxis and Directed Migration. Curr. Opin. Cell Biol. 2013, 25, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiang, Y.; Li, Y.; Wang, J.; Fan, L.; Scott, M.J.; Xiao, G.; Li, S.; Billiar, T.R.; Wilson, M.A.; et al. TLR4 Signaling Augments Monocyte Chemotaxis by Regulating G Protein-Coupled Receptor Kinase 2 Translocation. J. Immunol. 2013, 191, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Arnon, T.I.; Xu, Y.; Lo, C.; Pham, T.; An, J.; Coughlin, S.; Dorn, G.W.; Cyster, J.G. GRK2-Dependent S1PR1 Desensitization Is Required for Lymphocytes to Overcome Their Attraction to Blood. Science 2011, 333, 1898–1903. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, B.; Malik, A.B.; Tang, H.; Yang, T.; Sun, B.; Wang, G.; Minshall, R.D.; Li, Y.; Zhao, Y.; et al. Bidirectional Regulation of Neutrophil Migration by Mitogen-Activated Protein Kinases. Nat. Immunol. 2012, 13, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Penela, P.; Nogués, L.; Mayor, F. Role of G Protein-Coupled Receptor Kinases in Cell Migration. Curr. Opin. Cell Biol. 2014, 27, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Lafarga, V.; Mayor, J.F.; Penela, P. The Interplay between G Protein-Coupled Receptor Kinase 2 (GRK2) and Histone Deacetylase 6 (HDAC6) at the Crossroads of Epithelial Cell Motility. Cell Adhes. Migr. 2012, 6, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Otten, J.J.T.; De Jager, S.C.A.; Kavelaars, A.; Seijkens, T.; Bot, I.; Wijnands, E.; Beckers, L.; Westra, M.M.; Bot, M.; Busch, M.; et al. Hematopoietic G-Protein-Coupled Receptor Kinase 2 Deficiency Decreases Atherosclerotic Lesion Formation in LDL Receptor-Knockout Mice. FASEB J. 2013, 27, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Arraes, S.M.A.; Freitas, M.S.; Da Silva, S.V.; Neto, H.A.D.P.; Alves-Filho, J.C.; Martins, M.A.; Basile-Filho, A.; Tavares-Murta, B.M.; Barja-Fidalgo, C.; Cunha, F.Q. Impaired Neutrophil Chemotaxis in Sepsis Associates with GRK Expression and Inhibition of Actin Assembly and Tyrosine Phosphorylation. Blood 2006, 108, 2906–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoratti, F.M.D.S.; Trevelin, S.C.; Cunha, F.Q.; Rocha, B.C.; Costa, P.A.C.; Gravina, H.D.; Tada, M.S.; Pereira, D.B.; Golenbock, D.T.; Antonelli, L.R.D.V.; et al. Neutrophil Paralysis in Plasmodium vivax Malaria. PLoS Negl. Trop. Dis. 2012, 6, e1710. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Ribas, C.; Aymerich, I.; Eijkelkamp, N.; Barreiro, O.; Heijnen, C.J.; Kavelaars, A.; Sánchez-Madrid, F.; Mayor, F., Jr. G Protein-Coupled Receptor Kinase 2 Positively Regulates Epithelial Cell Migration. EMBO J. 2008, 27, 1206–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cant, S.H.; Pitcher, J.A. G Protein-Coupled Receptor Kinase 2-Mediated Phosphorylation of Ezrin Is Required for G Protein-Coupled Receptor-Dependent Reorganization of the Actin Cytoskeleton. Mol. Biol. Cell 2005, 16, 3088–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahsai, A.W.; Zhu, S.; Fenteany, G. G Protein-Coupled Receptor Kinase 2 Activates Radixin, Regulating Membrane Protrusion and Motility in Epithelial Cells. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1803, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.-H.; Zhang, L.; Fanaroff, A.C.; Cai, X.; Sharma, K.C.; Brian, L.; Exum, S.T.; Shenoy, S.K.; Peppel, K.; Freedman, N.J. G Protein–Coupled Receptor Kinase-5 Attenuates Atherosclerosis by Regulating Receptor Tyrosine Kinases and 7-Transmembrane Receptors. Arter. Thromb. Vasc. Biol. 2012, 32, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Barker, B.L.; Benovic, J.L. G Protein-Coupled Receptor Kinase 5 Phosphorylation of Hip Regulates Internalization of the Chem-Okine Receptor CXCR4. Biochemistry 2011, 50, 6933–6941. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, F.; Long, H.; Chen, Y.; Wu, Z.; Ma, L. GRK5 Promotes F-Actin Bundling and Targets Bundles to Membrane Structures to Control Neuronal Morphogenesis. J. Cell Biol. 2011, 194, 905–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, P.K.; Zhang, Y.; Coomes, A.S.; Kim, W.-J.; Stupay, R.; Lynch, L.D.; Atkinson, T.; Kim, J.I.; Nie, Z.; Daaka, Y. G Protein–Coupled Receptor Kinase GRK5 Phosphorylates Moesin and Regulates Metastasis in Prostate Cancer. Cancer Res. 2014, 74, 3489–3500. [Google Scholar] [CrossRef] [Green Version]
- Raghuwanshi, S.K.; Su, Y.; Singh, V.; Haynes, K.; Richmond, A.; Richardson, R.M. The Chemokine Receptors CXCR1 and CXCR2 Couple to Distinct G Protein-Coupled Receptor Kinases to Mediate and Regulate Leukocyte Functions. J. Immunol. 2012, 189, 2824–2832. [Google Scholar] [CrossRef] [Green Version]
- Vroon, A.; Heijnen, C.J.; Raatgever, R.; Touw, I.P.; Ploemacher, R.E.; Premont, R.T.; Kavelaars, A. GRK6 Defi-Ciency Is Associated with Enhanced CXCR4-Mediated Neutrophil Chemotaxis in Vitro and Impaired Responsiveness to G-CSF In Vivo. J. Leukoc. Biol. 2004, 75, 698–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavelaars, A.; Vroon, A.; Raatgever, R.P.; Fong, A.M.; Premont, R.T.; Patel, D.D.; Lefkowitz, R.J.; Heijnen, C.J. Increased Acute Inflammation, Leukotriene B4-Induced Chemotaxis, and Signaling in Mice Deficient for G Protein-Coupled Receptor Kinase 6. J. Immunol. 2003, 171, 6128–6134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarrant, T.K.; Rampersad, R.R.; Esserman, D.; Rothlein, L.R.; Liu, P.; Premont, R.T.; Lefkowitz, R.J.; Lee, D.M.; Patel, D.D. Granulocyte Chemotaxis and Disease Expression Are Differentially Regulated by GRK Subtype in an Acute Inflammatory Arthritis Model (K/BxN). Clin. Immunol. 2008, 129, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, A.M.; Premont, R.T.; Richardson, R.M.; Yu, Y.R.; Lefkowitz, R.J.; Patel, D.D. Defective Lymphocyte Chem-Otaxis in Beta-arrestin2-and GRK6-Deficient Mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7478–7483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eijkelkamp, N.; Heijnen, C.J.; Lucas, A.; Premont, R.T.; Elsenbruch, S.; Schedlowski, M.; Kavelaars, A. G Protein-Coupled Receptor Kinase 6 Controls Chronicity and Severity of Dextran Sodium Sulphate-Induced Colitis in Mice. Gut 2007, 56, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Alves-Filho, J.C.; Sônego, F.; Souto, F.O.; Freitas, A.; Verri, W.A.; Auxiliadora-Martins, M.; Basile-Filho, A.; McKenzie, A.N.; Xu, D.; Cunha, F.Q.; et al. Interleukin-33 Attenuates Sepsis by Enhancing Neutrophil Influx to the Site of Infection. Nat. Med. 2010, 16, 708–712. [Google Scholar] [CrossRef]
