
The Effect of Oxidized Dopamine on the Structure and Molecular Chaperone Function of the Small Heat-Shock Proteins, αB-Crystallin and Hsp27



 and
and
Abstract
:1. Introduction
2. Results
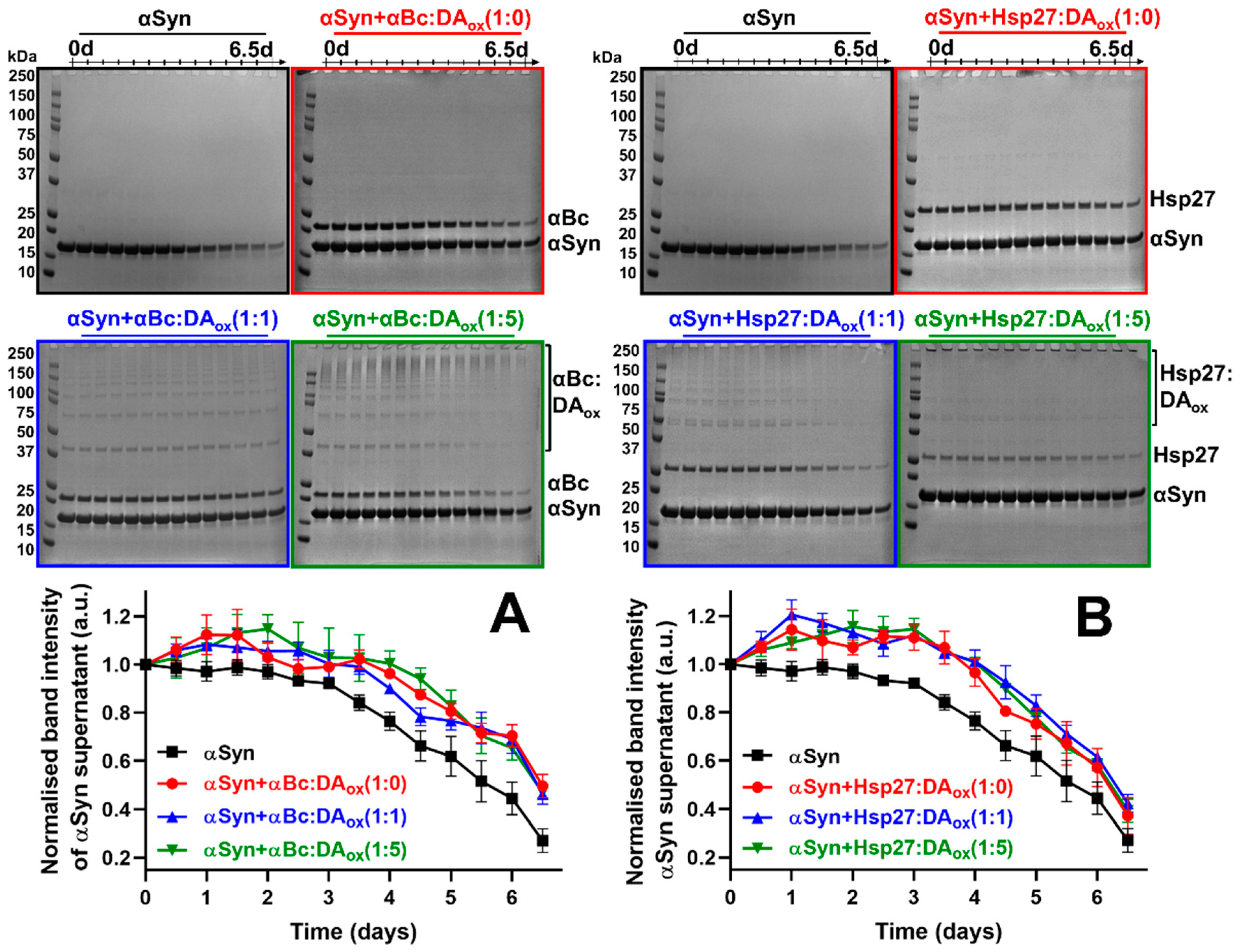
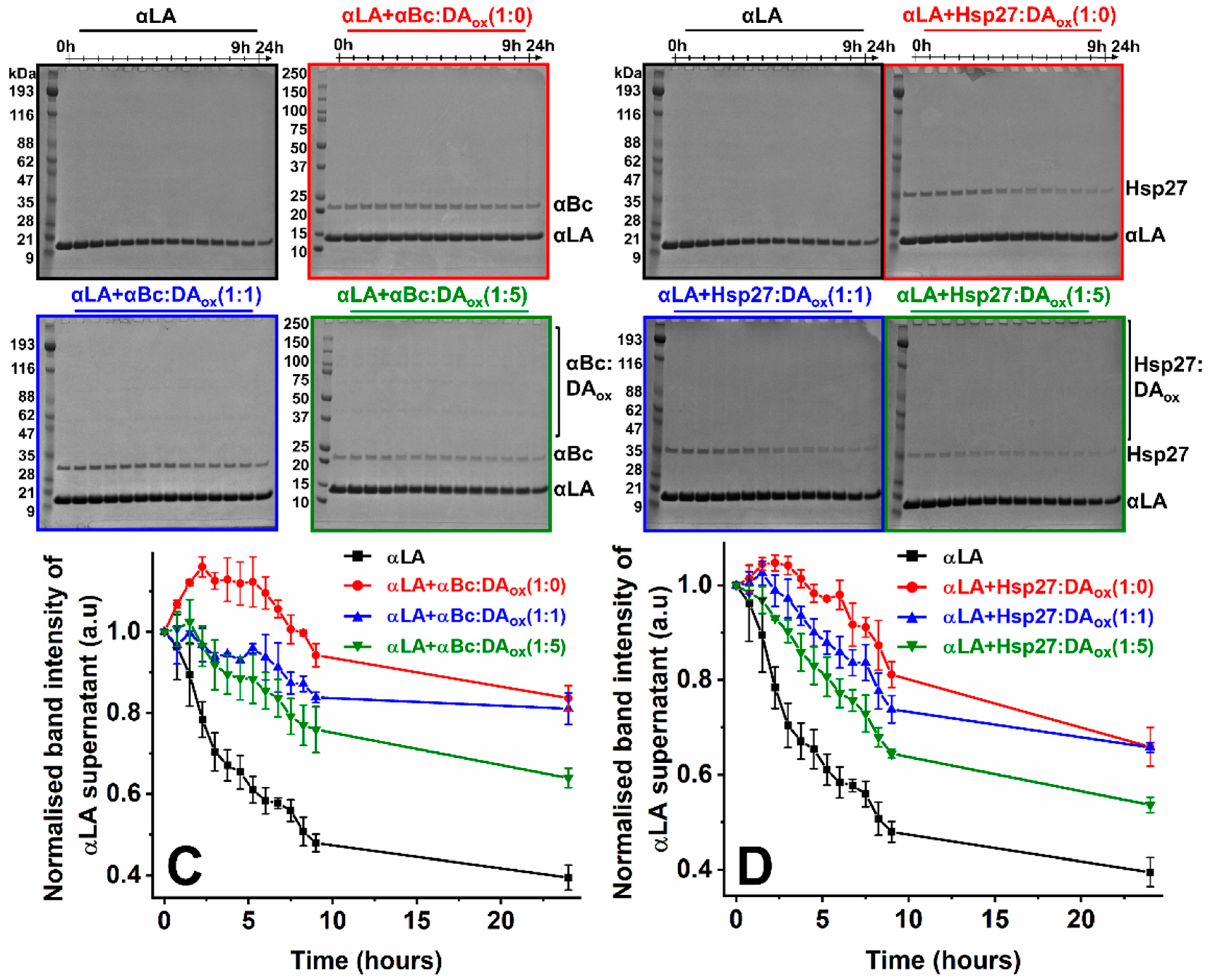
2.1. Oxidized Dopamine-Modified sHsps Prevent Amorphous and Amyloid Fibrillar Protein Aggregation
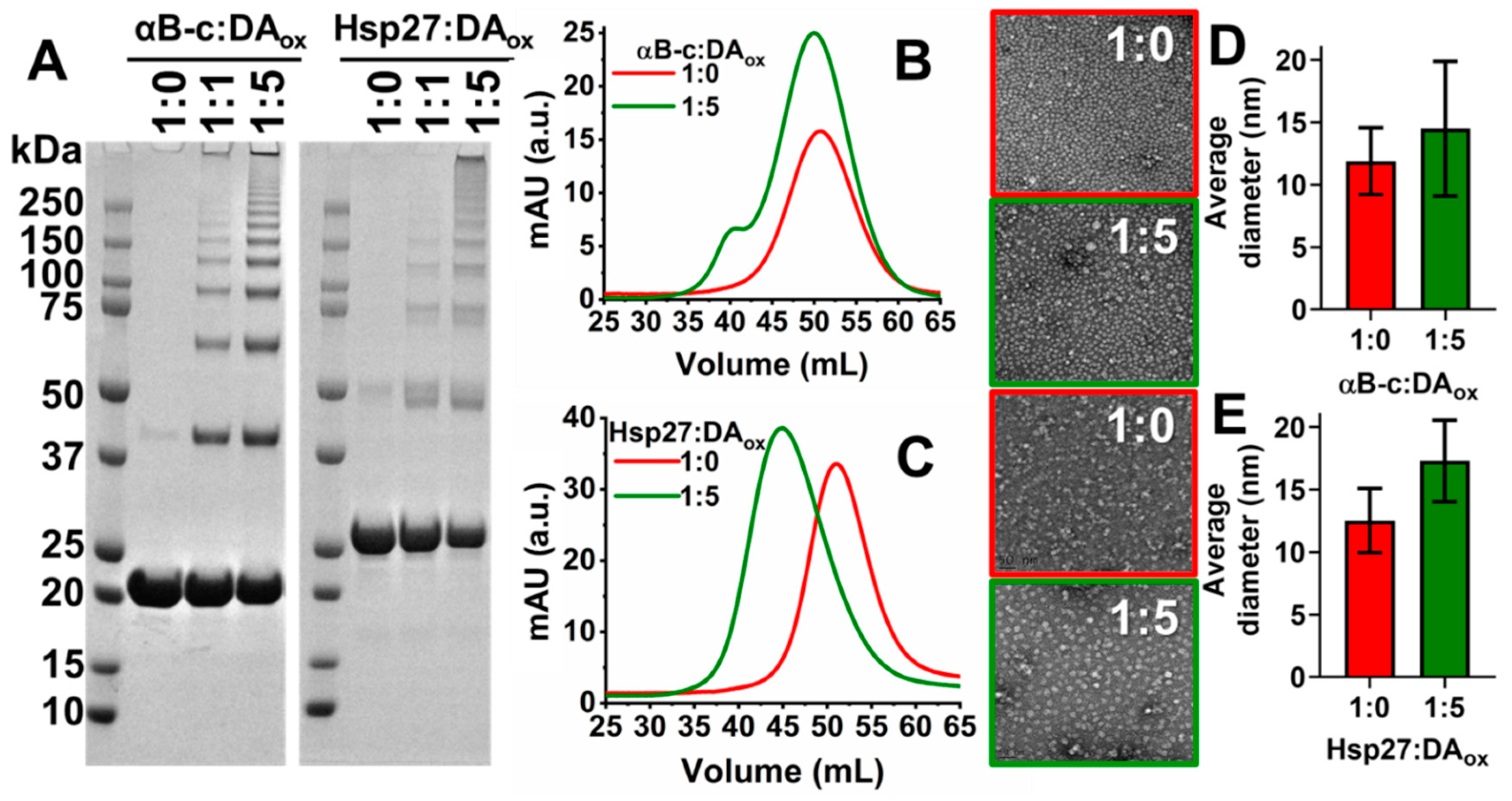
2.2. DAox Promotes Cross-Linking of sHsps
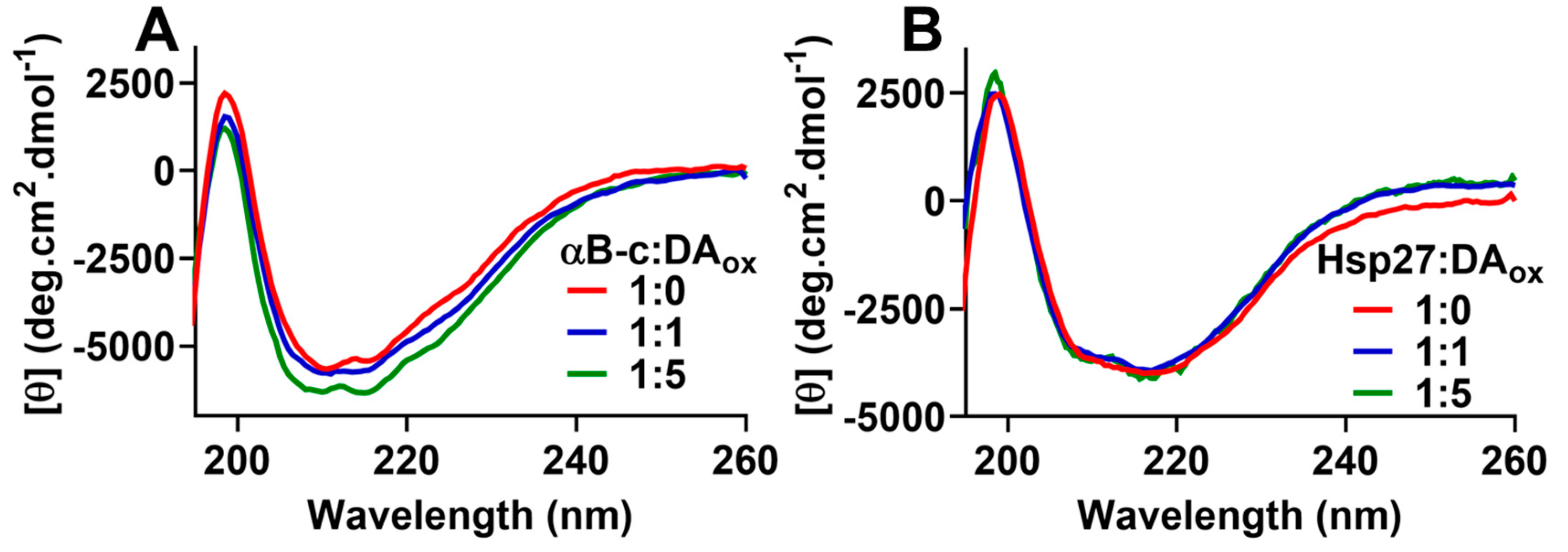
2.3. sHsps:DAox Retain Their β-Sheet Secondary Structure in the ACD
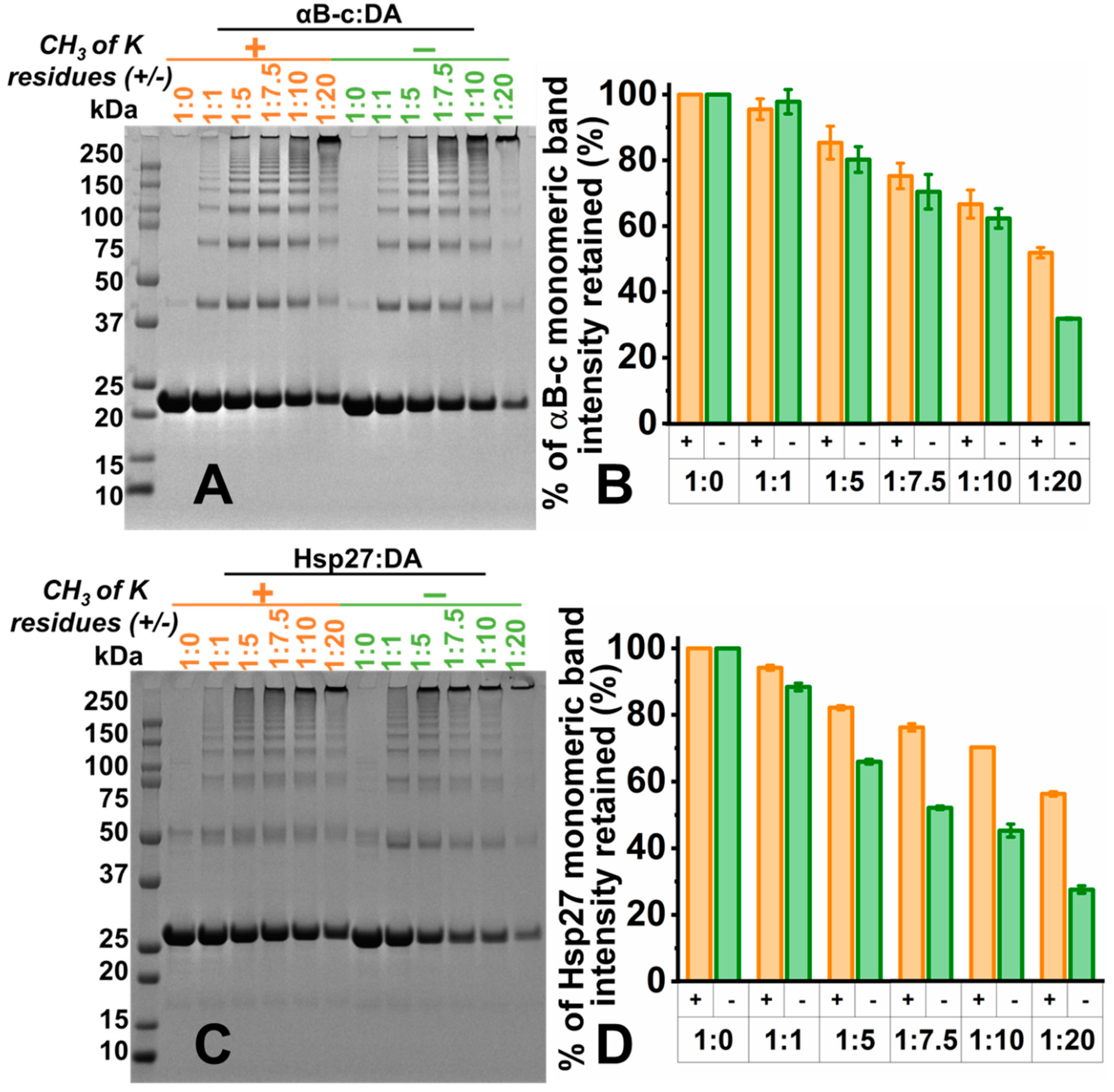
2.4. Lysine Residues Are Involved in the Formation of DAox-Induced HMW Species of sHsps
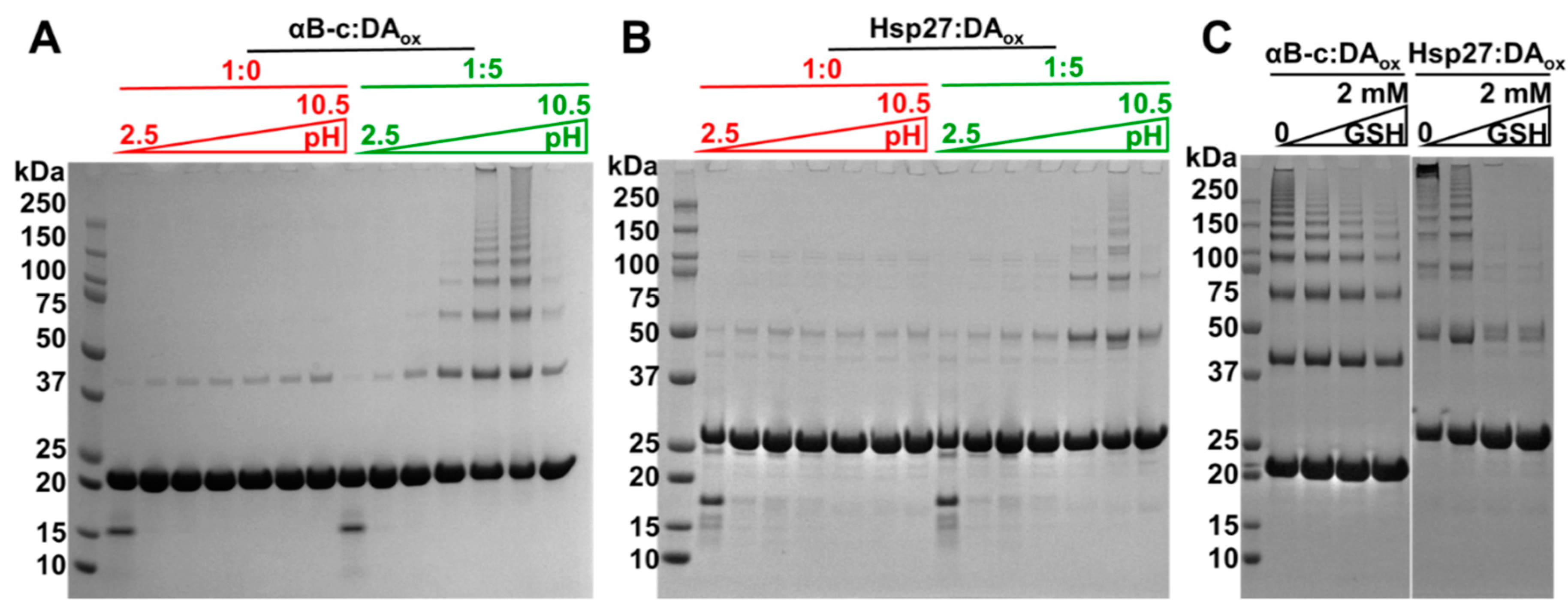
2.5. Acidic pH and the Antioxidant, Glutathione, Rescue sHsps from DAox-Induced Oligomerization
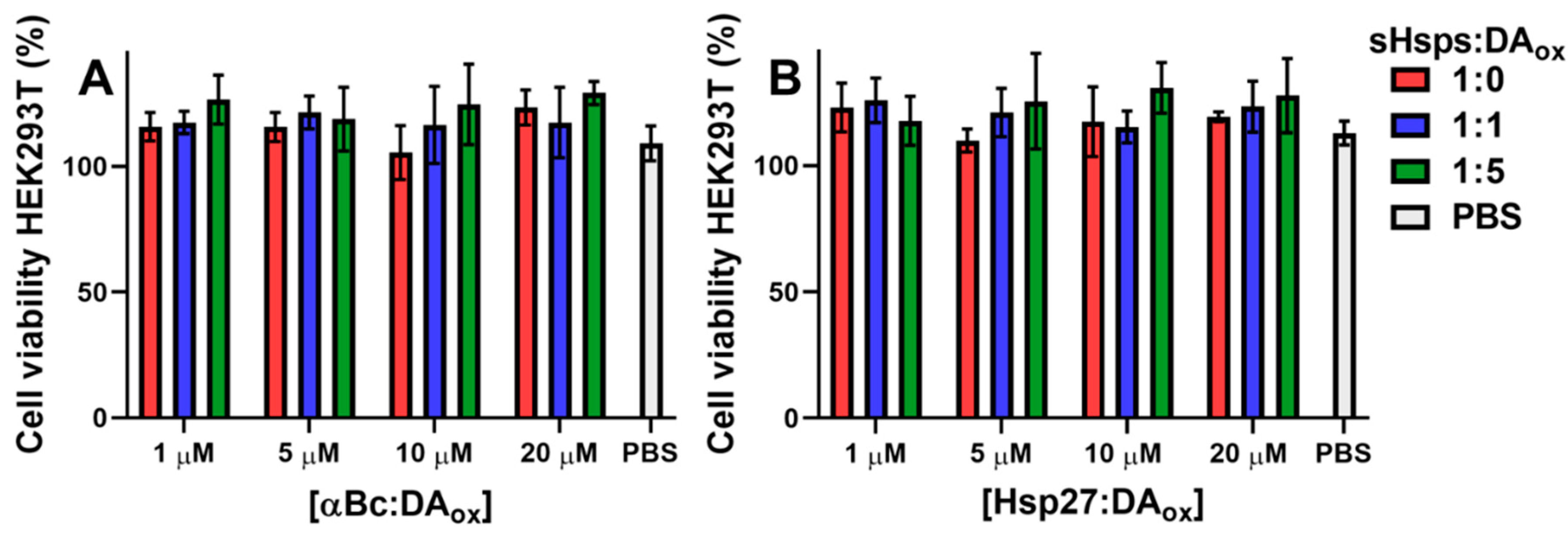
2.6. DAox-Modified sHsps Are Not Toxic to Cells
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Preparation of sHsps:DAox
4.3. Chaperone Assays
4.4. SDS-PAGE
4.5. Mass Determination of sHsp:DA Oligomers from SDS-PAGE
4.6. Size-Exclusion Chromatography
4.7. Circular Dichroism Spectroscopy
4.8. Cell Viability Assays
4.9. pH-Based Incubations
4.10. Glutathione Incubations
4.11. Selective Dimethylation of sHsp Lysine Residues via Reductive Alkylation
4.12. Mass Spectrometry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| αBc | αB-crystallin |
| ACD | α-crystallin domain |
| ANS | 8-anilino-1-napthalenesulfonic acid |
| αSyn | α-synuclein |
| CTR | C-terminal region |
| CV | Column volume |
| DA | Dopamine |
| DAox | Oxidized dopamine |
| DTT | Dithiothreitol |
| GSH | Glutathione |
| HMW | High molecular weight |
| Hsp27 | Heat-shock protein 27 |
| NTR | N-terminal region |
| PD | Parkinson’s disease |
| rcf | Relative centrifugal force |
| Rf | Relative motility |
| rpm | Revolutions per minute |
| TEM | Transmission electron microscopy |
References
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 2011, 3, a004440. [Google Scholar] [CrossRef] [Green Version]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Treweek, T.M.; Meehan, S.; Ecroyd, H.; Carver, J.A. Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell. Mol. Life Sci. 2014, 72, 429–451. [Google Scholar] [CrossRef]
