
Targeting GRK5 for Treating Chronic Degenerative Diseases


 ,
,  , and
, and 
Abstract
:1. Introduction
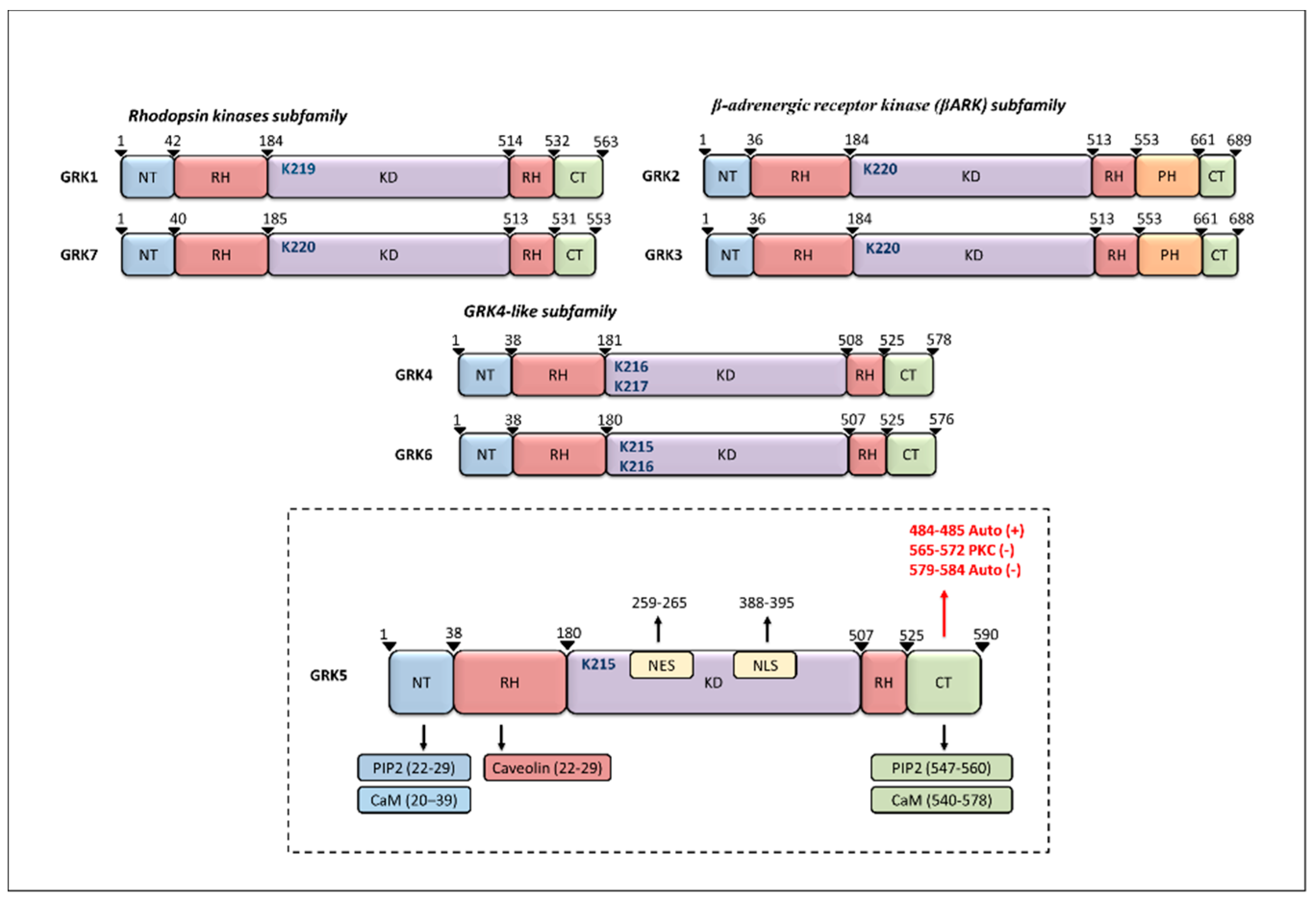
2. GRK5 Structure
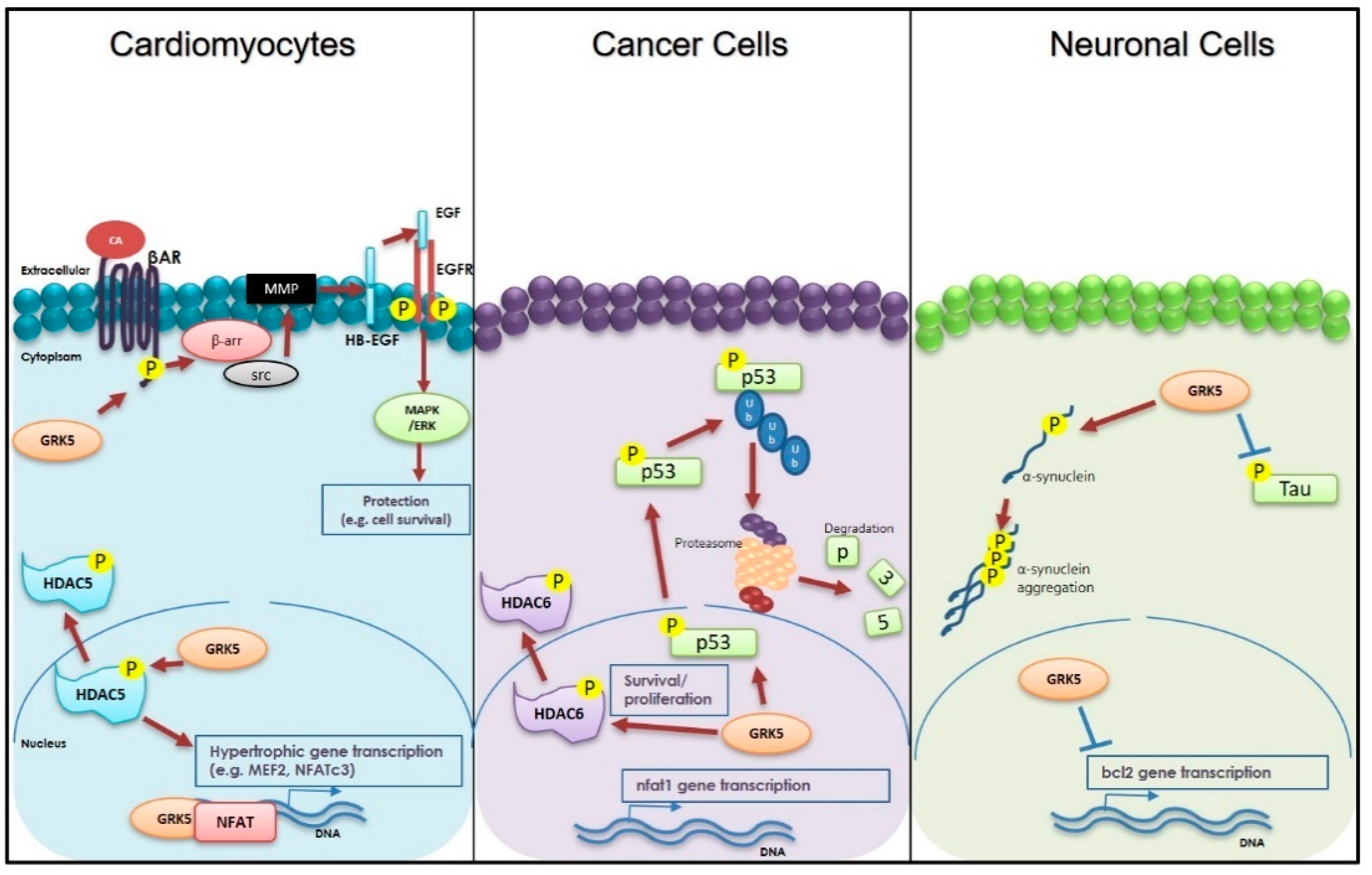
3. Cardiac Roles of GRK5
4. GRK5 and Neurodegenerative Diseases
5. GRK5 and Cancer
6. Targeting GRK5 as a Therapeutic Strategy for Chronic-Degenerative Disease
7. Conclusive Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar] [PubMed]
- Pierce, K.L.; Lefkowitz, R.J. Classical and new roles of β-arrestins in the regulation of G-PROTEIN-COUPLED receptors. Nat. Rev. Neurosci. 2001, 2, 727–733. [Google Scholar] [CrossRef]
- Watari, K.; Nakaya, M.; Kurose, H. Multiple functions of G protein-coupled receptor kinases. J. Mol. Signal. 2014, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, A.; Komici, K.; Bencivenga, L.; D’Amico, M.L.; Gambino, G.; Liccardo, D.; Ferrara, N.; Rengo, G. GRK2 as a therapeutic target for heart failure. Expert Opin. Ther. Targets 2018, 22, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T.; Inglese, J.; Lefkowitz, R.J. Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB J. 1995, 9, 175–182. [Google Scholar] [CrossRef]
- Sato, P.Y.; Chuprun, J.K.; Schwartz, M.; Koch, W.J. The Evolving Impact of G Protein-Coupled Receptor Kinases in Cardiac Health and Disease. Physiol. Rev. 2015, 95, 377–404. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, S.M.; Koch, W.J. Noncanonical Roles of G Protein-coupled Receptor Kinases in Cardiovascular Signaling. J. Cardiovasc. Pharmacol. 2017, 70, 129–141. [Google Scholar] [CrossRef]
- Weiss, E.R.; Raman, D.; Shirakawa, S.; Ducceschi, M.H.; Bertram, P.T.; Wong, F.; Kraft, T.W.; Osawa, S. The cloning of GRK7, a candidate cone opsin kinase, from cone- and rod-dominant mammalian retinas. Mol. Vis. 1998, 4, 27. [Google Scholar]
- Weiss, E.R.; Ducceschi, M.H.; Horner, T.J.; Li, A.; Craft, C.M.; Osawa, S. Species-Specific Differences in Expression of G-Protein-Coupled Receptor Kinase (GRK) 7 and GRK1 in Mammalian Cone Photoreceptor Cells: Implications for Cone Cell Phototransduction. J. Neurosci. 2001, 21, 9175–9184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Premont, R.T.; Macrae, A.D.; Stoffel, R.H.; Chung, N.; Pitcher, J.A.; Ambrose, C.; Inglese, J.; Macdonald, M.E.; Lefkowitz, R.J. Characterization of the G Protein-coupled Receptor Kinase GRK4. J. Biol. Chem. 1996, 271, 6403–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virlon, B.; Firsov, D.; Cheval, L.; Reiter, E.; Troispoux, C.; Guillou, F.; Elalouf, J.-M. Rat G Protein-Coupled Receptor Kinase GRK4: Identification, Functional Expression, and Differential Tissue Distribution of Two Splice Variants. Endocrinology 1998, 139, 2784–2795. [Google Scholar] [CrossRef]
- Sallese, M.; Salvatore, L.; D’Urbano, E.; Sala, G.; Storto, M.; Launey, T.; de Blasi, A.; Nicoletti, F.; Knopfel, T. The G-protein-coupled receptor kinase GRK4 mediates homologous desensitization of metabotropic glutamate receptor 1. FASEB J. 2000, 14, 2569–2580. [Google Scholar] [CrossRef]
- Felder, R.A.; Sanada, H.; Xu, J.; Yu, P.-Y.; Wang, Z.; Watanabe, H.; Asico, L.D.; Wang, W.; Zheng, S.; Yamaguchi, I.; et al. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc. Natl. Acad. Sci. USA 2002, 99, 3872–3877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenninkmeijer, C.B.; Price, S.A.; Bernal, A.L.; Phaneuf, S. Expression of G-protein-coupled receptor kinases in pregnant term and non-pregnant human myometrium. J. Endocrinol. 1999, 162, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Kunapuli, P.; Benovic, J.L. Cloning and expression of GRK5: A member of the G protein-coupled receptor kinase family. Proc. Natl. Acad. Sci. USA 1993, 90, 5588–5592. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.R.; Robinson, J.D.; Lester, K.N.; Pitcher, J.A. Distinct Structural Features of G Protein-Coupled Receptor Kinase 5 (GRK5) Regulate Its Nuclear Localization and DNA-Binding Ability. PLoS ONE 2013, 8, e62508. [Google Scholar] [CrossRef] [Green Version]
- Tesmer, V.M.; Kawano, T.; Shankaranarayanan, A.; Kozasa, T.; Tesmer, J.J.G. Snapshot of Activated G Proteins at the Membrane: The G q-GRK2-G Complex. Science 2005, 310, 1686–1690. [Google Scholar] [CrossRef]