- Packiriswamy, N.; Lee, T.; Raghavendra, P.B.; Durairaj, H.; Wang, H.; Parameswaran, N. G-Protein-Coupled Receptor Kinase-5 Mediates Inflammation but Does Not Regulate Cellular Infiltration or Bacterial Load in a Polymicrobial Sepsis Model in Mice. J. Innate Immun. 2013, 5, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Parvataneni, S.; Gonipeta, B.; Packiriswamy, N.; Lee, T.; Durairaj, H.; Parameswaran, N. Role of Myeloid-Specific G-Protein Coupled Receptor Kinase-2 in Sepsis. Int. J. Clin. Exp. Med. 2011, 4, 320–330. [Google Scholar] [PubMed]
- Patial, S.; Saini, Y.; Parvataneni, S.; Appledorn, D.M.; Dorn, G.W., 2nd; Lapres, J.J.; Amalfitano, A.; Senagore, P.; Parameswaran, N. Myeloid-specific GPCR Kinase-2 Negatively Regulates NF-κB1p105-ERK Pathway and Limits Endotoxemic Shock in Mice. J. Cell. Physiol. 2011, 226, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Patial, S.; Shahi, S.; Saini, Y.; Lee, T.; Packiriswamy, N.; Appledorn, D.M.; LaPres, J.J.; Amalfitano, A.; Parameswaran, N. G-Protein Coupled Receptor Kinase 5 Mediates LipopolysacchariDe-induced NFκB Activation in Primary Macrophages and Mod-Ulates Inflammation in Vivo in Mice. J. Cell. Physiol. 2011, 226, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, N.; Pao, C.S.; Leonhard, K.S.; Kang, D.S.; Kratz, M.; Ley, S.C.; Benovic, J.L. Arrestin-2 and G Protein-Coupled Receptor Kinase 5 Interact with NFκb1 p105 and Negatively Regulate Lipopolysaccharide-stimulated ERK1/2 Activation in Macrophages. J. Biol. Chem. 2006, 281, 34159–34170. [Google Scholar] [CrossRef] [Green Version]
- Patial, S.; Luo, J.; Porter, K.J.; Benovic, J.L.; Parameswaran, N. G-Protein Coupled Receptor Kinases Mediate TNF Alpha-Induced NF KappaB Signaling via Direct Interaction with and Phosphor-Ylation of I Kappa B Alpha (38.15). Am. Assoc. Immnol. 2009, 425, 169–178. [Google Scholar]
- Peregrin, S.; Jurado-Pueyo, M.; Campos, P.M.; Sanz-Moreno, V.; Ruiz-Gomez, A.; Crespo, P.; Mayor, F., Jr.; Murga, C. Phos-Phorylation of p38 by GRK2 at the Docking Groove Unveils a Novel Mechanism for Inactivating p38MAPK. Curr. Biol. 2006, 16, 2042–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemen, H.L.; Eijkelkamp, N.; Wang, H.; Dantzer, R.; Dorn II, G.W.; Kelley, K.W.; Heijnen, C.J.; Kavelaars, A. Microglial/Macrophage GRK2 Determines Duration of Peripheral IL-1β-Induced Hyperalgesia: Contribution of Spinal Cord CX3CR1, p38 and IL-1 signaling. Pain 2010, 150, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Jurado-Pueyo, M.; Campos, P.M.; Mayor, F.; Murga, C. GRK2-Dependent Desensitization Downstream of G Proteins. J. Recept. Signal Transduct. 2008, 28, 59–70. [Google Scholar] [CrossRef]
- Liu, S.; Premont, R.T.; Kontos, C.D.; Zhu, S.; Rockey, D.C. A Crucial Role for GRK2 in Regulation of Endothelial Cell Nitric Oxide Synthase Function in Portal Hypertension. Nat. Med. 2005, 11, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Kobayashi, T.; Takenouchi, Y.; Matsumoto, T.; Kamata, K. Angiotensin II Causes Endothelial Dysfunction via the GRK2/Akt/eNOS Pathway in Aortas from a Murine Type 2 Diabetic Model. Pharmacol. Res. 2011, 64, 535–546. [Google Scholar] [CrossRef]
- Premont, R.T.; Claing, A.; Vitale, N.; Freeman, J.L.R.; Pitcher, J.A.; Patton, W.A.; Moss, J.; Vaughan, M.; Lefkowitz, R.J. 2-Adrenergic Receptor Regulation by GIT1, a G Protein-Coupled Receptor Kinase-Associated ADP Ribosylation Factor GTPase-Activating Protein. Proc. Natl. Acad. Sci. USA 1998, 95, 14082–14087. [Google Scholar] [CrossRef] [Green Version]
- Lafarga, V.; Aymerich, I.; Tapia, O.; Mayor, F., Jr.; Penela, P. A Novel GRK2/HDAC6 Interaction Modulates Cell Spreading and Motility. EMBO J. 2012, 31, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Benovic, J.L. G Protein-coupled Receptor Kinase Interaction with Hsp90 Mediates Kinase Maturation. J. Biol. Chem. 2003, 278, 50908–50914. [Google Scholar] [CrossRef] [Green Version]
- De Jager, S.C.A.; Bermúdez, B.; Bot, I.; Koenen, R.R.; Bot, M.; Kavelaars, A.; De Waard, V.; Heijnen, C.J.; Muriana, F.J.G.; Weber, C.; et al. Growth Differentiation Factor 15 Deficiency Protects against Atherosclerosis by Attenuating CCR2-Mediated Macrophage Chemotaxis. J. Exp. Med. 2011, 208, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; Cocolakis, E.; Dumas, V.M.; Posner, B.I.; Laporte, S.A.; Lebrun, J.J. The G protein-coupled Receptor Kinase-2 is a TGFbeta-Inducible Antagonist of TGFbeta Signal Transduction. EMBO J. 2005, 24, 3247–3258. [Google Scholar] [CrossRef] [Green Version]