- Carver, J.A.; Ecroyd, H.; Truscott, R.J.W.; Thorn, D.C.; Holt, C. Proteostasis and the regulation of intra- and extracellular protein aggregation by ATP-independent molecular chaperones: Lens α-crystallins and milk caseins. Acc. Chem. Res. 2018, 51, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, J.; Carver, J.A. The multifaceted nature of αB-crystallin. Cell Stress Chaperones 2020, 25, 639–654. [Google Scholar] [CrossRef]
- Haley, D.A.; Horwitz, J.; Stewart, P.L. The small heat-shock protein, αB-crystallin, has a variable quaternary structure. J. Mol. Biol. 1998, 277, 27–35. [Google Scholar] [CrossRef]
- Alderson, T.R.; Roche, J.; Gastall, H.Y.; Dias, D.M.; Pritišanac, I.; Ying, J.; Bax, A.; Benesch, J.L.P.; Baldwin, A.J. Local unfolding of the HSP27 monomer regulates chaperone activity. Nat. Commun. 2019, 10, 1068. [Google Scholar] [CrossRef] [Green Version]
- Aquilina, J.A.; Benesch, J.L.P.; Bateman, O.A.; Slingsby, C.; Robinson, C.V. Polydispersity of a mammalian chaperone: Mass spectrometry reveals the population of oligomers in αB-crystallin. Proc. Natl. Acad. Sci. USA 2003, 100, 10611–10616. [Google Scholar] [CrossRef] [Green Version]
- Jehle, S.; Vollmar, B.S.; Bardiaux, B.; Dove, K.K.; Rajagopal, P.; Gonen, T.; Oschkinat, H.; Klevit, R.E. N-terminal domain of αB-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proc. Natl. Acad. Sci. USA 2011, 108, 6409–6414. [Google Scholar] [CrossRef] [Green Version]
- Mainz, A.; Peschek, J.; Stavropoulou, M.; Back, K.C.; Bardiaux, B.; Asami, S.; Prade, E.; Peters, C.; Weinkauf, S.; Buchner, J.; et al. The chaperone αB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 2015, 22, 898–905. [Google Scholar] [CrossRef]
- Hochberg, G.K.A.; Ecroyd, H.; Liu, C.; Cox, D.; Cascio, D.; Sawaya, M.R.; Collier, M.P.; Stroud, J.; Carver, J.A.; Baldwin, A.J.; et al. The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, E1562–E1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, I.K.; Malfois, M.; Hendle, J.; van der Zandt, H.; Svergun, D.I. A novel quaternary structure of the dimeric α-crystallin domain with chaperone-like activity. J. Biol. Chem. 2001, 276, 12024–12029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, J.G.; Shenoy, A.K.; Clark, J.I. N- and C-terminal motifs in human αB crystallin play an important role in the recognition, selection, and solubilization of substrates. Biochemistry 2006, 45, 13847–13854. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Estrada, M.R.; Clark, J.I. Interactive domains for chaperone activity in the small heat shock protein, human αB crystallin. Biopolymers 2005, 44, 14854–14869. [Google Scholar] [CrossRef]
