
ABCG2 Is Overexpressed on Red Blood Cells in Ph-Negative Myeloproliferative Neoplasms and Potentiates Ruxolitinib-Induced Apoptosis

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
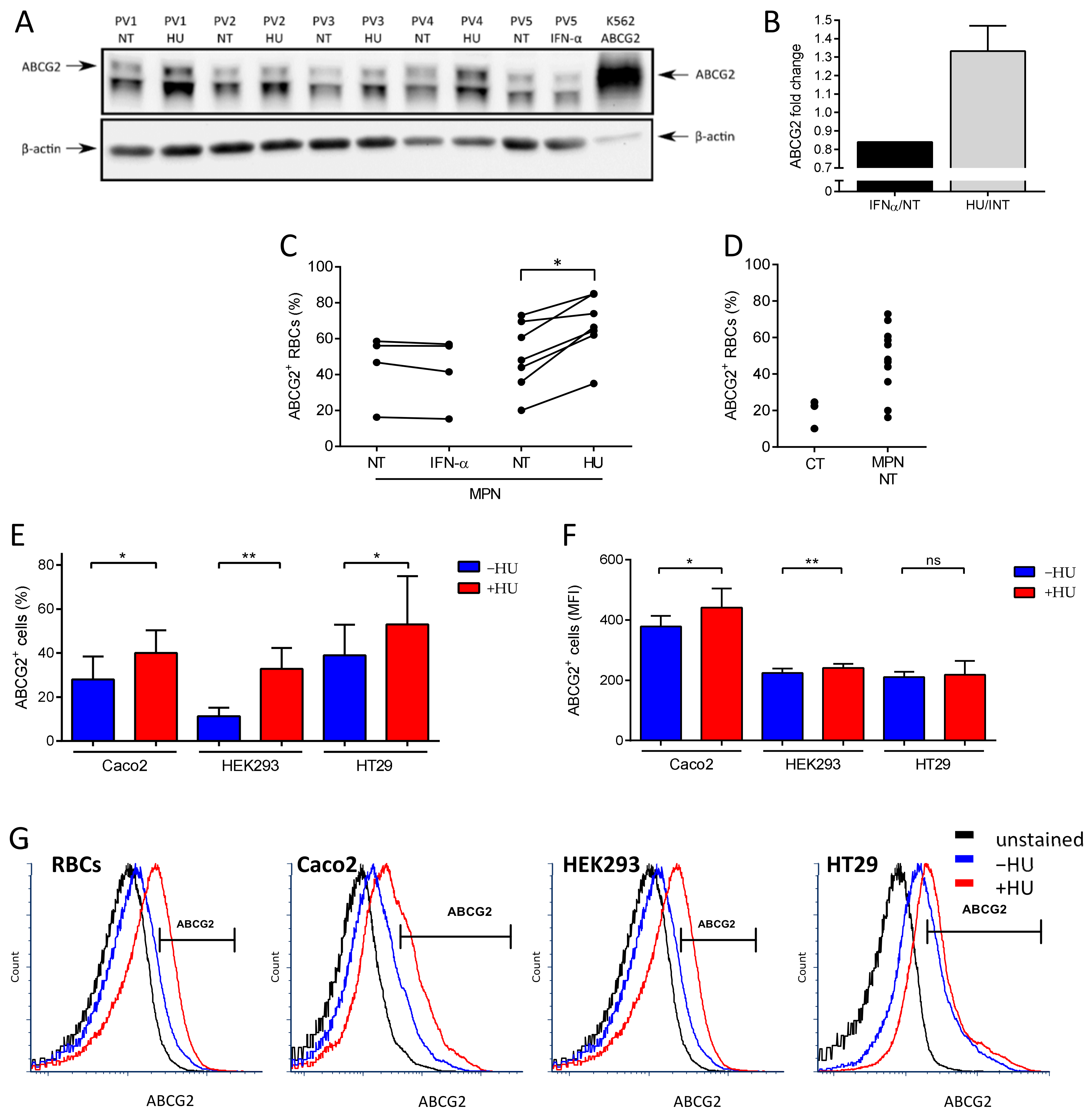
2.1. HU Treatment Increases ABCG2 Levels on the RBC Surface and on the Surface of ABCG2-Expressing Cell Lines
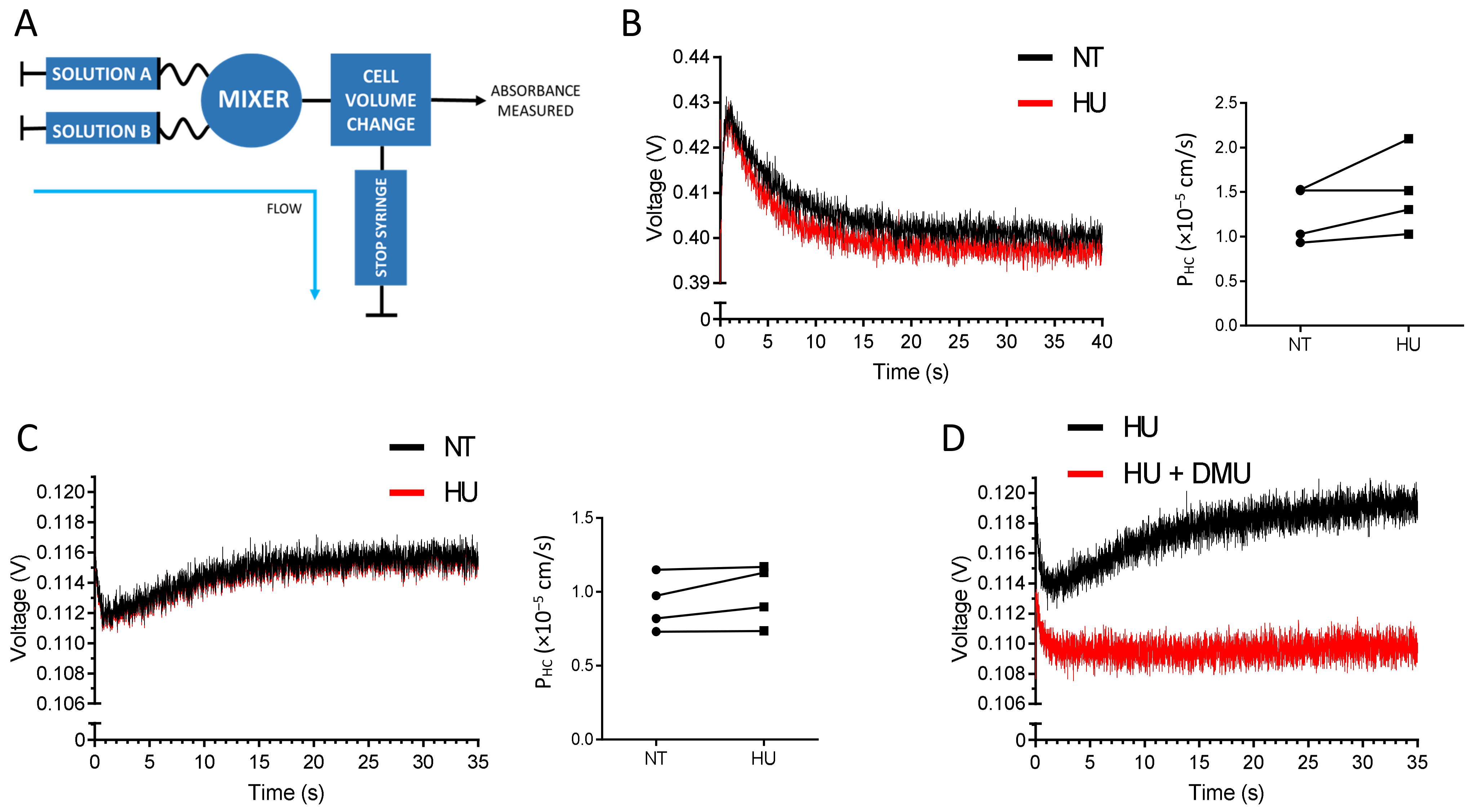
2.2. HU Is not a Substrate of ABCG2
2.3. ABCG2 Inhibition Reduces Ruxolitinib-Induced PS Exposure on MPN RBCs
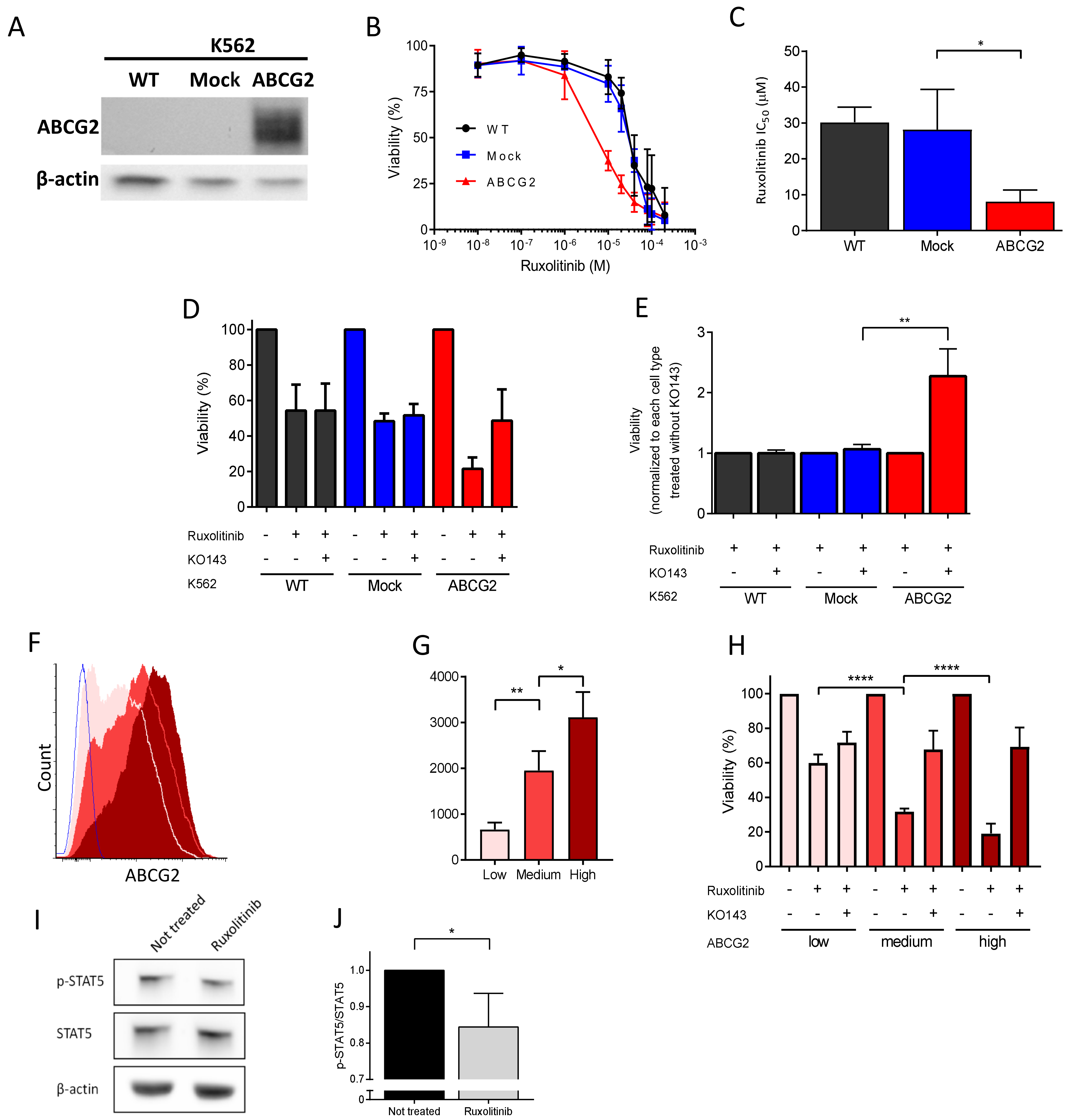
2.4. ABCG2 Inhibition Reduces Ruxolitinib-Induced Apoptosis in K562 Cells
2.5. ABCG2 Inhibition Rescues Ruxolitinib-Induced Apoptosis during Human In Vitro Terminal Erythroid Differentiation
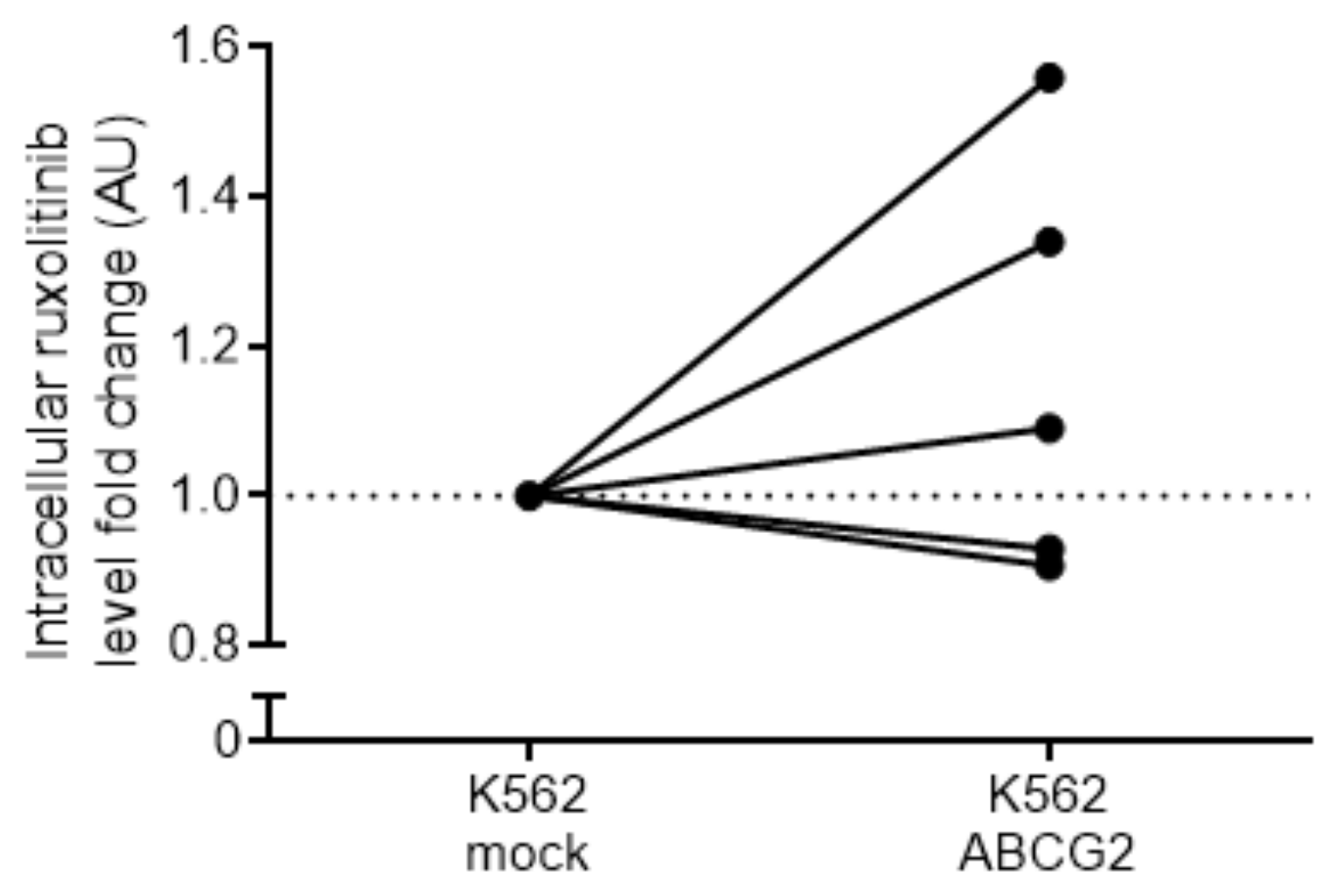
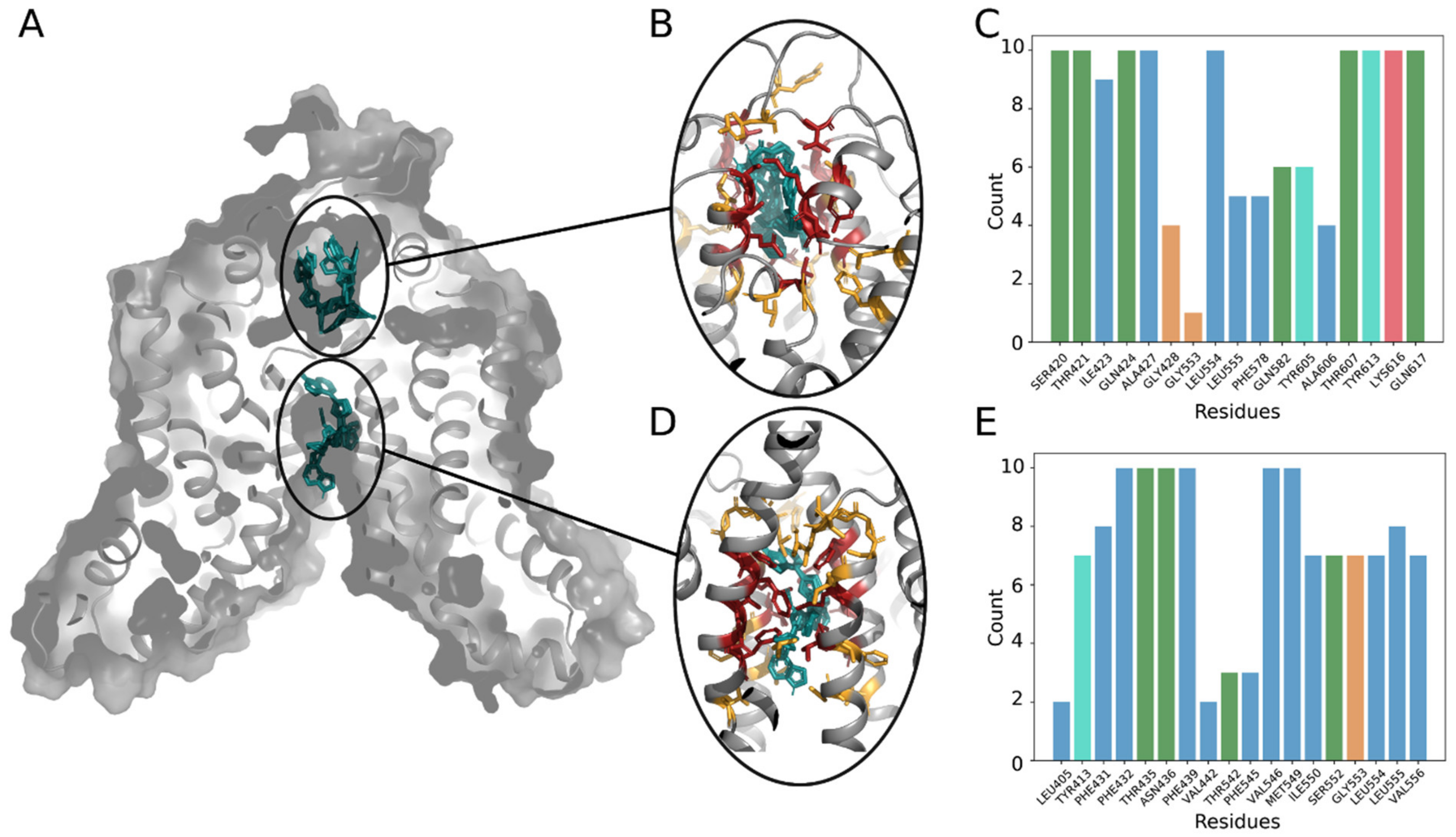
2.6. Analysis of Ruxolitinib Transport Ex Vivo and In Silico
3. Discussion
4. Materials and Methods
4.1. Blood Samples
4.2. Cell Lines
4.3. In Vitro Erythroid Differentiation
4.4. Western Blots
4.5. Stopped-Flow Assays
4.6. Flow Cytometry
4.7. Cytotoxicity and Apoptosis Assays
4.8. Ruxolitinib Measurements by LC–MS
4.9. Molecular Dynamics of ABCG2 in Membrane Environment
4.10. Trajectory Analysis
4.11. Ruxolitinib Docking Procedure
4.12. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nangalia, J.; Grinfeld, J.; Green, A.R. Pathogenesis of Myeloproliferative Disorders. Annu. Rev. Pathol. 2016, 11, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.M.; Zhang, L.; Giacomini, K.M. The International Transporter Consortium: A collaborative group of scientists from academia, industry, and the FDA. Clin. Pharmacol. Ther. 2010, 87, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Kosztyu, P.; Bukvova, R.; Dolezel, P.; Mlejnek, P. Resistance to daunorubicin, imatinib, or nilotinib depends on expression levels of ABCB1 and ABCG2 in human leukemia cells. Chem. Biol. Interact. 2014, 219, 203–210. [Google Scholar] [CrossRef]