- Inglese, J.; Freedman, N.; Koch, W.; Lefkowitz, R. Structure and mechanism of the G protein-coupled receptor kinases. J. Biol. Chem. 1993, 268, 23735–23738. [Google Scholar] [CrossRef]
- Fusco, A.; Santulli, G.; Sorriento, D.; Cipolletta, E.; Garbi, C.; Dorn, G.W.; Trimarco, B.; Feliciello, A.; Iaccarino, G. Mitochondrial localization unveils a novel role for GRK2 in organelle biogenesis. Cell. Signal. 2012, 24, 468–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Sato, P.Y.; Chuprun, J.K.; Peroutka, R.J.; Otis, N.J.; Ibetti, J.; Pan, S.; Sheu, S.-S.; Gao, E.; Koch, W.J.; et al. Prodeath signaling of G protein-coupled receptor kinase 2 in cardiac myocytes after ischemic stress occurs via extracellular signal-regulated kinase-dependent heat shock protein 90-mediated mitochondrial targeting. Circ. Res. 2013, 112, 1121–1134. [Google Scholar] [CrossRef] [Green Version]
- Siderovski, D.P.; Hessel, A.; Chung, S.; Mak, T.W.; Tyers, M. A new family of regulators of G-protein-coupled receptors? Curr. Biol. 1996, 6, 211–212. [Google Scholar] [CrossRef] [Green Version]
- Ribas, C.; Penela, P.; Murga, C.; Salcedo, A.; García-Hoz, C.; Jurado-Pueyo, M.; Aymerich, I.; Mayor, F. The G protein-coupled receptor kinase (GRK) interactome: Role of GRKs in GPCR regulation and signaling. Biochim. Biophys. Acta Biomembr. 2007, 1768, 913–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inglese, J.; Koch, W.J.; Caron, M.G.; Lefkowitz, R.J. Isoprenylation in regulation of signal transduction by G-protein-coupled receptor kinases. Nat. Cell Biol. 1992, 359, 147–150. [Google Scholar] [CrossRef]
- Pitcher, J.A.; Inglese, J.; Higgins, J.B.; Arriza, J.L.; Casey, P.J.; Kim, C.; Benovic, J.L.; Kwatra, M.M.; Caron, M.G.; Lefkowitz, R.J. Role of beta gamma subunits of G proteins in targeting the beta-adrenergic receptor kinase to membrane-bound receptors. Science 1992, 257, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Koch, W.; Inglese, J.; Stone, W.; Lefkowitz, R. The binding site for the beta gamma subunits of heterotrimeric G proteins on the beta-adrenergic receptor kinase. J. Biol. Chem. 1993, 268, 8256–8260. [Google Scholar] [CrossRef]
- Deb-Burman, S.K.; Ptasienski, J.; Benovic, J.L.; Hosey, M.M. G Protein-coupled Receptor Kinase GRK2 Is a Phospholipid-dependent Enzyme That Can Be Conditionally Activated by G Protein βγ Subunits. J. Biol. Chem. 1996, 271, 22552–22562. [Google Scholar] [CrossRef] [Green Version]
- Stoffel, R.H.; Randall, R.R.; Premont, R.T.; Lefkowitz, R.J.; Inglese, J. Palmitoylation of G protein-coupled receptor kinase, GRK6. Lipid modification diversity in the GRK family. J. Biol. Chem. 1994, 269, 27791–27794. [Google Scholar] [CrossRef]
- Jiang, X.; Benovic, J.L.; Wedegaertner, P.B. Plasma Membrane and Nuclear Localization of G Protein–coupled Receptor Kinase 6A. Mol. Biol. Cell 2007, 18, 2960–2969. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, M.M.; Stracquatanio, R.P.; Pronin, A.N.; Evanko, D.S.; Benovic, J.L.; Wedegaertner, P.B. A Predicted Amphipathic Helix Mediates Plasma Membrane Localization of GRK5. J. Biol. Chem. 2004, 279, 17989–17995. [Google Scholar] [CrossRef] [Green Version]
- Kunapuli, P.; Gurevich, V.; Benovic, J. Phospholipid-stimulated autophosphorylation activates the G protein-coupled receptor kinase GRK5. J. Biol. Chem. 1994, 269, 10209–10212. [Google Scholar] [CrossRef]
- Ding, B.; Glukhova, A.; Sobczyk-Kojiro, K.; Mosberg, H.I.; Tesmer, J.J.G.; Chen, Z. Unveiling the Membrane-Binding Properties of N-Terminal and C-Terminal Regions of G Protein-Coupled Receptor Kinase 5 by Combined Optical Spectroscopies. Langmuir 2014, 30, 823–831. [Google Scholar] [CrossRef]
- Yang, P.; Glukhova, A.; Tesmer, J.J.G.; Chen, Z. Membrane Orientation and Binding Determinants of G Protein-Coupled Receptor Kinase 5 as Assessed by Combined Vibrational Spectroscopic Studies. PLoS ONE 2013, 8, e82072. [Google Scholar] [CrossRef] [Green Version]
- Pronin, A.N.; Satpaev, D.K.; Slepak, V.Z.; Benovic, J.L. Regulation of G Protein-coupled Receptor Kinases by Calmodulin and Localization of the Calmodulin Binding Domain. J. Biol. Chem. 1997, 272, 18273–18280. [Google Scholar] [CrossRef] [Green Version]
