
Hepatic Senescence Accompanies the Development of NAFLD in Non-Aged Mice Independently of Obesity

 ,
,  , ,
, , 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
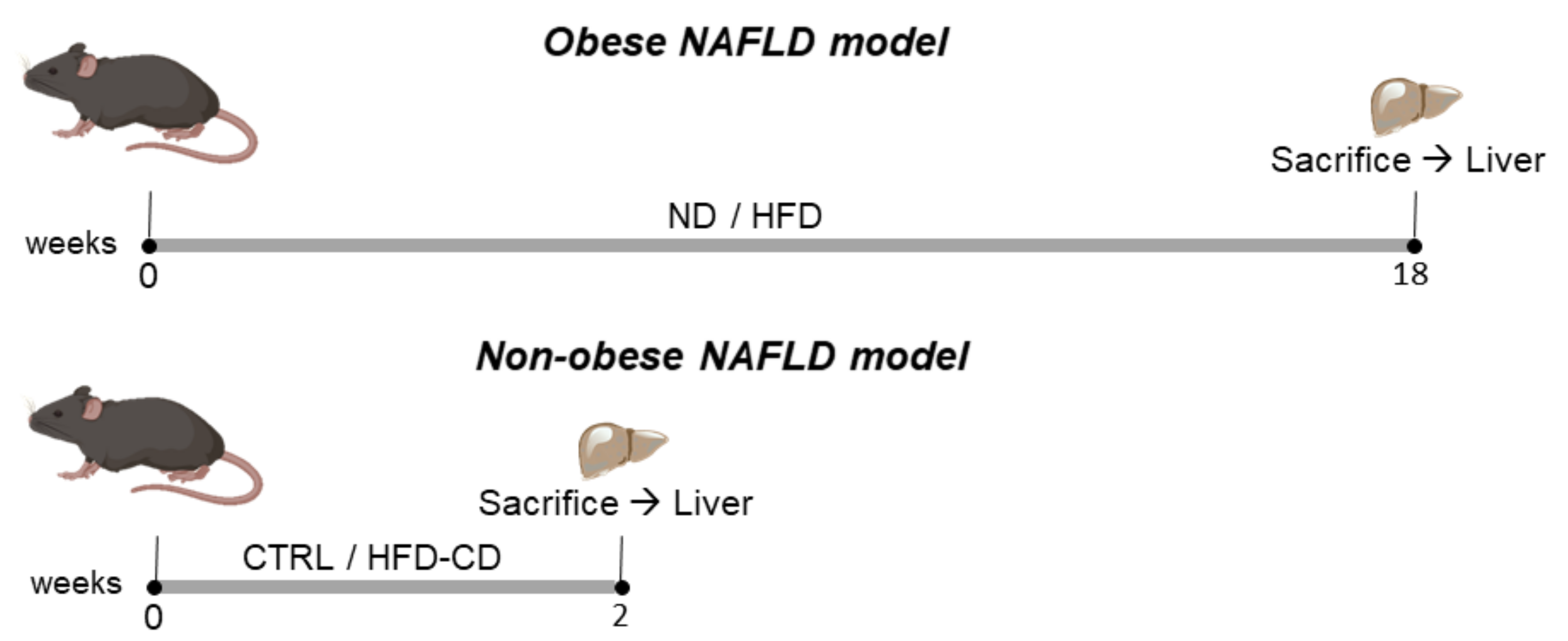
2.1. Animal Models of NAFLD in Non-Aged Mice
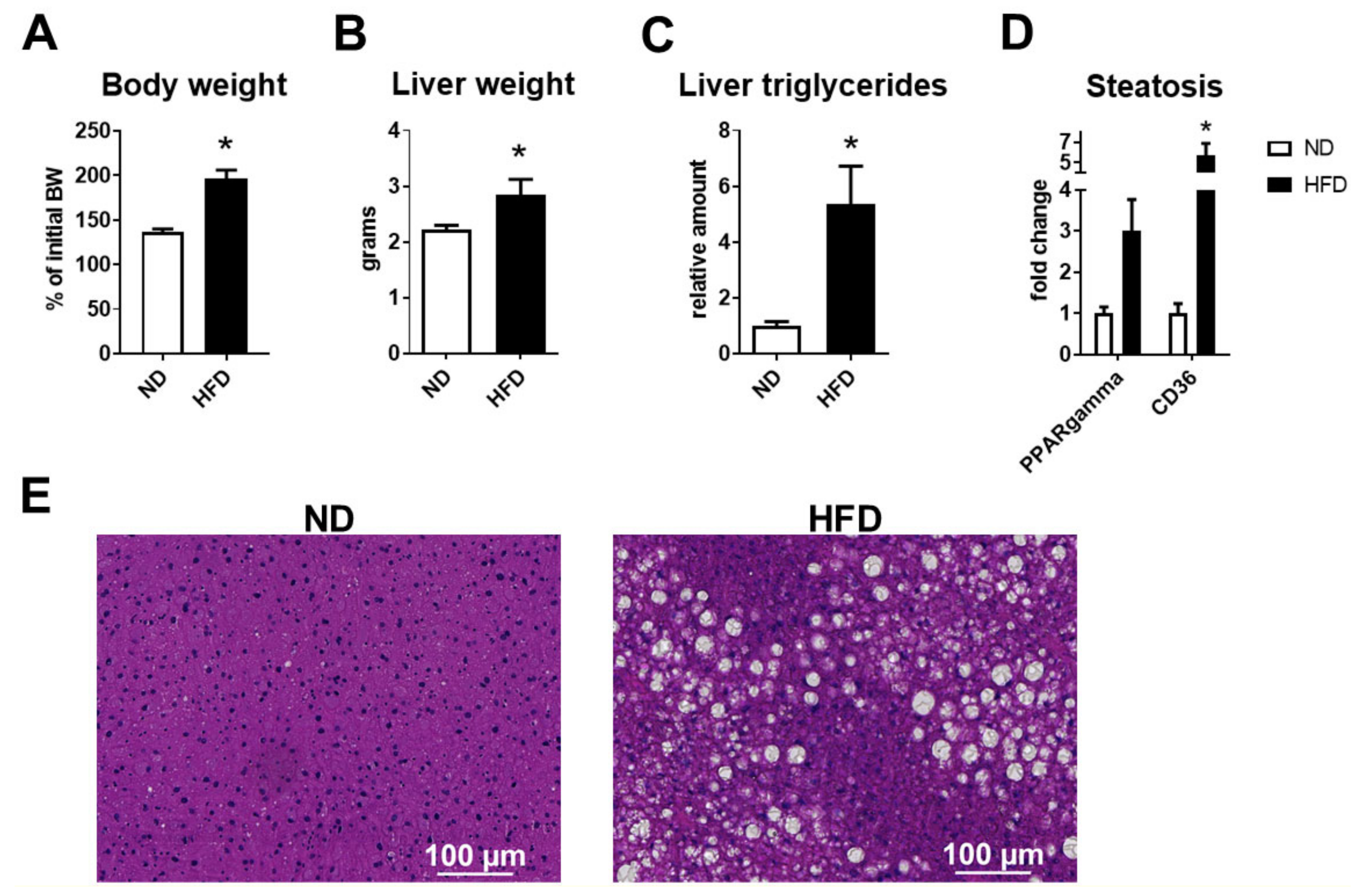
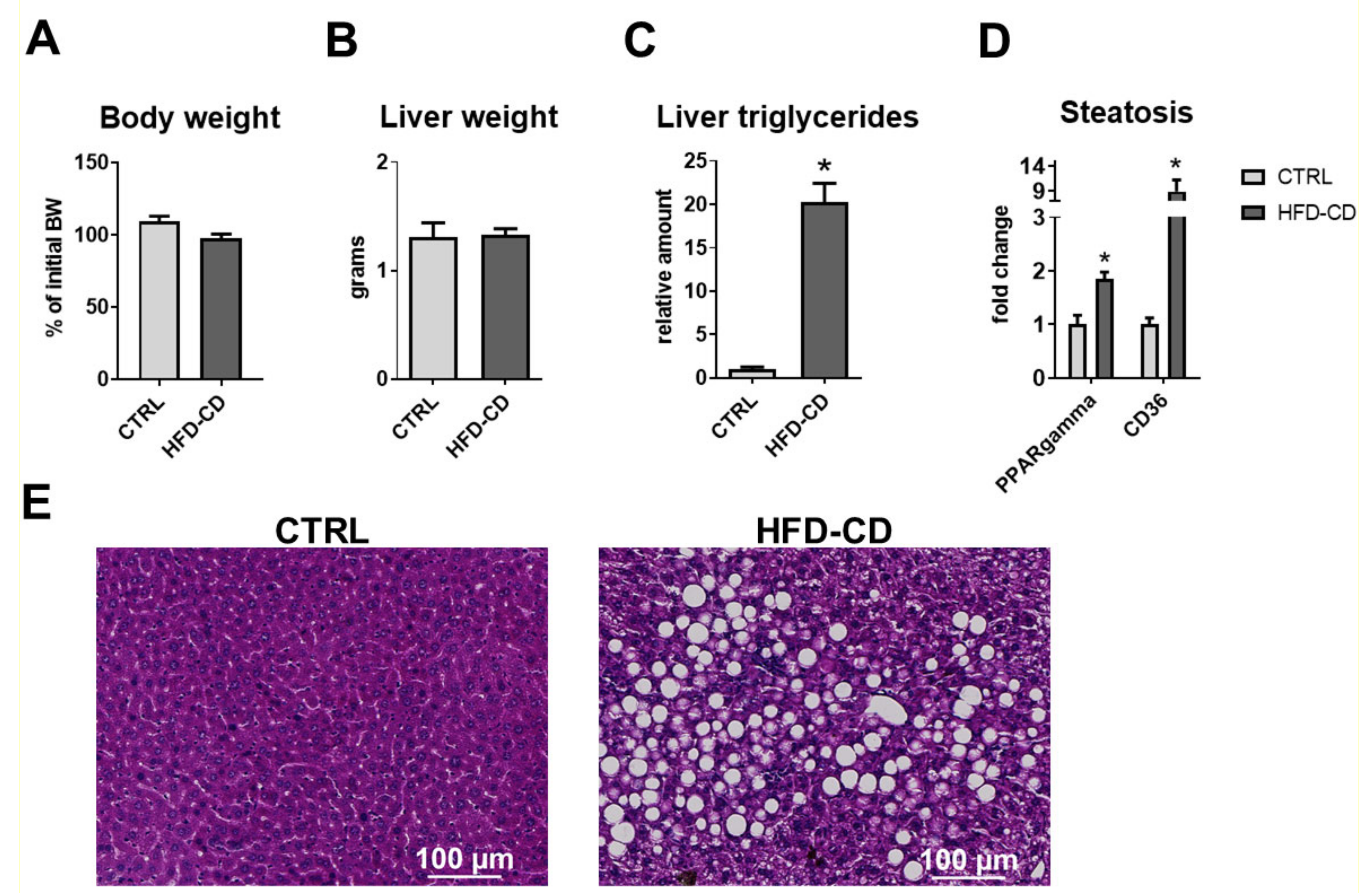
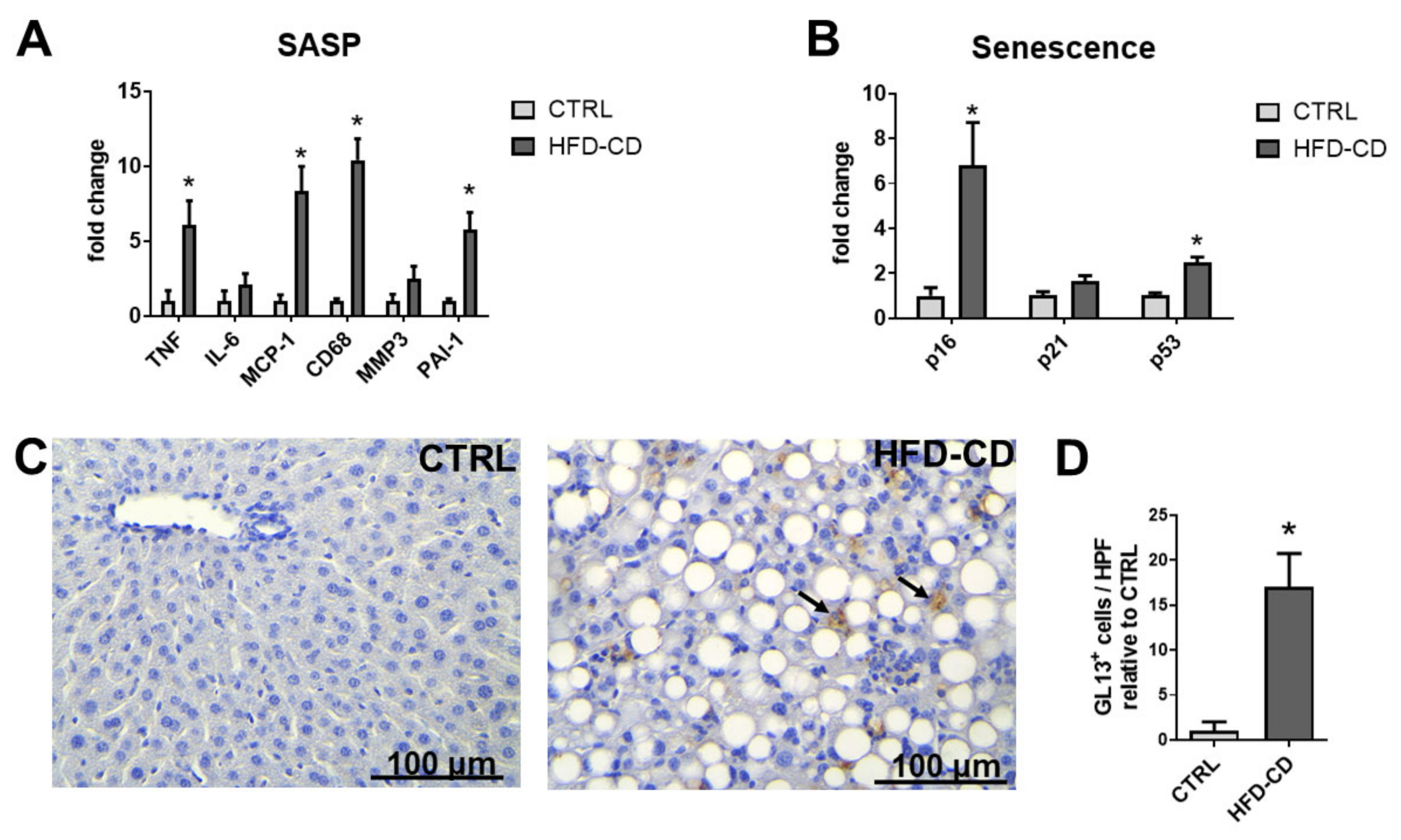
2.2. Senescence Accompanied Steatosis during NAFLD Development
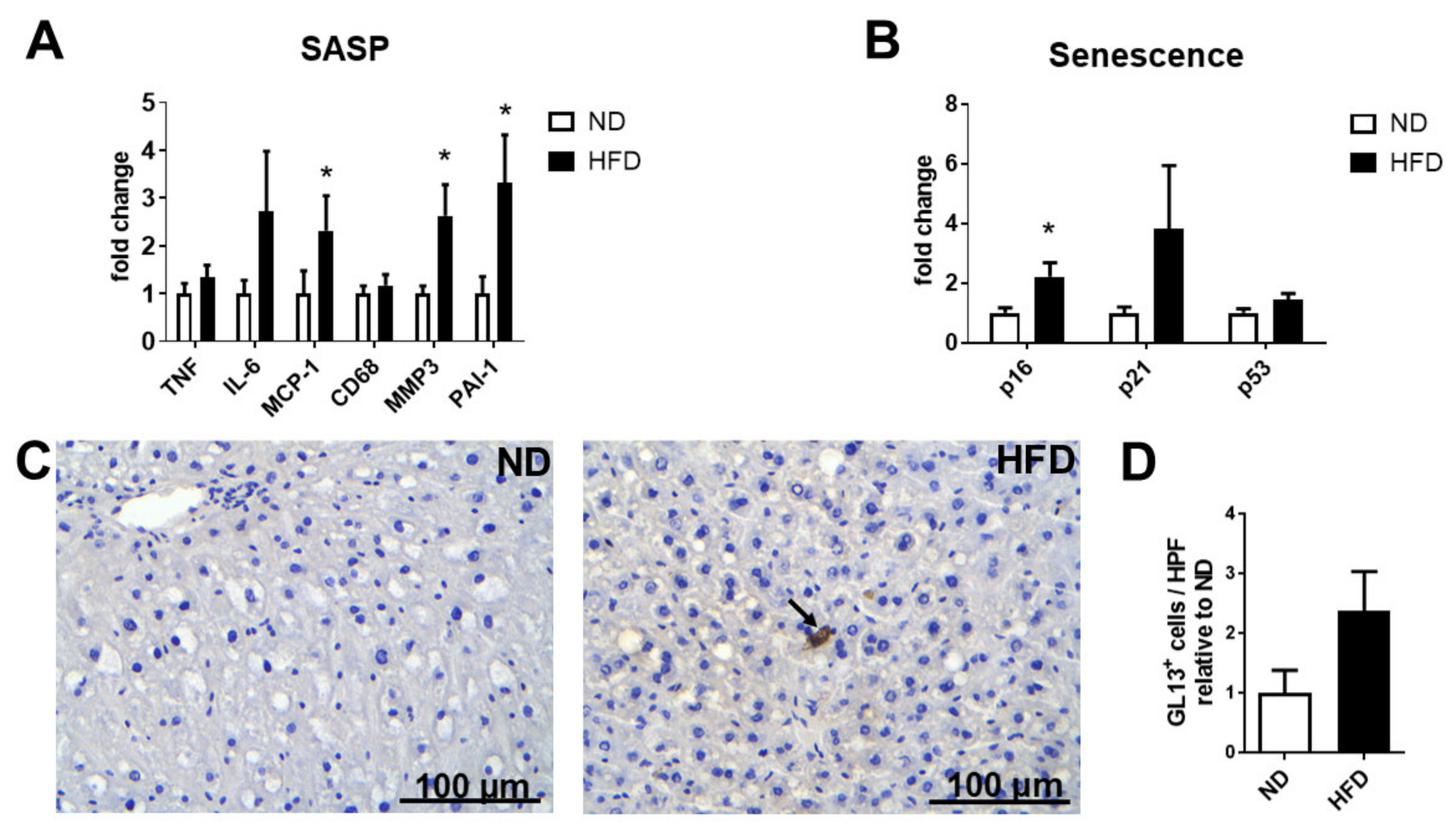
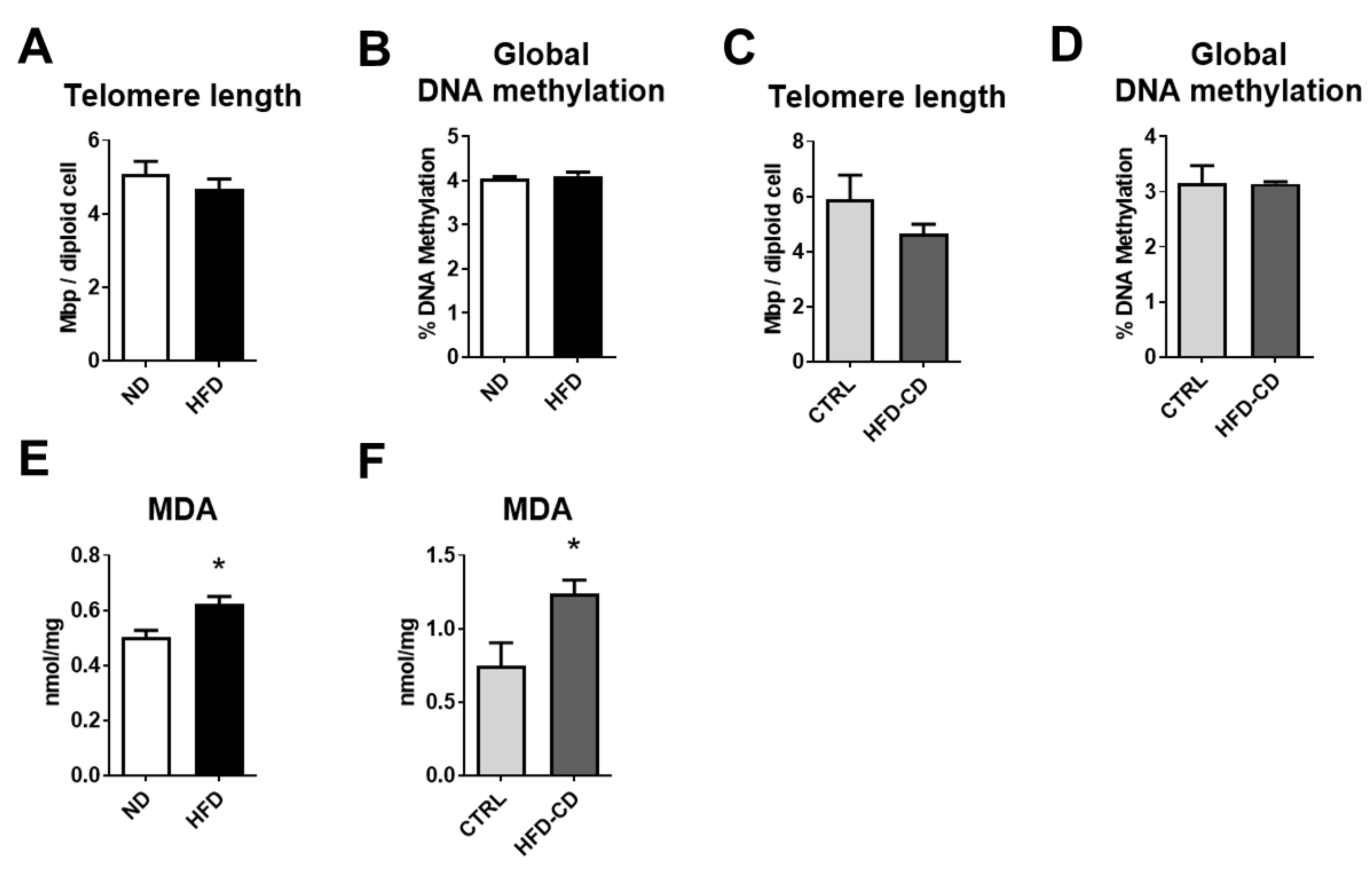
2.3. NAFLD of Non-Aged Mice Is Characterized by Stress-Induced Senescence
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Measurement of Liver Triglycerides
4.3. RNA Isolation and qPCR
4.4. Histology and Immunohistochemistry
4.5. Determination of Telomere Length and Global DNA Methylation
4.6. Measurement of Lipid Peroxidation in Liver
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; Van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Orjalo, A.V.; Desprez, P.-Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatheodoridi, A.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Palmer, A.K.; Tchkonia, T.; Lebrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Tessari, R.; Zenari, L.; Day, C.; Arcaro, G. Prevalence of Nonalcoholic Fatty Liver Disease and Its Association with Cardiovascular Disease Among Type 2 Diabetic Patients. Diabetes Care 2007, 30, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, N.C.; Salles, G.F.; Araujo, A.L.E.; Villela-Nogueira, C.A.; Cardoso, C.R.L. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 2009, 29, 113–119. [Google Scholar] [CrossRef]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Touros, A.; Kim, W.R. Nonalcoholic Fatty Liver Disease and Metabolic Syndrome. Clin. Liver Dis. 2018, 22, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Chatzigeorgiou, A.; Kandaraki, E.; Papavassiliou, A.G.; Koutsilieris, M. Peripheral targets in obesity treatment: A comprehensive update. Obes. Rev. 2014, 15, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2020, 1–16. [Google Scholar] [CrossRef]
- Katsarou, A.; Moustakas, I.; Pyrina, I.; Lembessis, P.; Koutsilieris, M.; Chatzigeorgiou, A. Metabolic inflammation as an instigator of fibrosis during non-alcoholic fatty liver disease. World J. Gastroenterol. 2020, 26, 1993–2011. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Khan, M.A.; Yoo, E.R.; Cholankeril, G.; Kim, D.; Ahmed, A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 8263–8276. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zou, B.; Yeo, Y.H.; Li, J.; Huang, D.Q.; Wu, Y.; Yang, H.; Liu, C.; Kam, L.Y.; Tan, X.X.E.; et al. Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 739–752. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, G.B.; Zhou, D.; Pan, Y.-X. High-fat diet modifies expression of hepatic cellular senescence gene p16(INK4a) through chromatin modifications in adult male rats. Genes Nutr. 2018, 13, 1–12. [Google Scholar] [CrossRef]
- Aravinthan, A.; Scarpini, C.; Tachtatzis, P.; Verma, S.; Penrhyn-Lowe, S.; Harvey, R.; Davies, S.E.; Allison, M.; Coleman, N.; Alexander, G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatol. 2013, 58, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Valanejad, L.; Cast, A.; Wright, M.; Garcia, J.M.; El-Serag, H.B.; Karns, R.; Timchenko, N.A. Elimination of Age-Associated Hepatic Steatosis and Correction of Aging Phenotype by Inhibition of cdk4-C/EBPalpha-p300 Axis. Cell Rep. 2018, 24, 1597–1609. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, T.; Moriguchi, M.; Katagishi, T.; Sekoguchi, S.; Nishikawa, T.; Takashima, H.; Kimura, H.; Minami, M.; Itoh, Y.; Kagawa, K.; et al. Premature telomere shortening and impaired regenerative response in hepatocytes of individuals with NAFLD. Liver Int. 2005, 26, 23–31. [Google Scholar] [CrossRef]
- Chrysavgis, L.; Ztriva, E.; Protopapas, A.; Tziomalos, K.; Cholongitas, E. Nonalcoholic fatty liver disease in lean subjects: Prognosis, outcomes and management. World J. Gastroenterol. 2020, 26, 6514–6528. [Google Scholar] [CrossRef]