- Carver, J.A.; Aquilina, J.; Truscott, R.J.; Ralston, G.B. Identification by 1H NMR spectroscopy of flexible C-terminal extensions in bovine lens α-crystallin. FEBS Lett. 1992, 311, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Carver, J.A.; Esposito, G.; Schwedersky, G.; Gaestel, M. 1H NMR spectroscopy reveals that mouse Hsp25 has a flexible C-terminal extension of 18 amino acids. FEBS Lett. 1995, 369, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Triarhou, L. Dopamine and Parkinson’s disease. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J.; Paris, I. Mechanisms of dopamine oxidation and Parkinson’s disease. In Handbook of Neurotoxicity; Springer: New York, NY, USA, 2014; pp. 865–883. [Google Scholar]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Park. Dis. 2012, 2012. [Google Scholar] [CrossRef]
- Perry, T.L.; Godin, D.V.; Hansen, S. Parkinson’s disease: A disorder due to nigral glutathione deficiency? Neurosci. Lett. 1982, 33, 305–310. [Google Scholar] [CrossRef]
- Forooshani, P.K.; Lee, B.P. Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 9–33. [Google Scholar] [CrossRef]
- Lavoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005, 11, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Mammi, S.; Bubacco, L. Kinetic and structural analysis of the early oxidation products of dopamine. J. Biol. Chem. 2007, 282, 15597–15605. [Google Scholar] [CrossRef] [Green Version]
- Paris, I.; Perez-Pastene, C.; Cardenas, S.; Iturra, P.; Muñoz, P.; Couve, E.; Caviedes, P.; Segura-Aguilar, J. Aminochrome induces disruption of actin, alpha-, and beta-tubulin cytoskeleton networks in substantia-nigra-derived cell line. Neurotox. Res. 2010, 18, 82–92. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nat. Cell Biol. 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Condroyer, C.; Pollak, P.; Durif, F.; Dupuits, C.; Viallet, F.; Lohmann, E.; Corvol, J.