- Penela, P.; Ribas, C.; Mayor, F. Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell. Signal. 2003, 15, 973–981. [Google Scholar] [CrossRef]
- Pronin, A.N.; Carman, C.V.; Benovic, J.L. Structure-Function Analysis of G Protein-coupled Receptor Kinase-5. J. Biol. Chem. 1998, 273, 31510–31518. [Google Scholar] [CrossRef] [Green Version]
- Carman, C.V.; Lisanti, M.P.; Benovic, J.L. Regulation of G Protein-coupled Receptor Kinases by Caveolin. J. Biol. Chem. 1999, 274, 8858–8864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.R.; Scott, M.G.H.; Pitcher, J.A. G Protein-Coupled Receptor Kinase 5 Contains a DNA-Binding Nuclear Localization Sequence. Mol. Cell. Biol. 2004, 24, 10169–10179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traynham, C.J.; Hullmann, J.; Koch, W.J. Canonical and non-canonical actions of GRK5 in the heart. J. Mol. Cell. Cardiol. 2016, 92, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, J.; Franco, A.; del Giudice, C.; Fiordelisi, A.; Cipolletta, E.; Ciccarelli, M.; Trimarco, B.; Iaccarino, G.; Sorriento, D. Dual role of GRK5 in cancer development and progression. Transl. Med. UniSa 2016, 14, 28–37. [Google Scholar]
- Hendrickx, J.O.; van Gastel, J.; Leysen, H.; Santos-Otte, P.; Premont, R.T.; Martin, B.; Maudsley, S. GRK5—A Functional Bridge Between Cardiovascular and Neurodegenerative Disorders. Front. Pharmacol. 2018, 9, 1484. [Google Scholar] [CrossRef]
- Cannavo, A.; Liccardo, D.; Koch, W.J. Targeting cardiac β-adrenergic signaling via GRK2 inhibition for heart failure therapy. Front. Physiol. 2013, 4, 264. [Google Scholar] [CrossRef] [Green Version]
- Rockman, H.A.; Choi, D.J.; Rahman, N.U.; Akhter, S.A.; Lefkowitz, R.J.; Koch, W.J. Receptor-specific in vivo desensitization by the G protein-coupled receptor kinase-5 in transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9954–9959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaccarino, G.; Barbato, E.; Cipolletta, E.; de Amicis, V.; Margulies, K.B.; Leosco, D.; Trimarco, B.; Koch, W.J. Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. Eur. Hear. J. 2005, 26, 1752–1758. [Google Scholar] [CrossRef] [Green Version]
- Agüero, J.; Almenar, L.; Montó, F.; Oliver, E.; Sánchez-Lázaro, I.; Vicente, D.; Martínez-Dolz, L.; D’Ocon, P.; Rueda, J.; Salvador, A. Myocardial G Protein Receptor–Coupled Kinase Expression Correlates With Functional Parameters and Clinical Severity in Advanced Heart Failure. J. Card. Fail. 2012, 18, 53–61. [Google Scholar] [CrossRef]
- Dzimiri, N.; Muiya, P.; Andres, E.; Al-Halees, Z. Differential functional expression of human myocardial G protein receptor kinases in left ventricular cardiac diseases. Eur. J. Pharmacol. 2004, 489, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Dzimiri, N.; Basco, C.; Moorji, A.; Afrane, B.; Al-Halees, Z. Characterization of Lymphocyte beta2-Adrenoceptor Signalling In Patients With Left Ventricular Volume Overload Disease. Clin. Exp. Pharmacol. Physiol. 2002, 29, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Martini, J.S.; Raake, P.; Vinge, L.E.; DeGeorge, B.R.; Chuprun, J.K.; Harris, D.M.; Gao, E.; Eckhart, A.D.; Pitcher, J.A.; Koch, W.J. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 12457–12462. [Google Scholar] [CrossRef] [Green Version]
- Gold, J.I.; Gao, E.; Shang, X.; Premont, R.T.; Koch, W.J. Determining the Absolute Requirement of G Protein–Coupled Receptor Kinase 5 for Pathological Cardiac Hypertrophy. Circ. Res. 2012, 111, 1048–1053. [Google Scholar] [CrossRef]
- Liggett, S.B.; Cresci, S.; Kelly, R.J.; Syed, F.M.; Matkovich, S.J.; Hahn, H.S.; Diwan, A.; Martini, J.S.; Sparks, L.; Parekh, R.R.; et al. A GRK5 polymorphism that inhibits β-adrenergic receptor signaling is protective in heart failure. Nat. Med. 2008, 14, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Noma, T.; Lemaire, A.; Prasad, S.V.N.; Barki-Harrington, L.; Tilley, D.G.; Chen, J.; Le Corvoisier, P.; Violin, J.D.; Wei, H.; Lefkowitz, R.J.; et al. β-Arrestin–mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Investig. 2007, 117, 2445–2458. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Tesmer, J.J.G.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hullmann, J.; Traynham, C.J.; Coleman, R.C.; Koch, W.J. The expanding GRK interactome: Implications in cardiovascular disease and potential for therapeutic development. Pharmacol. Res. 2016, 110, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Traynham, C.J.; Cannavo, A.; Zhou, Y.; Vouga, A.G.; Woodall, B.P.; Hullmann, J.; Ibetti, J.; Gold, J.I.; Chuprun, J.K.; Gao, E.; et al. Differential Role of G Protein–Coupled Receptor Kinase 5 in Physiological Versus Pathological Cardiac Hypertrophy. Circ. Res. 2015, 117, 1001–1012. [Google Scholar] [CrossRef] [Green Version]