- Kim, D.; Kim, W.R. Nonobese Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2017, 15, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Thangapandi, V.R.; Knittelfelder, O.; Brosch, M.; Patsenker, E.; Vvedenskaya, O.; Buch, S.; Hinz, S.; Hendricks, A.; Nati, M.; Herrmann, A.; et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Guarasci, F.; D’Aquila, P.; Mandalà, M.; Garasto, S.; Lattanzio, F.; Corsonello, A.; Passarino, G.; Bellizzi, D. Aging and nutrition induce tissue-specific changes on global DNA methylation status in rats. Mech. Ageing Dev. 2018, 174, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Ore, A.; Akinloye, O. Oxidative Stress and Antioxidant Biomarkers in Clinical and Experimental Models of Non-Alcoholic Fatty Liver Disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.G. The Mouse in Biomedical Research, 2nd ed.; Elsevier AP: Boston, MA, USA, 2007. [Google Scholar]
- Kondo, Y.; Hasegawa, G.; Okada, H.; Senmaru, T.; Fukui, M.; Nakamura, N.; Sawada, M.; Kitawaki, J.; Okanoue, T.; Kishimoto, Y.; et al. Lepr(db/db) Mice with senescence marker protein-30 knockout (Lepr(db/db)Smp30(Y/-)) exhibit increases in small dense-LDL and severe fatty liver despite being fed a standard diet. PLoS ONE 2013, 8, e65698. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; White, T.A.; Evans, G.; Tonne, J.M.; Verzosa, G.C.; Stout, M.B.; Mazula, D.L.; Palmer, A.K.; Baker, D.J.; Jensen, M.D.; et al. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes 2016, 65, 1606–1615. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhou, D.; Strakovsky, R.; Zhang, Y.; Pan, Y.-X. Hepatic cellular senescence pathway genes are induced through histone modifications in a diet-induced obese rat model. Am. J. Physiol. Liver Physiol. 2012, 302, G558–G564. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Lugari, S.; Nascimbeni, F. Telomere shortening: An innocent bystander at the crossroad of NASH with ageing and cardiometabolic risk? Liver Int. 2018, 38, 1730–1732. [Google Scholar] [CrossRef]
- Alves-Paiva, R.M.; Kajigaya, S.; Feng, X.; Chen, J.; Desierto, M.; Wong, S.; Townsley, D.M.; Donaires, F.S.; Bertola, A.; Gao, B.; et al. Telomerase enzyme deficiency promotes metabolic dysfunction in murine hepatocytes upon dietary stress. Liver Int. 2017, 38, 144–154. [Google Scholar] [CrossRef]
- Lai, Z.; Chen, J.; Ding, C.; Wong, K.; Chen, X.; Pu, L.; Huang, Q.; Chen, X.; Cheng, Z.; Liu, Y.; et al. Association of Hepatic Global DNA Methylation and Serum One-Carbon Metabolites with Histological Severity in Patients with NAFLD. Obesity 2019, 28, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-J.; Zhang, H.-W.; Zhou, J.-Y.; Liu, Y.; Yang, Y.; Chen, X.-L.; Zhu, C.-H.; Zheng, R.-D.; Ling, W.-H.; Zhu, H.