-C.; Honoré, A.; et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 2010, 20, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.E.; Chesi, A.; Paleologou, K.E.; Schmid, A.; Munoz, A.; Vendruscolo, M.; Gustincich, S.; Lashuel, H.A.; Carloni, P. Inhibition of α-synuclein fibrillization by dopamine is mediated by interactions with five C-terminal residues and with E83 in the NAC region. PLoS ONE 2008, 3, e3394. [Google Scholar] [CrossRef]
- Norris, E.H.; Giasson, B.I.; Hodara, R.; Xu, S.; Trojanowski, J.Q.; Ischiropoulos, H.; Lee, V.M.-Y. Reversible inhibition of α-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J. Biol. Chem. 2005, 280, 21212–21219. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.A.; Rochet, J.-C.; Bieganski, R.M.; Lansbury, P.T. Kinetic Stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Leong, S.L.; Pham, C.L.; Galatis, D.; Fodero-Tavoletti, M.T.; Perez, K.; Hill, A.F.; Masters, C.L.; Ali, F.E.; Barnham, K.J.; Cappai, R. Formation of dopamine-mediated α-synuclein-soluble oligomers requires methionine oxidation. Free Radic. Biol. Med. 2009, 46, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, Q.; Tang, M.; Fu, N.; Shao, W.; Zhang, S.; Yin, Y.; Zeng, R.; Wang, X.; Hu, G.; et al. Upregulation of alphaB-crystallin expression in the substantia nigra of patients with Parkinson’s disease. Neurobiol. Aging 2015, 36, 1686–1691. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; Landon, M.; Pike, I.; Spendlove, I.; McDermott, H.; Mayer, R.J. Dementia with β-amyloid deposition: Involvement of αB-crystallin supports two main diseases. Lancet 1990, 336, 515–516. [Google Scholar] [CrossRef]
- McLean, P.J.; Kawamata, H.; Shariff, S.; Hewett, J.W.; Sharma, N.; Ueda, K.; Breakefield, X.O.; Hyman, B.T. TorsinA and heat shock proteins act as molecular chaperones: Suppression of α-synuclein aggregation. J. Neurochem. 2002, 83, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Rekas, A.; Adda, C.G.; Aquilina, J.A.; Barnham, K.J.; Sunde, M.; Galatis, D.; Williamson, N.A.; Masters, C.L.; Anders, R.F.; Robinson, C.V.; et al. Interaction of the molecular chaperone αB-crystallin with α-synuclein: Effects on amyloid fibril formation and chaperone activity. J. Mol. Biol. 2004, 340, 1167–1183. [Google Scholar] [CrossRef]