- Luerman, G.C.; Powell, D.W.; Uriarte, S.M.; Cummins, T.D.; Merchant, M.L.; Ward, R.A.; McLeish, K.R. Identification of Phosphoproteins Associated with Human Neutrophil Granules Following Chemotactic Peptide Stimulation. Mol. Cell. Proteom. 2011, 10, M110.001552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiedemann, R.E.; Zhu, Y.X.; Schmidt, J.; Yin, H.; Shi, C.-X.; Que, Q.; Basu, G.; Azorsa, D.; Perkins, L.M.; Braggio, E.; et al. Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood 2010, 115, 1594–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, C.; Tavernier, G.; Charpentier, F.; Langin, D.; Le Marec, H. Functional beta3-Adrenoceptor in the Human Heart. J. Clin. Investig. 1996, 98, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Vinge, L.E.; von Lueder, T.G.; Aasum, E.; Qvigstad, E.; Gravning, J.A.; How, O.J.; Edvardsen, T.; Bjornerheim, R.; Ahmed, M.S.; Mikkelsen, B.W.; et al. Cardiac-Restricted Expression of the Carboxyl-Terminal Fragment of GRK3 Uncovers Distinct Functions of GRK3 in Regulation of Cardiac Contractility and Growth: GR3 Controls Cardiac alpha1-Adrenergic Receptor Responsiveness. J. Biol. Chem. 2008, 283, 10601–10610. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Matkovich, S.J.; Duan, X.; Gold, J.I.; Koch, W.J.; Dorn, G.W. Nuclear Effects of G-Protein Receptor Kinase 5 on Histone Deacetylase 5–Regulated Gene Transcription in Heart Failure. Circ. Heart Fail. 2011, 4, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.P.; Bittner, H.B.; Akhter, S.A.; Koch, W.J.; Davis, R. Myocardial Function in Hearts with Transgenic Overexpression of the G Protein-Coupled Receptor Kinase 5. Ann. Thorac. Surg. 2001, 71, 1320–1324. [Google Scholar] [CrossRef]
- Ungerer, M.; Böhm, M.; Elce, J.S.; Erdmann, E.; Lohse, M.J. Altered Expression of Beta-Adrenergic Receptor Kinase and Beta 1-Adrenergic Receptors in the Failing Human Heart. Circulation 1993, 87, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Ungerer, M.; Kessebohm, K.; Kronsbein, K.; Lohse, M.J.; Richardt, G. Activation of Beta-Adrenergic Receptor Kinase during Myocardial Ischemia. Circ. Res. 1996, 79, 455–460. [Google Scholar] [CrossRef]
- Gros, R.; Benovic, J.L.; Tan, C.M.; Feldman, R.D. G-Protein-Coupled Receptor Kinase Activity Is Increased in Hypertension. J. Clin. Investig. 1997, 99, 2087–2093. [Google Scholar] [CrossRef]
- Choi, D.J.; Koch, W.J.; Hunter, J.J.; Rockman, H.A. Mechanism of Beta-Adrenergic Receptor Desensitization in Cardiac Hy-Pertrophy Is Increased Beta-Adrenergic Receptor Kinase. J. Biol. Chem. 1997, 272, 17223–17229. [Google Scholar] [PubMed] [Green Version]
- Raake, P.W.; Vinge, L.E.; Gao, E.; Boucher, M.; Rengo, G.; Chen, X.; DeGeorge, B.R., Jr.; Matkovich, S.; Houser, S.R.; Most, P.; et al. G Protein-Coupled Receptor Kinase 2 Ablation in Cardiac Myocytes before or after Myocardial Infarction Prevents Heart Failure. Circ. Res. 2008, 103, 413–422. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Pesant, S.; Cohn, H.I.; Soltys, S.; Koch, W.J.; Eckhart, A.D. Negative Regulation of VEGF Signaling in Human Coronary Artery Endothelial Cells by G Protein-Coupled Receptor Kinase 5. Clin. Transl. Sci. 2009, 2, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Brinks, H.; Boucher, M.; Gao, E.; Chuprun, J.K.; Pesant, S.; Raake, P.W.; Huang, Z.M.; Wang, X.; Qiu, G.; Gumpert, A.; et al. Level of G Protein-Coupled Receptor Kinase-2 Determines Myocardial Ische-Mia/Reperfusion Injury via Pro-and Anti-apoptotic Mechanisms. Circ. Res. 2010, 107, 1140–1149. [Google Scholar] [CrossRef] [Green Version]
- Suo, W.Z.; Li, L. Dysfunction of G Protein-Coupled Receptor Kinases in Alzheimer’s Disease. Sci. World J. 2010, 10, 1667–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vroon, A.; Kavelaars, A.; Limmroth, V.; Lombardi, M.S.; Goebel, M.U.; Van Dam, A.-M.; Caron, M.G.; Schedlowski, M.; Heijnen, C.J. G Protein-Coupled Receptor Kinase 2 in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. J. Immunol. 2005, 174, 4400–4406. [Google Scholar] [CrossRef] [Green Version]
- Bychkov, E.R.; Gurevich, V.V.; Joyce, J.N.; Benovic, J.L.; Gurevich, E.V. Arrestins and Two Receptor Kinases Are Upregulated in Parkinson’s Disease with Dementia. Neurobiol. Aging 2008, 29, 379–396. [Google Scholar] [CrossRef] [Green Version]
- Obrenovich, M.E.; Palacios, H.H.; Gasimov, E.; Leszek, J.; Aliev, G. The GRK2 Overexpression Is a Primary Hallmark of Mitochondrial Lesions during Early Alzheimer Disease. Cardiovasc. Psychiatry Neurol. 2009, 2009, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, S.D.; Conn, P.J. G Protein-Coupled Receptor Kinases Regulate Metabotropic Glutamate Receptor 5 Function and Ex-pression. Neuropharmacology 2003, 44, 699–706. [Google Scholar] [CrossRef]
- Degos, V.; Peineau, S.; Nijboer, C.; Kaindl, A.M.; Sigaut, S.; Favrais, G.; Plaisant, F.; Teissier, N.; Gouadon, E.; Lombet, A.; et al. G Protein-Coupled Receptor Kinase 2 and Group I Metabotropic Glutamate Receptors Mediate Inflammation-Induced Sensitization to Excitotoxic Neurodegeneration. Ann. Neurol. 2013, 73, 667–678. [Google Scholar] [CrossRef]
- Eijkelkamp, N.; Heijnen, C.J.; Willemen, H.L.; Deumens, R.; Joosten, E.A.; Kleibeuker, W.; den Hartog, I.J.; van Velthoven, C.T.; Nijboer, C.; Nassar, M.A.; et al. GRK2: A Novel Cell-Specific Regulator of Severity and Duration of Inflammatory Pain. J. Neurosci. 2010, 30, 2138–2149. [Google Scholar] [CrossRef]
- Ferrari, L.F.; Bogen, O.; Alessandri–Haber, N.; Levine, E.; Gear, R.W.; Levine, J.D. Transient Decrease in Nociceptor grk2 Expression Produces Long-Term Enhancement in Inflammatory Pain. Neuroscience 2012, 222, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Rasul, I.; Sun, Y.; Wu, G.; Li, L.; Premont, R.T.; Suo, W.Z. GRK5 Deficiency Leads to Reduced Hippocampal Acetylcholine Level via Impaired Presynaptic M2/M4 Autoreceptor Desensitization. J. Biol. Chem. 2009, 284, 19564–19571. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Tajima, M.; Kosako, H.; Nakaya, T.; Hashimoto, A.; Watari, K.; Nishihara, H.; Ohba, M.; Komiya, S.; Tani, N.; et al. GRK6 Deficiency in Mice Causes Autoimmune Disease Due to Impaired Apoptotic Cell Clearance. Nat. Commun. 2013, 4, 1532. [Google Scholar] [CrossRef] [Green Version]