-L. Betaine attenuates hepatic steatosis by reducing methylation of the MTTP promoter and elevating genomic methylation in mice fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Lyall, M.J.; Thomson, J.P.; Cartier, J.; Ottaviano, R.; Kendall, T.J.; Meehan, R.R.; Drake, A.J. Non-alcoholic fatty liver disease (NAFLD) is associated with dynamic changes in DNA hydroxymethylation. Epigenetics 2020, 15, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Henkel, J.; Kanuri, G.; Blank, D.; Püschel, G.P.; Bischoff, S.C.; Bergheim, I. Female Mice Are More Susceptible to Nonalcoholic Fatty Liver Disease: Sex-Specific Regulation of the Hepatic AMP-Activated Protein Kinase-Plasminogen Activator Inhibitor 1 Cascade, but Not the Hepatic Endotoxin Response. Mol. Med. 2012, 18, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Kim, S.H.; Kim, S.-N.; Kwon, H.-J.; Kim, J.-D.; Oh, J.Y.; Jung, Y.-S. Sex-specific metabolic interactions between liver and adipose tissue in MCD diet-induced non-alcoholic fatty liver disease. Oncotarget 2016, 7, 46959–46971. [Google Scholar] [CrossRef] [Green Version]
- Chmelař, J.; Chatzigeorgiou, A.; Chung, K.-J.; Prucnal, M.; Voehringer, D.; Roers, A.; Chavakis, T. No Role for Mast Cells in Obesity-Related Metabolic Dysregulation. Front. Immunol. 2016, 7, 524. [Google Scholar] [CrossRef] [Green Version]