- Ebert, C.; Perner, F.; Wolleschak, D.; Schnoder, T.M.; Fischer, T.; Heidel, F.H. Expression and function of ABC-transporter protein ABCB1 correlates with inhibitory capacity of Ruxolitinib in vitro and in vivo. Haematologica 2016, 101, e81–e85. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Zong, Y.; Ney, P.A.; Nair, G.; Stewart, C.F.; Sorrentino, B.P. Increased expression of the Abcg2 transporter during erythroid maturation plays a role in decreasing cellular protoporphyrin IX levels. Blood 2005, 105, 2571–2576. [Google Scholar] [CrossRef]
- An, X.; Schulz, V.P.; Li, J.; Wu, K.; Liu, J.; Xue, F.; Hu, J.; Mohandas, N.; Gallagher, P.G. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 2014, 123, 3466–3477. [Google Scholar] [CrossRef] [Green Version]
- Brusson, M.; Cochet, S.; Leduc, M.; Guillonneau, F.; Mayeux, P.; Peyrard, T.; Chomienne, C.; Le Van Kim, C.; Cassinat, B.; Kiladjian, J.-J.; et al. Enhanced calreticulin expression in red cells of polycythemia vera patients harboring the JAK2V617F mutation. Haematologica 2017, 102, e241–e244. [Google Scholar] [CrossRef] [Green Version]
- Brusson, M.; De Grandis, M.; Cochet, S.; Bigot, S.; Marin, M.; Leduc, M.; Guillonneau, F.; Mayeux, P.; Peyrard, T.; Chomienne, C.; et al. Impact of hydroxycarbamide and interferon-alpha on red cell adhesion and membrane protein expression in polycythemia vera. Haematologica 2018, 103, 972–981. [Google Scholar] [CrossRef]