- Cox, D.; Selig, E.; Griffin, M.D.W.; Carver, J.A.; Ecroyd, H. Small heat-shock proteins prevent α-synuclein aggregation via transient interactions and their efficacy is affected by the rate of aggregation. J. Biol. Chem. 2016, 291, 22618–22629. [Google Scholar] [CrossRef] [Green Version]
- Augusteyn, R. Dissociation is not required for α-crystallin’s chaperone function. Exp. Eye Res. 2004, 79, 781–784. [Google Scholar] [CrossRef]
- Hudson, S.A.; Ecroyd, H.; Kee, T.W.; Carver, J.A. The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 2009, 276, 5960–5972. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Carver, J.A.; Lindner, R.A.; Lyon, C.; Canet, D.; Hernandez, H.; Dobson, C.M.; Redfield, C. The interaction of the molecular chaperone α-crystallin with unfolding α-lactalbumin: A structural and kinetic spectroscopic study. J. Mol. Biol. 2002, 318, 815–827. [Google Scholar] [CrossRef]
- Cox, D.; Carver, J.A.; Ecroyd, H. Preventing α-synuclein aggregation: The role of the small heat-shock molecular chaperone proteins. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1830–1843. [Google Scholar] [CrossRef] [Green Version]
- Hill, E.K.; Krebs, B.; Goodall, D.G.; Howlett, G.J.; Dunstan, D.E. Shear flow induces amyloid fibril formation. Biomacromolecules 2005, 7, 10–13. [Google Scholar] [CrossRef]
- Carver, J.A.; Grosas, A.B.; Ecroyd, H.; Quinlan, R.A. The functional roles of the unstructured N- and C-terminal regions in αB-crystallin and other mammalian small heat-shock proteins. Cell Stress Chaperon. 2017, 22, 627–638. [Google Scholar] [CrossRef]
- Garvey, M.; Ecroyd, H.; Ray, N.J.; Gerrard, J.A.; Carver, J.A. Functional Amyloid Protection in the Eye Lens: Retention of α-crystallin molecular chaperone activity after modification into amyloid fibrils. Biomolecules 2017, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Rekas, A.; Knott, R.B.; Sokolova, A.; Barnham, K.J.; Perez, K.A.; Masters, C.L.; Drew, S.C.; Cappai, R.; Curtain, C.C.; Pham, C.L.L. The structure of dopamine induced α-synuclein oligomers. Eur. Biophys. J. 2010, 39, 1407–1419. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Treweek, T.M.; Rekas, A.; Walker, M.J.; Carver, J.A. A quantitative NMR spectroscopic examination of the flexibility of the C-terminal extensions of the molecular chaperones, αA- and αB-crystallin. Exp. Eye Res. 2010, 91, 691–699. [Google Scholar] [CrossRef]