- Chatzigeorgiou, A.; Chung, K.-J.; Garcia-Martin, R.; Alexaki, V.-I.; Ameln, A.K.-V.; Phieler, J.; Sprott, D.; Kanczkowski, W.; Tzanavari, T.; Bdeir, M.; et al. Dual role of B7 costimulation in obesity-related nonalcoholic steatohepatitis and metabolic dysregulation. Hepatology 2014, 60, 1196–1210. [Google Scholar] [CrossRef] [Green Version]
- García-Martín, R.; Alexaki, V.I.; Qin, N.; Rubin De Celis, M.F.; Economopoulou, M.; Ziogas, A.; Gercken, B.; Kotlabova, K.; Phieler, J.; Ehrhart-Bornstein, M.; et al. Adipocyte-Specific Hypoxia-Inducible Factor 2alpha Deficiency Exacerbates Obesity-Induced Brown Adipose Tissue Dysfunction and Metabolic Dysregulation. Mol. Cell. Biol. 2016, 36, 376–393. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kouadjo, K.; Nishida, Y.; Cadrin-Girard, J.F.; Yoshioka, M.; St-Amand, J. Housekeeping and tissue-specific genes in mouse tissues. BMC Genom. 2007, 8, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troullinaki, M.; Chen, L.; Witt, A.; Pyrina, I.; Phieler, J.; Kourtzelis, I.; Chmelar, J.; Sprott, D.; Gercken, B.; Koutsilieris, M.; et al. Robo4-mediated pancreatic endothelial integrity decreases inflammation and islet destruction in autoimmune diabetes. FASEB J. 2020, 34, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2016, 16, 192–197. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moustakas, I.I.; Katsarou, A.; Legaki, A.-I.; Pyrina, I.; Ntostoglou, K.; Papatheodoridi, A.-M.; Gercken, B.; Pateras, I.S.; Gorgoulis, V.G.; Koutsilieris, M.; et al. Hepatic Senescence Accompanies the Development of NAFLD in Non-Aged Mice Independently of Obesity. Int. J. Mol. Sci. 2021, 22, 3446. https://doi.org/10.3390/ijms22073446