- Edwards-Moulds, J.; Kasschau, M.R. The effect of 2 molar urea on Jk (a-b-) red cells. Vox Sang. 1988, 55, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, O.; Macey, R.I.; Edwards-Moulds, J.; Gargus, J.J.; Gunn, R.B. Urea transport deficiency in Jk(a-b-) erythrocytes. Am. J. Physiol. 1991, 260, C778–C783. [Google Scholar] [CrossRef] [PubMed]
- Olives, B.; Mattei, M.G.; Huet, M.; Neau, P.; Martial, S.; Cartron, J.P.; Bailly, P. Kidd blood group and urea transport function of human erythrocytes are carried by the same protein. J. Biol. Chem. 1995, 270, 15607–15610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azouzi, S.; Gueroult, M.; Ripoche, P.; Genetet, S.; Colin Aronovicz, Y.; Le Van Kim, C.; Etchebest, C.; Mouro-Chanteloup, I. Energetic and molecular water permeation mechanisms of the human red blood cell urea transporter B. PLoS ONE 2013, 8, e82338. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.L.; Franke, R.M.; Sparreboom, A.; Ware, R.E. Transcellular movement of hydroxyurea is mediated by specific solute carrier transporters. Exp. Hematol. 2011, 39, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Briglia, M.; Fazio, A.; Faggio, C.; Laufer, S.; Alzoubi, K.; Lang, F. Triggering of Suicidal Erythrocyte Death by Ruxolitinib. Cell Physiol. Biochem. 2015, 37, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Cardama, A.; Vaddi, K.; Liu, P.; Manshouri, T.; Li, J.; Scherle, P.A.; Caulder, E.; Wen, X.; Li, Y.; Waeltz, P.; et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010, 115, 3109–3117. [Google Scholar] [CrossRef]
- Hu, J.; Liu, J.; Xue, F.; Halverson, G.; Reid, M.; Guo, A.; Chen, L.; Raza, A.; Galili, N.; Jaffray, J.; et al. Isolation and functional characterization of human erythroblasts at distinct stages: Implications for understanding of normal and disordered erythropoiesis in vivo. Blood 2013, 121, 3246–3253. [Google Scholar] [CrossRef] [Green Version]