- Gold, J.I.; Martini, J.S.; Hullmann, J.; Gao, E.; Chuprun, J.K.; Lee, L.; Tilley, D.G.; Rabinowitz, J.E.; Bossuyt, J.; Bers, D.M.; et al. Nuclear Translocation of Cardiac G Protein-Coupled Receptor Kinase 5 Downstream of Select Gq-Activating Hypertrophic Ligands Is a Calmodulin-Dependent Process. PLoS ONE 2013, 8, e57324. [Google Scholar] [CrossRef] [Green Version]
- Hullmann, J.E.; Grisanti, L.A.; Makarewich, C.A.; Gao, E.; Gold, J.I.; Chuprun, J.K.; Tilley, D.G.; Houser, S.R.; Koch, W.J. GRK5-Mediated Exacerbation of Pathological Cardiac Hypertrophy Involves Facilitation of Nuclear NFAT Activity. Circ. Res. 2014, 115, 976–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maning, J.; McCrink, K.A.; Pollard, C.M.; Desimine, V.L.; Ghandour, J.; Perez, A.; Cora, N.; Ferraino, K.E.; Parker, B.M.; Brill, A.R.; et al. Antagonistic Roles of GRK2 and GRK5 in Cardiac Aldosterone Signaling Reveal GRK5-Mediated Cardioprotection via Mineralocorticoid Receptor Inhibition. Int. J. Mol. Sci. 2020, 21, 2868. [Google Scholar] [CrossRef]
- Cannavo, A.; Liccardo, D.; Eguchi, A.; Elliott, K.J.; Traynham, C.J.; Ibetti, J.; Eguchi, S.; Leosco, D.; Ferrara, N.; Rengo, D.L.N.F.G.; et al. Myocardial pathology induced by aldosterone is dependent on non-canonical activities of G protein-coupled receptor kinases. Nat. Commun. 2016, 7, 10877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, T.; Yamamoto, T.; Kato, T.; Uchinoumi, H.; Fukui, G.; Hamada, Y.; Nanno, T.; Ishiguchi, H.; Nakamura, Y.; Okamoto, Y.; et al. Nuclear translocation of calmodulin in pathological cardiac hypertrophy originates from ryanodine receptor bound calmodulin. J. Mol. Cell. Cardiol. 2018, 125, 87–97. [Google Scholar] [CrossRef]
- Yeh, S.-T.; Zambrano, C.M.; Koch, W.J.; Purcell, N.H. PH domain leucine-rich repeat protein phosphatase 2 (PHLPP2) regulates G-protein–coupled receptor kinase 5 (GRK5)-induced cardiac hypertrophy in vitro. J. Biol. Chem. 2018, 293, 8056–8064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Premont, R.T.; Gainetdinov, R.R. Physiological Roles of G Protein–Coupled Receptor Kinases and Arrestins. Annu. Rev. Physiol. 2007, 69, 511–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdtmann-Vourliotis, M.; Mayer, P.; Ammon, S.; Riechert, U.; Höllt, V. Distribution of G-protein-coupled receptor kinase (GRK) isoforms 2, 3, 5 and 6 mRNA in the rat brain. Mol. Brain Res. 2001, 95, 129–137. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, F.; Long, H.; Chen, Y.; Wu, Z.; Ma, L. GRK5 promotes F-actin bundling and targets bundles to membrane structures to control neuronal morphogenesis. J. Cell Biol. 2011, 194, 905–920. [Google Scholar] [CrossRef] [Green Version]
- Kunapuli, P.; Onorato, J.; Hosey, M.; Benovic, J. Expression, purification, and characterization of the G protein-coupled receptor kinase GRK5. J. Biol. Chem. 1994, 269, 1099–1105. [Google Scholar] [CrossRef]
- Bubser, M.; Byun, N.; Wood, M.R.; Jones, C.K. Muscarinic Receptor Pharmacology and Circuitry for the Modulation of Cognition. Handb. Exp. Pharmacol. 2011, 121–166. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Bohn, L.M.; Walker, J.K.; Laporte, S.A.; Macrae, A.D.; Caron, M.G.; Lefkowitz, R.J.; Premont, R.T. Muscarinic Supersensitivity and Impaired Receptor Desensitization in G Protein–Coupled Receptor Kinase 5–Deficient Mice. Neuron 1999, 24, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Niu, B.; Liu, P.; Shen, M.; Liu, C.; Wang, L.; Wang, F.; Ma, L. GRK5 Regulates Social Behavior Via Suppression of mTORC1 Signaling in Medial Prefrontal Cortex. Cereb. Cortex 2017, 28, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Peng, W.; Zhang, Q.; Ding, X.; Suo, W.Z. GRK5 deficiency leads to susceptibility to intermittent hypoxia-induced cognitive impairment. Behav. Brain Res. 2016, 302, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Suo, Z.; Wu, M.; Citron, B.A.; Wong, G.T.; Festoff, B.W. Abnormality of G-Protein-Coupled Receptor Kinases at Prodromal and Early Stages of Alzheimer’s Disease: An Association with Early -Amyloid Accumulation. J. Neurosci. 2004, 24, 3444–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suo, Z.; Cox, A.A.; Bartelli, N.; Rasul, I.; Festoff, B.W.; Premont, R.T.; Arendash, G.W. GRK5 deficiency leads to early Alzheimer-like pathology and working memory impairment. Neurobiol. Aging 2007, 28, 1873–1888. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [CrossRef]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging. 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Liccardo, D.; Marzano, F.; Carraturo, F.; Guida, M.; Femminella, G.D.; Bencivenga, L.; Agrimi, J.; Addonizio, A.; Melino, I.; Valletta, A.; et al. Potential Bidirectional Relationship between Periodontitis and Alzheimer’s Disease. Front. Physiol. 2020, 11, 683. [Google Scholar] [CrossRef]
- Pakaski, M.; Kalman, J. Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem. Int. 2008, 53, 103–111. [Google Scholar] [CrossRef]
- Rossner, S.; Ueberham, U.; Schliebs, R.; Perez-Polo, J.R.; Bigl, V.; Roßner, S. The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Prog. Neurobiol. 1998, 56, 541–569. [Google Scholar] [CrossRef]
- Li, L.; Liu, J.; Suo, W.Z. GRK5 deficiency exaggerates inflammatory changes in TgAPPsw mice. J. Neuroinflammation 2008, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Rasul, I.; Liu, J.; Zhao, B.; Tang, R.; Premont, R.T.; Suo, W.Z. Augmented axonal defects and synaptic degenerative changes in female GRK5 deficient mice. Brain Res. Bull. 2009, 78, 145–151. [Google Scholar] [CrossRef]
- Liu, J.; Rasul, I.; Sun, Y.; Wu, G.; Li, L.; Premont, R.T.; Suo, W.Z. GRK5 Deficiency Leads to Reduced Hippocampal Acetylcholine Level via Impaired Presynaptic M2/M4 Autoreceptor Desensitization. J. Biol. Chem. 2009, 284, 19564–19571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Li, L.; He, S.; Liu, J.; Sun, Y.; He, M.; Grasing, K.; Premont, R.T.; Suo, W.Z. GRK5 Deficiency Accelerates β-Amyloid Accumulation in Tg2576 Mice via Impaired Cholinergic Activity. J. Biol. Chem. 2010, 285, 41541–41548. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Singh, P.; Cheng, S.; Zhang, Q.; Peng, W.; Ding, X.; Li, L.; Liu, J.; Premont, R.T.; Morgan, D.; et al. GRK5 Deficiency Leads to Selective Basal Forebrain Cholinergic Neuronal Vulnerability. Sci. Rep. 2016, 6, 26116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, L.; Shen, G.; Zhao, Q.; Shangguan, L.; He, M. GRK5 dysfunction accelerates tau hyperphosphorylation in APP (swe) mice through impaired cholinergic activity. NeuroReport 2014, 25, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, X.; Chen, X.; Cai, Y.; Wang, Y.; Sun, W.; Mai, H.; Yang, J.; Fan, W.; Tang, P.; et al. GRK5 influences the phosphorylation of tau via GSK3β and contributes to Alzheimer’s disease. J. Cell. Physiol. 2019, 234, 10411–10420. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, J.; Yin, M.; Cai, Y.; Liu, S.; Wang, Y.; Zhang, X.; Cao, H.; Chen, T.; Huang, P.; et al. The influence of two functional genetic variants of GRK5 on tau phosphorylation and their association with Alzheimer’s disease risk. Oncotarget 2017, 8, 72714–72726. [Google Scholar] [CrossRef] [Green Version]
- Arawaka, S.; Wada, M.; Goto, S.; Karube, H.; Sakamoto, M.; Ren, C.-H.; Koyama, S.; Nagasawa, H.; Kimura, H.; Kawanami, T.; et al. The Role of G-Protein-Coupled Receptor Kinase 5 in Pathogenesis of Sporadic Parkinson’s Disease. J. Neurosci. 2006, 26, 9227–9238. [Google Scholar] [CrossRef]