- Lappano, R.; Maggiolini, M. G Protein-Coupled Receptors: Novel Targets for Drug Discovery in Cancer. Nat. Rev. Drug Discov. 2010, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-Protein-Coupled Receptors and Cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Penela, P.; Murga, C.; Ribas, C.; Lafarga, V.; Mayor, F., Jr. The Complex G Protein-Coupled Receptor Kinase 2 (GRK2) Interactome Unveils New Physiopathological Targets. Br. J. Pharmacol. 2010, 160, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G Protein-Coupled Receptor Kinases: More Than Just Kinases and Not Only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef] [Green Version]
- Métayé, T.; Menet, E.; Guilhot, J.; Kraimps, J.-L. Expression and Activity of G Protein-Coupled Receptor Kinases in Differentiated Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2002, 87, 3279–3286. [Google Scholar] [CrossRef]
- Ma, Y.; Han, C.-C.; Huang, Q.; Sun, W.-Y.; Wei, W. GRK2 Overexpression Inhibits IGF1-Induced Proliferation and Migration of Human Hepatocellular Carcinoma Cells by Downregulating EGR1. Oncol. Rep. 2016, 35, 3068–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Hurtt, R.; Gu, T.; Bodzin, A.S.; Koch, W.J.; Doria, C. GRK2 Negatively Regulates IGF-1R Signaling Pathway and Cyclins’ Expression in HepG2 Cells. J. Cell. Physiol. 2013, 228, 1897–1901. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, M.-Y.; Liang, Z.-Y.; Zhou, W.-X.; You, L.; Pan, B.-J.; Liao, Q.; Zhao, Y.-P. G-Protein-Coupled Receptor Kinase 2 in Pancreatic Cancer: Clinicopathologic and Prognostic Significance. Hum. Pathol. 2016, 56, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Bin, W.Z. Expression of GRK2 and β-Arrestin2 in the Dorsal Root Ganglion Neurons and the Regulated Effect by Nerve Growth Factor in Rats with Bone Cancer. Pain. Chin. J. Cancer Prevent. Treat. 2011, 18, 4. [Google Scholar]
- Hu, M.; Wang, C.; Li, W.; Lu, W.; Bai, Z.; Qin, D.; Yan, Q.; Zhu, J.; Krueger, B.J.; Renne, R.; et al. A KSHV microRNA Directly Targets G Protein-Coupled Receptor Kinase 2 to Promote the Migration and Invasion of Endothelial Cells by Inducing CXCR2 and Activating AKT Signaling. PLoS Pathog. 2015, 11, e1005171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogues, L.; Salcedo, A.; Mendiola, M.; Hardission, D.; Mayor, F.; Penela, P. G Protein-Coupled Receptor Kinase 2 (GRK2) Con-Tributes to Cellular Transformation and Breast Tumor Progression. Eur. J. Cancer 2012, 48, S80–S81. [Google Scholar] [CrossRef]
- Nogués, L.; Reglero, C.; Rivas, V.; Salcedo, A.; Lafarga, V.; Neves, M.; Ramos, P.; Mendiola, M.; Berjón, A.; Stamatakis, K.; et al. G Protein-coupled Receptor Kinase 2 (GRK2) Promotes Breast Tumorigenesis Through a HDAC6-Pin1 Axis. EBioMedicine 2016, 13, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayáns, M.P.; Somoza-Martín, J.; Barros-Angueira, F.; Gayoso-Diz, P.; Otero-Rey, E.; Gándra-Rey, J.; Garcia-Garcia, A. Activity of β2-Adrenergic Receptor in Oral Squamous Cell Carcinoma Is Mediated by Overexpression of the ADRBK2 Gene: A Pilot Study. Biotech. Histochem. 2011, 87, 179–186. [Google Scholar] [CrossRef]
- Nakata, H.; Kinoshita, Y.; Kishi, K.; Fukuda, H.; Kawanami, C.; Matsushima, Y.; Asahara, M.; Okada, A.; Maekawa, T.; Chiba, T. Involvement of Beta-Adrenergic Receptor Kinase-1 in Homologous Desensitization of Histamine H2 Receptors in Human Gastric Carcinoma Cell Line MKN-45. Digestion 1996, 57, 406–410. [Google Scholar] [CrossRef]
- Rivas, V.; Chapuli, R.; Mayor, F.; Penela, P. 346 Role of GRK2 in Developing Vasculature and Pathological Angiogenesis. Eur. J. Cancer 2012, 48, S84. [Google Scholar] [CrossRef]
- Robinson, J.D.; Pitcher, J.A. G Protein-Coupled Receptor Kinase 2 (GRK2) Is a Rho-Activated Scaffold Protein for the ERK MAP Kinase Cascade. Cell Signal. 2013, 25, 2831–2839. [Google Scholar] [CrossRef] [Green Version]
- Prowatke, I.; Devens, F.; Benner, A.; Gröne, E.F.; Mertens, D.; Gröne, H.J.; Lichter, P.; Joos, S. Expression Analysis of Imbal-Anced Genes in Prostate Carcinoma Using Tissue Microarrays. Br. J. Cancer 2007, 96, 82–88. [Google Scholar] [CrossRef]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Woerner, B.M.; Luo, J.; Brown, K.R.; Jackson, E.; Dahiya, S.M.; Mischel, P.; Benovic, J.L.; Piwnica-Worms, D.; Rubin, J.B. Suppression of G-protein–Coupled Receptor Kinase 3 Expression Is a Feature of Classical GBM That Is Required for Maximal Growth. Mol. Cancer Res. 2012, 10, 156–166. [Google Scholar] [CrossRef] [Green Version]
- King, D.W.; Steinmetz, R.; Wagoner, H.A.; Hannon, T.S.; Chen, L.Y.; Eugster, E.A.; Pescovitz, O.H. Differential Expression of GRK Isoforms in Nonmalignant and Malignant Human Granulosa Cells. Endocrine 2003, 22, 135–142. [Google Scholar] [CrossRef]
- Miyagawa, Y.; Ohguro, H.; Odagiri, H.; Maruyama, I.; Maeda, T.; Maeda, A.; Sasaki, M.; Nakazawa, M. Aberrantly Expressed Recoverin Is Functionally Associated with G-Protein-Coupled Receptor Kinases in Cancer Cell Lines. Biochem. Biophys. Res. Commun. 2003, 300, 669–673. [Google Scholar] [CrossRef]
- Sánchez-Más, J.; Guillo, L.A.; Zanna, P.; Jiménez-Cervantes, C.; García-Borrón, J.C. Role of G Protein-Coupled Receptor Kinases in the Homologous Desensitization of the Human and Mouse Melanocortin 1 Receptors. Mol. Endocrinol. 2005, 19, 1035–1048. [Google Scholar] [CrossRef] [Green Version]
- Fitzhugh, D.; McGinnis, M.; Timoshchenko, R.; Lininger, R.; DeMore, N.; Serody, J.; Tarrant, T. G Protein Coupled Receptor Kinase 3 (GRK3) Negatively Regulates CXCL12/CXCR4 Signaling and Tumor Migration in Breast Cancer. J. Allergy Clin. Immunol. 2011, 127, AB230. [Google Scholar] [CrossRef]
- Li, W.; Ai, N.; Wang, S.; Bhattacharya, N.; Vrbanac, V.; Collins, M.; Signoretti, S.; Hu, Y.; Boyce, F.M.; Gravdal, K.; et al. GRK3 is Essential for Metastatic Cells and Promotes Prostate Tumor Progression. Proc. Natl. Acad. Sci. USA 2014, 111, 1521–1526. [Google Scholar] [CrossRef] [Green Version]