- Khunweeraphong, N.; Szollosi, D.; Stockner, T.; Kuchler, K. The ABCG2 multidrug transporter is a pump gated by a valve and an extracellular lid. Nat. Commun. 2019, 10, 5433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.J.; Bonito, C.A.; Cordeiro, M.; Ferreira, M.U.; Dos Santos, D. Structure-function relationships in ABCG2: Insights from molecular dynamics simulations and molecular docking studies. Sci. Rep. 2017, 7, 15534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.M.; Dalvi, P.; Lu, X.; Yang, M.; Riddick, D.S.; Matthews, J.; Clevenger, C.V.; Ross, D.D.; Harper, P.A.; Ito, S. Induction of multidrug resistance transporter ABCG2 by prolactin in human breast cancer cells. Mol. Pharmacol. 2013, 83, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillinder, K.R.; Tuckey, H.; Bell, C.C.; Magor, G.W.; Huang, S.; Ilsley, M.D.; Perkins, A.C. Direct targets of pSTAT5 signalling in erythropoiesis. PLoS ONE 2017, 12, e0180922. [Google Scholar] [CrossRef]
- Cokic, V.P.; Smith, R.D.; Beleslin-Cokic, B.B.; Njoroge, J.M.; Miller, J.L.; Gladwin, M.T.; Schechter, A.N. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J. Clin. Investig. 2003, 111, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Bartolucci, P.; Chaar, V.; Picot, J.; Bachir, D.; Habibi, A.; Fauroux, C.; Galacteros, F.; Colin, Y.; Le Van Kim, C.; El Nemer, W. Decreased sickle red blood cell adhesion to laminin by hydroxyurea is associated with inhibition of Lu/BCAM protein phosphorylation. Blood 2010, 116, 2152–2159. [Google Scholar] [CrossRef] [Green Version]