- Manthey, M.; Pyne, S.; Truscott, R. Addition of aliphatic and aromatic amines to catechol in aqueous solution under oxidizing conditions. Aust. J. Chem. 1989, 42, 365–373. [Google Scholar] [CrossRef]
- Yang, J.; Stuart, M.A.C.; Kamperman, M. Jack of all trades: Versatile catechol crosslinking mechanisms. Chem. Soc. Rev. 2014, 43, 8271–8298. [Google Scholar] [CrossRef]
- Zhu, Y.; Carvey, P.M.; Ling, Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006, 1090, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Pasupuleti, N.; Gangadhariah, M.; Padmanabha, S.; Santhoshkumar, P.; Nagaraj, R.H. The role of the cysteine residue in the chaperone and anti-apoptotic functions of human Hsp27. J. Cell. Biochem. 2010, 110, 408–419. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef] [Green Version]
- Plum, S.; Steinbach, S.; Attems, J.; Keers, S.; Riederer, P.; Gerlach, M.; May, C.; Marcus, K. Proteomic characterization of neuromelanin granules isolated from human substantia nigra by laser-microdissection. Sci. Rep. 2016, 6, 37139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucca, F.A.; Vanna, R.; Cupaioli, F.A.; Bellei, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Grassi, S.; Prinetti, A.; Casella, L.; et al. Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson’s disease. NPJ Park. Dis. 2018, 4, 1–23. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nat. Cell Biol. 1988, 334, 345–348. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Graybiel, A.M.; Agid, Y. Selective vulnerability of pigmented dopaminergic neurons in Parkinson’s disease. Acta Neurol. Scand. 1989, 80, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Briceño, A.; Muñoz, P.; Brito, P.; Huenchuguala, S.; Segura-Aguilar, J.; Paris, I.B. Aminochrome toxicity is mediated by inhibition of microtubules polymerization through the formation of adducts with tubulin. Neurotox. Res. 2015, 29, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Cappai, R.; Leck, S.; Tew, D.J.; Williamson, N.A.; Smith, D.P.; Galatis, D.; Sharpies, R.A.; Curtain, C.C.; Ali, F.E.; Cherny, R.A.; et al. Dopamine promotes α-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 2005, 19, 1377–1379. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Tosatto, L.; Munari, F.; Tessari, I.; De Laureto, P.P.; Mammi, S.; Bubacco, L. Dopamine quinones interact with α-synuclein to form unstructured adducts. Biochem. Biophys. Res. Commun. 2010, 394, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.L.; Leong, S.L.; Ali, F.E.; Kenche, V.B.; Hill, A.F.; Gras, S.L.; Barnham, K.J.; Cappai, R. Dopamine and the dopamine oxidation product 5,6-dihydroxylindole promote distinct on-pathway and off-pathway aggregation of α-synuclein in a pH-dependent manner. J. Mol. Biol. 2009, 387, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Puangmalai, N.; Bhatt, N.; Garcia, S.; Zhao, Y.; Kayed, R. Polymorphic α-synuclein strains modified by dopamine and docosahexaenoic acid interact differentially with tau protein. Mol. Neurobiol. 2020, 57, 2741–2765. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Mishizen, A.J.; Cascio, M.; Hastings, T.G. Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol. Dis. 2009, 34, 487–500. [Google Scholar] [CrossRef] [Green Version]
- Touchette, J.C.; Breckenridge, J.M.; Wilken, G.H.; MacArthur, H. Direct intranigral injection of dopaminochrome causes degeneration of dopamine neurons. Neurosci. Lett. 2016, 612, 178–184. [Google Scholar] [CrossRef] [Green Version]
- de Araújo, F.M.; Ferreira, R.S.; Souza, C.S.; Dos Santos, C.C.; Rodrigues, T.L.; e Silva, J.H.; Gasparotto, J.; Gelain, D.P.; El-Bachá, R.S.; Maria de Fátima, D.C.; et al. Aminochrome decreases NGF, GDNF and induces neuroinflammation in organotypic midbrain slice cultures. NeuroToxicology 2018, 66, 98–106. [Google Scholar] [CrossRef]
- Garvey, M.; Griesser, S.S.; Griesser, H.J.; Thierry, B.; Nussio, M.R.; Shapter, J.G.; Ecroyd, H.; Giorgetti, S.; Bellotti, V.; Gerrard, J.A.; et al. Enhanced molecular chaperone activity of the small heat-shock protein αB-crystallin following covalent immobilization onto a solid-phase support. Biopolymers 2011, 95, 376–389. [Google Scholar] [CrossRef]
- Mehlen, P.; Arrigo, A.-P. The serum-induced phosphorylation of mammalian hsp27 correlates with changes in its intracellular localization and levels of oligomerization. JBIC J. Biol. Inorg. Chem. 1994, 221, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Kretz-Remy, C.; Briolay, J.; Fostan, P.; Mirault, M.E.; Arrigo, A.P. Intracellular reactive oxygen species as apparent modulators of heat-shock protein 27 (hsp27) structural organization and phosphorylation in basal and tumour necrosis factor α-treated T47D human carcinoma cells. Biochem. J. 1995, 312, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, D.A.; Bova, M.P.; Huang, Q.