- Dautzenberg, F.M.; Braun, S.; Hauger, R.L. GRK3 Mediates Desensitization of CRF1 Receptors: A Potential Mechanism Regu-Lating Stress Adaptation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R935–R946. [Google Scholar] [CrossRef] [Green Version]
- Matsubayashi, J.; Takanashi, M.; Oikawa, K.; Fujita, K.; Tanaka, M.; Xu, M.; De Blasi, A.; Bouvier, M.; Kinoshita, M.; Kuroda, M.; et al. Expression of G Protein-Coupled Receptor Kinase 4 Is Associated with Breast Cancer Tumourigenesis. J. Pathol. 2008, 216, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Kim, J.; Kaur, R.; Tan, I.; Bloch, O.; Sun, M.Z.; Safaee, M.; Oh, M.C.; Sughrue, M.; Phillips, J.; et al. G-Protein Coupled Receptor Kinase (GRK)-5 Regulates Proliferation of Glioblastoma-Derived Stem Cells. J. Clin. Neurosci. 2013, 20, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhang, H.; Liu, J.; Rubin, J.B.; Cho, Y.J.; Shu, H.K.; Schniederjan, M.; MacDonald, T.J. Growth Factor Receptor-Src-Mediated Suppression of GRK6 Dysregulates CXCR4 Signaling and Promotes Medulloblastoma Migration. Mol. Cancer 2013, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Chakraborty, P.; Wang, Z.; Daaka, Y. G-Protein Coupled Receptor Kinase 5 Regulates Prostrate Tumor Growth. J. Urol. 2012, 187, 322–329. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, H.; Yuan, M.; Fu, J.; Zhou, Y.; Ma, L. G-protein-coupled Receptor Kinase 5 Phosphorylates p53 and Inhibits DNA Damage-induced Apoptosis. J. Biol. Chem. 2010, 285, 12823–12830. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Tsai, F.-M.; Shyu, R.-Y.; Tsai, Y.-M.; Wang, C.-H.; Jiang, S.-Y. G Protein-Coupled Receptor Kinase 5 Mediates Tazarotene-Induced Gene 1-Induced Growth Suppression of Human Colon Cancer Cells. BMC Cancer 2011, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Geras-Raaka, E.; Varma, A.; Clark-Lewis, I.; Gershengorn, M.C. Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Chemokine VMIP-II and Human SDF-1alpha Inhibit Signaling by KSHV G Protein-Coupled Receptor. Biochem. Biophys. Res. Commun. 1998, 253, 725–727. [Google Scholar] [CrossRef]
- Li, Y.-P. GRK6 Expression in Patients with Hepatocellular Carcinoma. Asian Pac. J. Trop. Med. 2013, 6, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Chen, J.; Zhang, Z.; You, Y.; Wang, Z. Aberrant GRK6 Promoter Methylation Is Associated with Poor Prognosis in Hypopharyngeal Squamous Cell Carcinoma. Oncol. Rep. 2016, 35, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Raghuwanshi, S.K.; Smith, N.; Rivers, E.J.; Thomas, A.J.; Sutton, N.; Hu, Y.; Mukhopadhyay, S.; Chen, X.L.; Leung, T.; Richardson, R.M.; et al. G Protein-Coupled Receptor Kinase 6 Deficiency Promotes Angiogenesis, Tumor Progression, and Metastasis. J. Immunol. 2013, 190, 5329–5336. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Zhong, L.; Liu, J.; Feng, J.; Bian, T.; Zhang, Q.; Chen, J.; Lv, X.; Chen, J.; Liu, Y. Prognostic Value of Decreased GRK6 Expression in Lung Adenocarcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 2541–2549. [Google Scholar] [CrossRef]
- Maeda, T.; Maeda, A.; Maruyama, I.; Ogawa, K.I.; Kuroki, Y.; Sahara, H.; Sato, N.; Ohguro, H. Mechanisms of Photoreceptor Cell Death in Cancer-Associated Retinopathy. Investig. Ophthalmol. Vis. Sci. 2001, 42, 705–712. [Google Scholar]
- Zhang, H.; Li, S.; Doan, T.; Rieke, F.; Detwiler, P.B.; Frederick, J.M.; Baehr, W. Deletion of PrBP/Delta Impedes Transport of GRK1 and PDE6 Catalytic Subunits to Photoreceptor Outer Segments. Proc. Natl. Acad. Sci. USA 2007, 104, 8857–8862. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Frederick, J.M.; Baehr, W. Unc119 Gene Deletion Partially Rescues the grk1 Transport Defect of PDE6d (-/-) Cones. Adv. Exp. Med. Biol. 2014, 801, 487–493. [Google Scholar]
- Mukherjee, D.; Zhao, J. The Role of Chemokine Receptor CXCR4 in Breast Cancer Metastasis. Am. J. Cancer Res. 2013, 3, 46–57. [Google Scholar]
- Chen, K.; Fu, C.; Chen, C.; Liu, L.; Ren, H.; Han, Y.; Yang, J.; He, D.; Zhou, L.; Yang, Z.; et al. Role of GRK4 in the Regulation of Arterial AT1Receptor in Hypertension. Hypertension 2014, 63, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Villar, V.A.; Jones, J.E.; Armando, I.; Palmes-Saloma, C.; Yu, P.; Pascua, A.M.; Keever, L.; Arnaldo, F.B.; Wang, Z.; Luo, Y.; et al. G Protein-Coupled Receptor Kinase 4 (GRK4) Regulates the Phosphorylation and Function of the Dopamine D3 Receptor. J. Biol. Chem. 2009, 284, 21425–21434. [Google Scholar] [CrossRef] [Green Version]
- Harvey, J.W. The Feline Blood Film. J. Feline Med. Surg. 2017, 19, 747–757. [Google Scholar] [CrossRef]
- Senzel, L.; Gnatenko, D.V.; Bahou, W.F. The Platelet Proteome. Curr. Opin. Hematol. 2009, 16, 329–333. [Google Scholar] [CrossRef]
- White, J.G.; Key, N.S.; King, R.A.; Vercellotti, G.M. The White Platelet Syndrome: A New Autosomal Dominant Platelet Disorderi. Structural Abnormalities. Platelets 2004, 15, 173–184. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kashiwagi, H.; Pampori, N. Integrin Signaling: The Platelet Paradigm. Blood 1998, 91, 2645–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling During Platelet Adhesion and Activation. Arter. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaffran, Y.; Meyer, S.C.; Negrescu, E.; Reddy, K.B.; Fox, J.E. Signaling across the Platelet Adhesion Receptor Glycoprotein Ib-IX Induces Alpha Iibbeta 3 Activation Both in Platelets and a Transfected Chinese Hamster Ovary Cell System. J. Biol. Chem. 2000, 275, 16779–16787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Kambayashi, J.; Okuma, M.; Tandon, N.N. Activation of the GP IIb-IIIa Complex Induced by Platelet Adhesion to Collagen Is Mediated by Both Alpha2Beta1 Integrin and GP VI. J. Biol. Chem. 1999, 274, 11897–11903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offermanns, S. Activation of Platelet Function Through G Protein–Coupled Receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Etherington, M.; Jamieson, S. Refractory State of Platelet Aggregation with Major Operations. Lancet 1971, 298, 741–743. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Kim, S.; Jee, Y.; Lee, S.-H.; Park, K.-M.; Kim, S. Role of GRK6 in the Regulation of Platelet Activation through Selective G Protein-Coupled Receptor (GPCR) Desensitization. Int. J. Mol. Sci. 2020, 21, 3932. [Google Scholar] [CrossRef]