- Odievre, M.H.; Bony, V.; Benkerrou, M.; Lapoumeroulie, C.; Alberti, C.; Ducrocq, R.; Jacqz-Aigrain, E.; Elion, J.; Cartron, J.P. Modulation of erythroid adhesion receptor expression by hydroxyurea in children with sickle cell disease. Haematologica 2008, 93, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Almeida, C.B.; Scheiermann, C.; Jang, J.E.; Prophete, C.; Costa, F.F.; Conran, N.; Frenette, P.S. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood 2012, 120, 2879–2888. [Google Scholar] [CrossRef]
- Chaar, V.; Laurance, S.; Lapoumeroulie, C.; Cochet, S.; De Grandis, M.; Colin, Y.; Elion, J.; Le Van Kim, C.; El Nemer, W. Hydroxycarbamide decreases sickle reticulocyte adhesion to resting endothelium by inhibiting endothelial lutheran/basal cell adhesion molecule (Lu/BCAM) through phosphodiesterase 4A activation. J. Biol. Chem. 2014, 289, 11512–11521. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Choi, Y.; Burla, B.; Kim, Y.Y.; Jeon, B.; Maeshima, M.; Yoo, J.Y.; Martinoia, E.; Lee, Y. The ABC transporter AtABCB14 is a malate importer and modulates stomatal response to CO2. Nat. Cell Biol. 2008, 10, 1217–1223. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Hwang, J.U.; Lee, M.; Kim, Y.Y.; Assmann, S.M.; Martinoia, E.; Lee, Y. PDR-type ABC transporter mediates cellular uptake of the phytohormone abscisic acid. Proc. Natl. Acad. Sci. USA 2010, 107, 2355–2360. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, D.L.; Lynch, J.; Wang, Y.; Fukuda, Y.; Nachagari, D.; Du, G.; Sun, D.; Fan, Y.; Tsurkan, L.; Potter, P.M.; et al. ATP-dependent mitochondrial porphyrin importer ABCB6 protects against phenylhydrazine toxicity. J. Biol. Chem. 2012, 287, 12679–12690. [Google Scholar] [CrossRef] [Green Version]