- Bychkov, E.; Gurevich, V.; Joyce, J.; Benovic, J. Arrestins and two receptor kinases are upregulated in Parkinson’s disease with dementia. Neurobiol. Aging 2008, 29, 379–396. [Google Scholar] [CrossRef] [Green Version]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s Disease. Cholesterol Binding and Cholesterol Transport. Proteins 2012, 65, 389–455. [Google Scholar] [CrossRef] [Green Version]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of Al-pha-Synuclein in Lewy Bodies of Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Liu, P.; Wang, X.; Gao, N.; Zhu, H.; Dai, X.; Xu, Y.; Ma, C.; Huang, L.; Liu, Y.; Qin, C. G protein-coupled receptor kinase 5, overexpressed in the α-synuclein up-regulation model of Parkinson’s disease, regulates bcl-2 expression. Brain Res. 2010, 1307, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, G.; de Luca, V.; Tarantino, P.; Gagliardi, M.; Iannello, G.; Novellino, F.; Morelli, M.; Annesi, G.; Quattrone, A. Role of G-protein coupled receptor kinase 5 gene in cognitive impairment in Parkinson’s disease. Psychiatry Res. 2015, 230, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; de Marco, E.V.; Annesi, G.; Rocca, F.E.; Annesi, F.; Civitelli, D.; Provenzano, G.; Scornaienchi, V.; Greco, V.; Colica, C.; et al. Lack of association between G-protein coupled receptor kinase 5 gene and Parkinson’s disease. Am. J. Med. Genet. Part. B Neuropsychiatr. Genet. 2010, 156, 104–107. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2010, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Michal, A.M.; So, C.H.; Beeharry, N.; Shankar, H.; Mashayekhi, R.; Yen, T.J.; Benovic, J.L. G Protein-coupled Receptor Kinase 5 Is Localized to Centrosomes and Regulates Cell Cycle Progression. J. Biol. Chem. 2012, 287, 6928–6940. [Google Scholar] [CrossRef] [Green Version]
- So, C.H.; Michal, A.M.; Mashayekhi, R.; Benovic, J.L. G Protein-coupled Receptor Kinase 5 Phosphorylates Nucleophosmin and Regulates Cell Sensitivity to Polo-like Kinase 1 Inhibition. J. Biol. Chem. 2012, 287, 17088–17099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Sorriento, D.; del Giudice, C.; Bertamino, A.; Ciccarelli, M.; Gomez-Monterrey, I.; Campiglia, P.; Novellino, E.; Illario, M.; Trimarco, B.; de Luca, N.; et al. New small molecules, ISA27 and SM13, inhibit tumour growth inducing mitochondrial effects of p53. Br. J. Cancer 2014, 112, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhu, H.; Yuan, M.; Fu, J.; Zhou, Y.; Ma, L. G-protein-coupled Receptor Kinase 5 Phosphorylates p53 and Inhibits DNA Damage-induced Apoptosis. J. Biol. Chem. 2010, 285, 12823–12830. [Google Scholar] [CrossRef] [Green Version]
- Okuda, M.; Nishimura, Y. Extended String Binding Mode of the Phosphorylated Transactivation Domain of Tumor Suppressor p53. J. Am. Chem. Soc. 2014, 136, 14143–14152. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Chakraborty, P.; Wang, Z.; Daaka, Y. G-Protein Coupled Receptor Kinase 5 Regulates Prostate Tumor Growth. J. Urol. 2012, 187, 322–329. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Zhang, Y.; Coomes, A.S.; Kim, W.-J.; Stupay, R.; Lynch, L.D.; Atkinson, T.; Kim, J.I.; Nie, Z.; Daaka, Y. G Protein–Coupled Receptor Kinase GRK5 Phosphorylates Moesin and Regulates Metastasis in Prostate Cancer. Cancer Res. 2014, 74, 3489–3500. [Google Scholar] [CrossRef] [Green Version]
- Kaur, G.; Kim, J.; Kaur, R.; Tan, I.; Bloch, O.; Sun, M.Z.; Safaee, M.; Oh, M.C.; Sughrue, M.; Phillips, J.; et al. G-protein coupled receptor kinase (GRK)-5 regulates proliferation of glioblastoma-derived stem cells. J. Clin. Neurosci. 2013, 20, 1014–1018. [Google Scholar] [CrossRef]
- Jiang, L.-P.; Fan, S.-Q.; Xiong, Q.-X.; Zhou, Y.-C.; Yang, Z.-Z.; Li, G.-F.; Huang, Y.-C.; Wu, M.-G.; Shen, Q.-S.; Liu, K.; et al. GRK5 functions as an oncogenic factor in non-small-cell lung cancer. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lagman, J.; Sayegh, P.; Lee, C.S.; Sulon, S.M.; Jacinto, A.Z.; Sok, V.; Peng, N.; Alp, D.; Benovic, J.L.; So, C.H. G protein-coupled receptor kinase 5 modifies cancer cell resistance to paclitaxel. Mol. Cell. Biochem. 2019, 461, 103–118. [Google Scholar] [CrossRef]
- Zhao, T.L.; Gan, X.X.; Bao, Y.; Wang, W.P.; Liu, B.; Wang, L.H. GRK5 promotes tumor progression in renal cell carcinoma. Neoplasma 2019, 66, 261–270. [Google Scholar] [CrossRef]