- Liggett, S.B.; Freedman, N.J.; Schwinn, D.A.; Lefkowitz, R.J. Structural Basis for Receptor Subtype-Specific Regulation Revealed by a Chimeric Beta 3/Beta 2-Adrenergic Receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 3665–3669. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gupta, S.; Cooper, M.; DeHelian, D.; Zhao, X.; Naik, M.U.; Wurtzel, J.G.T.; Stalker, T.J.; Goldfinger, L.E.; Benovic, J.; et al. GRK6 Regulates the Hemostatic Response to Injury through Its Rate-Limiting Effects on Gpcr Signaling in Platelets. Blood Adv. 2020, 4, 76–86. [Google Scholar] [CrossRef]
- Hardy, A.R.; Conley, P.B.; Luo, J.; Benovic, J.L.; Poole, A.W.; Mundell, S.J. P2Y1 and P2Y12 Receptors for ADP Desensitize by Distinct Kinase-Dependent Mechanisms. Blood 2005, 105, 3552–3560. [Google Scholar] [CrossRef]
- Li, D.; D’Angelo, L.; Chavez, M.; Woulfe, D.S. Arrestin-2 Differentially Regulates PAR4 and ADP Receptor Signaling in Platelets. J. Biol. Chem. 2011, 286, 3805–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaff, M.; Receveur, N.; Bourdon, C.; Ohlmann, P.; Lanza, F.; Gachet, C.; Mangin, P.H. β-Arrestin-1 Participates in Thrombosis and Regulates Integrin aIIbβ3 Signalling without Affecting P2y Receptors Desensitisation and Function. Thromb. Haemost. 2012, 107, 735–748. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, J.L.; Zhao, X.; Hill, R.; Mundell, S.J. Arrestin-3 Differentially Regulates Platelet GPCR Subsets. Platelets 2019, 31, 641–645. [Google Scholar] [CrossRef]
- Amable, P.R.; Carias, R.B.V.; Teixeira, M.V.T.; da Cruz Pacheco, I.; do Amaral, R.J.F.C.; Granjeiro, J.M.; Borojevic, R. Platelet-Rich Plasma Preparation for Regenerative Medicine: Optimization and Quantification of Cytokines and Growth Factors. Stem Cell Res. Ther. 2013, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Bambace, N.M.; Holmes, C.E. The Platelet Contribution to Cancer Progression. J. Thromb. Haemost. 2011, 9, 237–249. [Google Scholar] [CrossRef]
- Wagner, D.D.; Frenette, P.S. The Vessel Wall and Its Interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef]
- Golebiewska, E.M.; Poole, A.W. Platelet Secretion: From Haemostasis to Wound Healing and Beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Ware, J.; Corken, A.; Khetpal, R. Platelet Function beyond Hemostasis and Thrombosis. Curr. Opin. Hematol. 2013, 20, 451–456. [Google Scholar] [CrossRef] [Green Version]
- McMorran, B.J.; Wieczorski, L.; Drysdale, K.E.; Chan, J.-A.; Huang, H.M.; Smith, C.; Mitiku, C.; Beeson, J.G.; Burgio, G.; Foote, S.J. Platelet Factor 4 and Duffy Antigen Required for Platelet Killing of Plasmodium falciparum. Science 2012, 338, 1348–1351. [Google Scholar] [CrossRef]
- Yeaman, M.R. Platelets in Defense against Bacterial Pathogens. Cell. Mol. Life Sci. 2009, 67, 525–544. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The Interaction of Bacterial Pathogens with Platelets. Nat. Rev. Genet. 2006, 4, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh-Cognasse, H.; Damien, P.; Chabert, A.; Pozzetto, B.; Cognasse, F.; Garraud, O. Platelets and Infections—Complex Interactions with Bacteria. Front. Immunol. 2015, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, P.; Flaumenhaft, R. Platelet α-granules: Basic Biology and Clinical Correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.-Q.; Yeaman, M.R.; Selsted, M.E. Antimicrobial Peptides from Human Platelets. Infect. Immun. 2002, 70, 6524–6533. [Google Scholar] [CrossRef] [Green Version]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.J.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.J.; et al. Platelet Gene Expression and Function in Patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, T.; Langer, H.F. Platelets and Neurovascular Inflammation. Thromb. Haemost. 2013, 110, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Behari, M.; Shrivastava, M. Role of Platelets in Neurodegenerative Diseases: A Universal Pathophysiology. Int. J. Neurosci. 2013, 123, 287–299. [Google Scholar] [CrossRef]
- Klouche, M.; Klinger, M.F.; Kühnel, W.; Wilhelm, D. Endocytosis, Storage, and Release of IgE by Human Platelets: Differences in Patients with Type I Allergy and Nonatopic Subjects. J. Allergy Clin. Immunol. 1997, 100, 235–241. [Google Scholar] [CrossRef]
- Pitchford, S.C.; Yano, H.; Lever, R.; Riffo-Vasquez, Y.; Ciferri, S.; Rose, M.J.; Giannini, S.; Momi, S.; Spina, D.; O’Connor, B.; et al. Platelets Are Essential for Leukocyte Recruitment in Allergic Inflammation. J. Allergy Clin. Immunol. 2003, 112, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Pitchford, S.C.; Riffo-Vasquez, Y.; Sousa, A.; Momi, S.; Gresele, P.; Spina, D.; Page, C.P. Platelets Are Necessary for Airway Wall Remodeling in a Murine Model of Chronic Allergic Inflammation. Blood 2004, 103, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, A.S.; Kumar, N.; Lerner, J.; Wiles, A.A.; Foerster, M.; Teach, S.J.; Freishtat, R.J. Airway Platelet Activation Is Associated with Airway Eosinophilic Inflammation in Asthma. J. Investig. Med. 2010, 58, 987–990. [Google Scholar] [CrossRef]