- Cserepes, M.; Turk, D.; Toth, S.; Pape, V.F.S.; Gaal, A.; Gera, M.; Szabo, J.E.; Kucsma, N.; Varady, G.; Vertessy, B.G.; et al. Unshielding Multidrug Resistant Cancer through Selective Iron Depletion of P-Glycoprotein-Expressing Cells. Cancer Res. 2020, 80, 663–674. [Google Scholar] [CrossRef]
- Pugliese, N.; Giordano, C.; Nappi, D.; Luciano, L.; Cerchione, C.; Annunziata, M.; Casale, B.; Crisa, E.; Villa, M.R.; Pezzullo, L.; et al. Adding hydroxyurea in combination with ruxolitinib improves clinical responses in hyperproliferative forms of myelofibrosis. Cancer Med. 2019, 8, 2802–2809. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Verstovsek, S.; Guglielmelli, P.; Griesshammer, M.; Burn, T.C.; Naim, A.; Paranagama, D.; Marker, M.; Gadbaw, B.; Kiladjian, J.J. Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann. Hematol. 2017, 96, 1113–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Wang, Y.; Grimm, S. ATP-dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab. Dispos. 2006, 34, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, B.N.; He, Y.J.; Fan, L.; Li, Q.; Liu, Z.Q.; Wang, A.; Liu, Y.L.; Tan, Z.R.; Fen, J.; et al. Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin. Chim. Acta 2006, 373, 99–103. [Google Scholar] [CrossRef]
- Birmingham, B.K.; Bujac, S.R.; Elsby, R.; Azumaya, C.T.; Wei, C.; Chen, Y.; Mosqueda-Garcia, R.; Ambrose, H.J. Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: A class effect? Eur. J. Clin. Pharmacol. 2015, 71, 341–355. [Google Scholar] [CrossRef]