- Pham, T.; Robinson, K.; Vleeshouwer-Neumann, T.; Annis, J.E.; Chen, E.Y. Characterization of GRK5 as a novel regulator of rhabdomyosarcoma tumor cell growth and self-renewal. Oncotarget 2020, 11, 1448–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-C.; Tsai, F.-M.; Shyu, R.-Y.; Tsai, Y.-M.; Wang, C.-H.; Jiang, S.-Y. G protein-coupled receptor kinase 5 mediates Tazarotene-induced gene 1-induced growth suppression of human colon cancer cells. BMC Cancer 2011, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Tsai, F.-M.; Wu, C.-C.; Shyu, R.-Y.; Wang, C.-H.; Jiang, S.-Y. Tazarotene-induced gene 1 inhibits prostaglandin E2-stimulated HCT116 colon cancer cell growth. J. Biomed. Sci. 2011, 18, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Métayé, T.; Menet, E.; Guilhot, J.; Kraimps, J.-L. Expression and Activity of G Protein-Coupled Receptor Kinases in Differentiated Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2002, 87, 3279–3286. [Google Scholar] [CrossRef] [PubMed]
- Geras-Raaka, E.; Arvanitakis, L.; Bais, C.; Cesarman, E.; Mesri, E.A.; Gershengorn, M.C. Inhibition of Constitutive Signaling of Kaposi’s Sarcoma–associated Herpesvirus G Protein–Coupled Receptor by Protein Kinases in Mammalian Cells in Culture. J. Exp. Med. 1998, 187, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.-Y.; Chen, S.-C.; Leach, M.W.; Manfra, D.; Homey, B.; Wiekowski, M.; Sullivan, L.; Jenh, C.-H.; Narula, S.K.; Chensue, S.W.; et al. Transgenic Expression of the Chemokine Receptor Encoded by Human Herpesvirus 8 Induces an Angioproliferative Disease Resembling Kaposi’s Sarcoma. J. Exp. Med. 2000, 191, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The Structure and Function of G-Protein-Coupled Receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [Green Version]
- Pfleger, J.; Gresham, K.; Koch, W.J. G protein-coupled receptor kinases as therapeutic targets in the heart. Nat. Rev. Cardiol. 2019, 16, 612–622. [Google Scholar] [CrossRef]
- Casey, L.M.; Pistner, A.R.; Belmonte, S.L.; Migdalovich, D.; Stolpnik, O.; Nwakanma, F.E.; Vorobiof, G.; Dunaevsky, O.; Matavel, A.; Lopes, C.M.; et al. Small Molecule Disruption of Gβγ Signaling Inhibits the Progression of Heart Failure. Circ. Res. 2010, 107, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.M.; Homan, K.T.; Chen, J.; Wu, E.K.; Hinkle, P.M.; Huang, Z.M.; Chuprun, J.K.; Song, J.; Gao, E.; Cheung, J.Y.; et al. Paroxetine Is a Direct Inhibitor of G Protein-Coupled Receptor Kinase 2 and Increases Myocardial Contractility. ACS Chem. Biol. 2012, 7, 1830–1839. [Google Scholar] [CrossRef]
- Rowlands, R.A.; Cato, M.C.; Waldschmidt, H.V.; Bouley, R.A.; Chen, Q.; Avramova, L.; Larsen, S.D.; Tesmer, J.J.G.; White, A.D. Structure-Based Design of Selective, Covalent G Protein-Coupled Receptor Kinase 5 Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1628–1634. [Google Scholar] [CrossRef]
- Sorriento, D.; Ciccarelli, M.; Santulli, G.; Campanile, A.; Altobelli, G.G.; Cimini, V.; Galasso, G.; Astone, D.; Piscione, F.; Pastore, L.; et al. The G-protein-coupled receptor kinase 5 inhibits NFκB transcriptional activity by inducing nuclear accumulation of IκBα. Proc. Natl. Acad. Sci. USA 2008, 105, 17818–17823. [Google Scholar] [CrossRef] [Green Version]
- Sorriento, D.; Santulli, G.; Fusco, A.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Intracardiac Injection of AdGRK5-NT Reduces Left Ventricular Hypertrophy by Inhibiting NF-κB–Dependent Hypertrophic Gene Expression. Hypertension 2010, 56, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorriento, D.; Santulli, G.; Ciccarelli, M.; Maione, A.S.; Illario, M.; Trimarco, B.; Iaccarino, G. The Amino-Terminal Domain of GRK5 Inhibits Cardiac Hypertrophy through the Regulation of Calcium-Calmodulin Dependent Transcription Factors. Int. J. Mol. Sci. 2018, 19, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyett, T.S.; Fraley, A.E.; LaBudde, E.; Patra, D.; Coleman, R.C.; Eguchi, A.; Glukhova, A.; Chen, Q.; Williams, R.M.; Koch, W.J.; et al. Perturbation of the interactions of calmodulin with GRK5 using a natural product chemical probe. Proc. Natl. Acad. Sci. USA 2019, 116, 15895–15900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Seo, H.W.; Ryu, J.Y.; Lim, C.J.; Yi, K.Y.; Oh, K.-S.; Lee, B.H. KR-39038, a Novel GRK5 Inhibitor, Attenuates Cardiac Hypertrophy and Improves Cardiac Function in Heart Failure. Biomol. Ther. 2020, 28, 482–489. [Google Scholar] [CrossRef]