Moustakas II, Katsarou A, Legaki A-I, Pyrina I, Ntostoglou K, Papatheodoridi A-M, Gercken B, Pateras IS, Gorgoulis VG, Koutsilieris M, et al. Hepatic Senescence Accompanies the Development of NAFLD in Non-Aged Mice Independently of Obesity. International Journal of Molecular Sciences. 2021; 22(7):3446. https://doi.org/10.3390/ijms22073446
Chicago/Turabian StyleMoustakas, Ioannis I., Angeliki Katsarou, Aigli-Ioanna Legaki, Iryna Pyrina, Konstantinos Ntostoglou, Alkistis-Maria Papatheodoridi, Bettina Gercken, Ioannis S. Pateras, Vassilis G. Gorgoulis, Michael Koutsilieris, and et al. 2021. "Hepatic Senescence Accompanies the Development of NAFLD in Non-Aged Mice Independently of Obesity" International Journal of Molecular Sciences 22, no. 7: 3446. https://doi.org/10.3390/ijms22073446
APA StyleMoustakas, I. I., Katsarou, A., Legaki, A. -I., Pyrina, I., Ntostoglou, K., Papatheodoridi, A. -M., Gercken, B., Pateras, I. S., Gorgoulis, V. G., Koutsilieris, M., Chavakis, T., & Chatzigeorgiou, A. (2021). Hepatic Senescence Accompanies the Development of NAFLD in Non-Aged Mice Independently of Obesity. International Journal of Molecular Sciences, 22(7), 3446. https://doi.org/10.3390/ijms22073446