- Mirosevic Skvrce, N.; Macolic Sarinic, V.; Simic, I.; Ganoci, L.; Muacevic Katanec, D.; Bozina, N. ABCG2 gene polymorphisms as risk factors for atorvastatin adverse reactions: A case-control study. Pharmacogenomics 2015, 16, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Wang, G.; Li, T.; Xu, B.; Pei, Q.; Peng, Y.; Sun, H.; Cheng, L.; Zeng, Y.; Yang, G.; et al. Marked Alteration of Rosuvastatin Pharmacokinetics in Healthy Chinese with ABCG2 34G>A and 421C>A Homozygote or Compound Heterozygote. J. Pharmacol. Exp. Ther. 2015, 354, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Lelkens, C.C.; Noorman, F.; Koning, J.G.; Truijens-de Lange, R.; Stekkinger, P.S.; Bakker, J.C.; Lagerberg, J.W.; Brand, A.; Verhoeven, A.J. Stability after thawing of RBCs frozen with the high- and low-glycerol method. Transfusion 2003, 43, 157–164. [Google Scholar] [CrossRef]
- Saison, C.; Helias, V.; Ballif, B.A.; Peyrard, T.; Puy, H.; Miyazaki, T.; Perrot, S.; Vayssier-Taussat, M.; Waldner, M.; Le Pennec, P.Y.; et al. Null alleles of ABCG2 encoding the breast cancer resistance protein define the new blood group system Junior. Nat. Genet. 2012, 44, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; van Loevezijn, A.; Lakhai, J.M.; van der Valk, M.; van Tellingen, O.; Reid, G.; Schellens, J.H.; Koomen, G.J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar] [PubMed]
- Matsson, P.; Pedersen, J.M.; Norinder, U.; Bergstrom, C.A.; Artursson, P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm. Res. 2009, 26, 1816–1831. [Google Scholar] [CrossRef]
- Mackay, G.M.; Zheng, L.; van den Broek, N.J.; Gottlieb, E. Analysis of Cell Metabolism Using LC-MS and Isotope Tracers. Methods Enzymol. 2015, 561, 171–196. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Davila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tubiana, T.; Carvaillo, J.C.; Boulard, Y.; Bressanelli, S. TTClust: A Versatile Molecular Simulation Trajectory Clustering Program with Graphical Summaries. J. Chem. Inf. Model. 