- Montenont, E.; Echagarruga, C.; Allen, N.; Araldi, E.; Suarez, Y.; Berger, J.S. Platelet WDR1 Suppresses Platelet Activity and Is Associated with Cardiovascular Disease. Blood 2016, 128, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNicol, A.; Israels, S.J. Beyond Hemostasis: The Role of Platelets in Inflammation, Malignancy and Infection. Cardiovasc. Haematol. Dis. Drug Targets 2008, 8, 99–117. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Brummel-Ziedins, K.E.; Bouchard, B.A.; Holmes, C.E. Platelets in Tumor Progression: A Host Factor That Offers Multiple Potential Targets in the Treatment of Cancer. J. Cell. Physiol. 2014, 229, 1005–1015. [Google Scholar] [CrossRef]
- Bendas, G.; Borsig, L. Cancer Cell Adhesion and Metastasis: Selectins, Integrins, and the Inhibitory Potential of Heparins. Int. J. Cell Biol. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Dang, S.; Hong, T.; Tang, J.; Fan, J.; Bu, D.; Sun, Y.; Wang, Z.; Wisniewski, T. A Humanized Single-Chain Antibody against Beta 3 Integrin Inhibits Pulmonary Metastasis by Preferentially Fragmenting Activated Platelets in the Tumor Microen-Vironment. Blood 2012, 120, 2889–2898. [Google Scholar] [CrossRef] [Green Version]
- Ware, J. Fragmenting the Platelet to Reduce Metastasis. Blood 2012, 120, 2779–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of Daily Aspirin on Risk of Cancer Metastasis: A Study of Incident Cancers during Randomised Controlled Trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Hurlé, M.A. Changes in the Expression of G Protein-Coupled Receptor Kinases and Beta-Arrestin 2 in Rat Brain during Opioid Tolerance and Supersensitivity. J. Neurochem. 2001, 77, 486–492. [Google Scholar] [CrossRef]
- Díaz, A.; Pazos, A.; Flórez, J.; Ayesta, F.J.; Santana, V.; Hurlé, M.A. Regulation of Mu-Opioid Receptors, G-Protein-Coupled Receptor Kinases and Beta-Arrestin 2 in the Rat Brain after Chronic Opioid Receptor Antagonism. Neuroscience 2002, 112, 345–353. [Google Scholar] [CrossRef]
- Suo, Z.; Cox, A.A.; Bartelli, N.; Rasul, I.; Festoff, B.W.; Premont, R.T.; Arendash, G.W. GRK5 Deficiency Leads to Early Alz-heimer-Like Pathology and Working Memory Impairment. Neurobiol. Aging 2007, 28, 1873–1888. [Google Scholar] [CrossRef]
- Ungerer, M.; Parruti, G.; Böhm, M.; Puzicha, M.; DeBlasi, A.; Erdmann, E.; Lohse, M.J. Expression of Beta-Arrestins and Be-Ta-Adrenergic Receptor Kinases in the Failing Human Heart. Circ. Res. 1994, 74, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Grange-Midroit, M.; García-Sevilla, J.A.; Ferrer-Alcón, M.; La Harpe, R.; Huguelet, P.; Guimón, J. Regulation of GRK 2 and 6, Beta-Arrestin-2 and Associated Proteins in the Prefrontal Cortex of Drug-Free and Antidepressant Drug-Treated Subjects with Major Depression. Brain Res. Mol. Brain Res. 2003, 111, 31–41. [Google Scholar] [CrossRef]
- Tesmer, V.M.; Lennarz, S.; Mayer, G.; Tesmer, J.J.G. Molecular Mechanism for Inhibition of G Protein-Coupled Receptor Kinase 2 by a Selective RNA Aptamer. Structures 2012, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homan, K.T.; Tesmer, J.J.G. Molecular Basis for Small Molecule Inhibition of G Protein-Coupled Receptor Kinases. ACS Chem. Biol. 2015, 10, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Waldschmidt, H.V.; Homan, K.T.; Cruz-Rodríguez, O.; Cato, M.C.; Waninger-Saroni, J.; Larimore, K.M.; Cannavo, A.; Song, J.; Cheung, J.Y.; Kirchhoff, P.D.; et al. Structure-Based Design, Synthesis, and Biological Evaluation of Highly Selective and Potent G Protein-Coupled Receptor Kinase 2 Inhibitors. J. Med. Chem. 2016, 59, 3793–3807. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.M.; Yeow, R.Y.; Schoenau, C.; Huber, J.; Tesmer, J.J.G. Molecular Mechanism of Selectivity among G Protein-Coupled Receptor Kinase 2 Inhibitors. Mol. Pharmacol. 2011, 80, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thal, D.M.; Homan, K.T.; Chen, J.; Wu, E.K.; Hinkle, P.M.; Huang, Z.M.; Chuprun, J.K.; Song, J.; Gao, E.; Cheung, J.Y.; et al. Paroxetine Is a Direct Inhibitor of G Protein-Coupled Receptor Kinase 2 and Increases Myocardial Contractility. ACS Chem. Biol. 2012, 7, 1830–1839. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| GRK Isoform | Interacting Partner(s) | Associated Signalling Pathway/Cellular Response | References |
|---|---|---|---|
| GRK2 | NF-kB p105 subunit and inhibitor (IkB-α) phosphorylation | TLR4-induced and Tumor Necrosis Factor-α (TNF-α) pathways | [79,81,82] |
| p38 phosphorylation Raf1, MEK1, ERK2, RhoA, RKIP, GIT | P38 mitogen-activated protein kinases (MAPK) pathways Extracellular signal-regulated kinase (ERK) pathways | [83,84,85] | |
| Serine-threonine kinase Akt phosphorylation | Akt-nitric oxide (NO) pathways | [86,87] | |
| Ezrin/radixin/moesin phosphorylation | Actin cytoskeleton | [64,65] | |
| ADP ribosylation factor (ARF)-specific GTPase-activating proteins (GIT) | Focal adhesion dynamic | [63,88] | |
| Histone deacetylase 6 (HDAC6) phosphorylation | Microtubules network | [89] | |
| Heat shock protein 90 (Hsp90) | Regulation of GRK expression | [90] | |
| Receptor-regulated Smads (R-Smads) phosphorylation | Transforming growth factor β (TGF-β) pathways | [91,92] | |
| GRK3 | HSP90 | Regulation of GRK expression | [90] |
| GRK5 | ERM (moesin phosphorylation) | Actin cytoskeleton | [69] |
| GIT1 | Regulation of receptor endocytosis | [88] | |
| HSP90, HSP70 | Regulation of GRK expression and CXCR4 endocytosis | ||
| NF-kB p105 subunit and IkB-α phosphorylation | TLR4-induced and TNF-α pathways | [90,91] | |
| Src Tyrosine kinase | GRK phosphorylation and neutrophils exocytosis | [93] | |
| GRK6 | HSP90 | Regulation of GRK expression | [90,94] |
| GRK Subtype | Type of Cancer | Interacting Partner (s) | Molecular Mechanism | Function | Biological Model | References |
|---|---|---|---|---|---|---|