- Homan, K.T.; Wu, E.; Cannavo, A.; Koch, W.J.; Tesmer, J.J.G. Identification and Characterization of Amlexanox as a G Protein-Coupled Receptor Kinase 5 Inhibitor. Molecules 2014, 19, 16937–16949. [Google Scholar] [CrossRef] [Green Version]
- Sommer, A.-K.; Falcenberg, M.; Ljepoja, B.; Fröhlich, T.; Arnold, G.J.; Wagner, E.; Roidl, A. Downregulation of GRK5 hampers the migration of breast cancer cells. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.-H.; Zhang, L.; Fanaroff, A.C.; Cai, X.; Sharma, K.C.; Brian, L.; Exum, S.T.; Shenoy, S.K.; Peppel, K.; Freedman, N.J. G Protein–Coupled Receptor Kinase-5 Attenuates Atherosclerosis by Regulating Receptor Tyrosine Kinases and 7-Transmembrane Receptors. Arter. Thromb. Vasc. Biol. 2012, 32, 308–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, L.; Shen, M.; Ma, L. GRK5 deficiency decreases diet-induced obesity and adipogenesis. Biochem. Biophys. Res. Commun. 2012, 421, 312–317. [Google Scholar] [CrossRef]
- Wang, L.; Shen, M.; Wang, F.; Ma, L. GRK5 ablation contributes to insulin resistance. Biochem. Biophys. Res. Commun. 2012, 429, 99–104. [Google Scholar] [CrossRef]
- Xia, Z.; Yang, T.; Wang, Z.; Dong, J.; Liang, C. GRK5 Intronic (CA)n Polymorphisms Associated with Type 2 Diabetes in Chinese Hainan Island. PLoS ONE 2014, 9, e90597. [Google Scholar] [CrossRef] [Green Version]
- Lutz, S.Z.; Falcenberg, M.; Machicao, F.; Peter, A.; Kächele, M.; Randrianarisoa, E.; Lehn-Stefan, A.; Wagner, R.; Machann, J.; Schick, F.; et al. Single Nucleotide Polymorphisms in the G-Protein Coupled Receptor Kinase 5 (GRK5) Gene are associated with Plasma LDL-Cholesterol Levels in Humans. Sci. Rep. 2018, 8, 7745. [Google Scholar] [CrossRef]
- Franklin, J.M.; Carrasco, G.A.; Qin, B.; Gao, B.; Yu, J.; Yuan, J.; Lou, Z. G-protein Receptor Kinase 5 Regulates the Cannabinoid Receptor 2-induced Up-regulation of Serotonin 2A Receptors. J. Biol. Chem. 2013, 288, 15712–15724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-C.; Chung, R.-H.; Kuo, H.-W.; Liu, T.-H.; Fang, C.-P.; Liu, S.C.; Liu, C.-C.; Tsou, H.-H.; Chen, A.C.H.; Liu, Y.-L. GRK5 Is Associated with the Regulation of Methadone Dosage in Heroin Dependence. Int. J. Neuropsychopharmacol. 2018, 21, 910–917. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Wang, J.; Hu, W.; Feng, Z. The Regulation of Ferroptosis by Tumor Suppressor p53 and its Pathway. Int. J. Mol. Sci. 2020, 21, 8387. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| GRK5 Inhibitors | Type of Study | Structure |
|---|---|---|
| Compound 5 | In vitro (Rowlands et al., 2019) |  |
| Compound 16d | In vitro (Rowlands et al., 2019) |  |
| Malbrancheamide | In vitro (Beyett et al., 2019) |  |
| KR-39038 | In vitro/In vivo (Lee et al., 2020) |  |
| Amlexanox | In vitro (Homan et al., 2014) |  |
| CCG-215022 | In vitro/In vivo (Pham et al., 2020) |  |
| Sunitinib | In vitro (Sommer et al., 2019) |  |
| Adenovirus (Ad) GRK5-NT | In vitro/In vivo (Sorriento et al., 2010) |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzano, F.; Rapacciuolo, A.; Ferrara, N.; Rengo, G.; Koch, W.J.; Cannavo, A. Targeting GRK5 for Treating Chronic Degenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1920. https://doi.org/10.3390/ijms22041920
Marzano F, Rapacciuolo A, Ferrara N, Rengo G, Koch WJ, Cannavo A. Targeting GRK5 for Treating Chronic Degenerative Diseases. International Journal of Molecular Sciences. 2021; 22(4):1920. https://doi.org/10.3390/ijms22041920
Chicago/Turabian StyleMarzano, Federica, Antonio Rapacciuolo, Nicola Ferrara, Giuseppe Rengo, Walter J. Koch, and Alessandro Cannavo. 2021. "Targeting GRK5 for Treating Chronic Degenerative Diseases" International Journal of Molecular Sciences 22, no. 4: 1920. https://doi.org/10.3390/ijms22041920
APA StyleMarzano, F., Rapacciuolo, A., Ferrara, N., Rengo, G., Koch, W. J., & Cannavo, A. (2021). Targeting GRK5 for Treating Chronic Degenerative Diseases. International Journal of Molecular Sciences, 22(4), 1920. https://doi.org/10.3390/ijms22041920