2018, 58, 2178–2182. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buks, R.; Brusson, M.; Cochet, S.; Galochkina, T.; Cassinat, B.; Nemazanyy, I.; Peyrard, T.; Kiladjian, J.-J.; de Brevern, A.G.; Azouzi, S.; et al. ABCG2 Is Overexpressed on Red Blood Cells in Ph-Negative Myeloproliferative Neoplasms and Potentiates Ruxolitinib-Induced Apoptosis. Int. J. Mol. Sci. 2021, 22, 3530. https://doi.org/10.3390/ijms22073530
Buks R, Brusson M, Cochet S, Galochkina T, Cassinat B, Nemazanyy I, Peyrard T, Kiladjian J-J, de Brevern AG, Azouzi S, et al. ABCG2 Is Overexpressed on Red Blood Cells in Ph-Negative Myeloproliferative Neoplasms and Potentiates Ruxolitinib-Induced Apoptosis. International Journal of Molecular Sciences. 2021; 22(7):3530. https://doi.org/10.3390/ijms22073530
Chicago/Turabian StyleBuks, Ralfs, Mégane Brusson, Sylvie Cochet, Tatiana Galochkina, Bruno Cassinat, Ivan Nemazanyy, Thierry Peyrard, Jean-Jacques Kiladjian, Alexandre G. de Brevern, Slim Azouzi, and et al. 2021. "ABCG2 Is Overexpressed on Red Blood Cells in Ph-Negative Myeloproliferative Neoplasms and Potentiates Ruxolitinib-Induced Apoptosis" International Journal of Molecular Sciences 22, no. 7: 3530. https://doi.org/10.3390/ijms22073530
APA StyleBuks, R., Brusson, M., Cochet, S., Galochkina, T., Cassinat, B., Nemazanyy, I., Peyrard, T., Kiladjian, J. -J., de Brevern, A. G., Azouzi, S., & El Nemer, W. (2021). ABCG2 Is Overexpressed on Red Blood Cells in Ph-Negative Myeloproliferative Neoplasms and Potentiates Ruxolitinib-Induced Apoptosis. International Journal of Molecular Sciences, 22(7), 3530. https://doi.org/10.3390/ijms22073530