| GRK2 | Thyroid carcinoma | TSHR | ND | Decrease proliferation through rapid desensitization | Differentiated thyroid carcinoma patients and cell lines | [120] |
| Hepatocellular carcinoma cell | IGFI-R | Decrease proliferation and migration | [121] | |||
| Human hepatocellular carcinoma (HepG2) | IGFI-R | Decrease cell cycle progression | [122] | |||
| Pancreatic cancer | N/A | ND | --T-stage and poor survival rate, increased proliferation | Pancreatic carcinoma patients, ductal adenocarcinoma patients and cell lines | [123] | |
| Breast carcinosarcoma | NGFR | Decrease bone cancer pain | [124] | |||
| Kaposi’s sarcoma-associated herpesvirus infected tumor cell | CXCR2 | Desensitization and AKT signaling | Decrease migration and invasion | Patients and cell lines | [125] | |
| Basal breast cancer with Her-2 amplification/infiltrating ductal carcinoma | Her-2/ER-α | Increase the promoting of mitogenic, anti-apoptic activities- survival and progression | [126] | |||
| Luminal and basal breast cancer | HDAC6/Pin1 | AKT/ERK cascades | Increase sensitivity of breast cancer cells to traditional chemotherapeutic treatment | Invasive ductal carcinoma patients, cell lines orthotopic and xenograftmouse models | [127] | |
| Breast cancer | CXCR4 | Desensitization and signaling | Decrease metastasis | Breast cancer patients, cell lines orthotopic mouse models | [128] | |
| Human gastric carcinoma cell line (MKN-45) | H2 receptor | --poor differentiation | [129] | |||
| Human breast cancer | N/A | Increase tumor growth and decrease angiogenesis | [130,131] | |||
| Prostrate | ND | Differentiation | Adenocarcinoma patients | [132] | ||
| Prostrate | ND | ND | ND | Neuroendocrine prostrate and metastatic castrastion-resistant prostate cancer patients | [133] | |
| Glioblastoma | ND | ND | Mesenchymal glioblastoma patients. | [134] | ||
| GRK2/4 | Ovary | ND | ND | Granulosa cell cancer patients | [135] | |
| GRK2/5/6 | Gastric cancer (SSTW-2) | recoverin | --tumor progression, metastasis | [136] | ||
| GRK2/6 | melanoma | Melanocortin 1 receptor | --determinant for skin cancer | [137] | ||
| GRK3 | Breast cancers (MDA-MB-231, MDA-MB-468 | CXCR4 | Decrease metastasis increase migration | Breast cancer patients, cell lines orthotopic mouse models | [138] | |
| Prostate cancer (PC3) | N/A | Downmodulation of angiogenesis inhibitors | Increase metastasis, tumor progression, angiogenesis | Metastatic castration-resistant prostate cancer patients, cell lines and orthotopic mouse models | [139] | |
| Retinoblastoma (Y-79) | CRFI receptor | Increase stress adaptation | [140] | |||
| Oral squamous carcinoma | β2-adrenergic receptor | --tumor malignancy and invasion | [128] | |||
| Glioblastoma | CXCR4 desensitization and signaling | desensitization and signaling | Increased proliferation | Classical Glioblastoma patients | [134] | |
| GRK4 | Ovarian malignant granulosa cell tumor | FSHR | --benign and malignant transformation in tumor development | [135] | ||
| Breast cancer | Arrestin2 receptor | Mediated ERK & JNK signaling | Increase proliferation | Ductal carcinoma patients and cell lines | [141] | |
| GRK5 | glioblastoma | N/A | Proliferation rate and WHO grade | Glioblastoma multiform patients and cell lines | [142] | |
| Thyroid carcinoma | TSHR | TSHR desensitization and signaling | Decrease proliferation through slow desensitization, increase proliferation | Differentiated thyroid carcinoma patients | [120] | |
| Prostate cancer (PC3) | Cyclin D1 | G2/M progression | Decrease proliferation, cell cycle | Cell lines and xenograft mouse tumors | [143] | |
| Prostate cancer (PC3, DU145, LNCaP) | Moesin | Moesin phosphorylation | Decrease migration, invasion Increase cell adhesion | Cell lines and xenograft mouse tumors | [69] | |
| Prostate cancer | N/A | Increase tumor growth, invasion, and metastasis | [144] | |||
| Osteosarcoma (U2OS, Saos-2) | P53 | Phosphorylation and degradation | Decrease cell apoptosis and radiosensitivity | Cell lines | [145] | |
| Colon | PGE2 | Desensitization and signaling | Increased proliferation | Cell lines | [146] | |
| Kaposi’s sarcoma | KSHV-GPCR | Desensitization and signaling | Increased proliferation | Cell lines | [147] | |
| GRK6 | Heptocellular carcinoma | N/A | --proliferation maker in early diagnosis | [148] | ||
| Hypopharyngeal squamous cell carcinoma (FaDu) | Methyl transferase | Methylation of GRK6 | --cancer progression Decrease invasion | [149] | ||
| Medulloblastoma | CXCR4/ EGFR/ PDGFR-Src | Increase migration | [143] | |||
| Lung cancer | CXCR2 | Decrease cancer development | [150] | |||
| Lung | ND | ND | Decreased survival | Adenocarcinoma patients | [151] | |
| Medullo-Blastoma | CXCR4 | Desensitization and signaling | Increased migration | Medulloblastoma patients and cell lines | [143] | |
| Myeloma | STAT3 | phosphorylation | Increased survival | Primary multiple myeloma patients and cell lines | [94] | |
| GRK1/7 | recoverin | --cancer-associated retinopathy | [152] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, P.K.; Kim, S. The GRKs Reactome: Role in Cell Biology and Pathology. Int. J. Mol. Sci. 2021, 22, 3375. https://doi.org/10.3390/ijms22073375
Chaudhary PK, Kim S. The GRKs Reactome: Role in Cell Biology and Pathology. International Journal of Molecular Sciences. 2021; 22(7):3375. https://doi.org/10.3390/ijms22073375
Chicago/Turabian StyleChaudhary, Preeti Kumari, and Soochong Kim. 2021. "The GRKs Reactome: Role in Cell Biology and Pathology" International Journal of Molecular Sciences 22, no. 7: 3375. https://doi.org/10.3390/ijms22073375
APA StyleChaudhary, P. K., & Kim, S. (2021). The GRKs Reactome: Role in Cell Biology and Pathology. International Journal of Molecular Sciences, 22(7), 3375. https://doi.org/10.3390/ijms22073375