-L.; Mchaourab, H.S.; Stewart, P.L. Small heat-shock protein structures reveal a continuum from symmetric to variable assemblies. J. Mol. Biol. 2000, 298, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Treweek, T.M.; Rekas, A.; Lindner, R.A.; Walker, M.J.; Aquilina, J.A.; Robinson, C.V.; Horwitz, J.; Der Perng, M.; Quinlan, R.A.; Carver, J.A. R120G αB-crystallin promotes the unfolding of reduced α-lactalbumin and is inherently unstable. FEBS J. 2005, 272, 711–724. [Google Scholar] [CrossRef] [Green Version]
- Treweek, T.M.; Ecroyd, H.; Williams, D.M.; Meehan, S.; Carver, J.A.; Walker, M.J. Site-directed mutations in the C-terminal extension of human αB-crystallin affect chaperone function and block amyloid fibril formation. PLoS ONE 2007, 2, e1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakytny, R.; Carver, J.A.; Harding, J.J.; Kilby, G.W.; Sheil, M.M. A spectroscopic study of glycated bovine α-crystallin: Investigation of flexibility of the C-terminal extension, chaperone activity and evidence for diglycation. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1997, 1343, 299–315. [Google Scholar] [CrossRef]
- Cartier, E.A.; Parra, L.A.; Baust, T.B.; Quiroz, M.; Salazar, G.; Faundez, V.; Egaña, L.; Torres, G.E. A biochemical and functional protein complex involving dopamine synthesis and transport into synaptic vesicles. J. Biol. Chem. 2010, 285, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Huenchuguala, S.; Sjödin, B.; Mannervik, B.; Segura-Aguilar, J. Novel Alpha-synuclein oligomers formed with the aminochrome-glutathione conjugate are not neurotoxic. Neurotox. Res. 2018, 35, 432–440. [Google Scholar] [CrossRef]
- Cuevas, C.; Huenchuguala, S.; Muñoz, P.; Villa, M.; Paris, I.; Mannervik, B.; Segura-Aguilar, J. Glutathione transferase-M2-2 secreted from glioblastoma cell protects SH-SY5Y cells from aminochrome neurotoxicity. Neurotox. Res. 2014, 27, 217–228. [Google Scholar] [CrossRef]
- Jewett, M.; Dickson, E.; Brolin, K.; Negrini, M.; Jimenez-Ferrer, I.; Swanberg, M. Glutathione S-transferase alpha 4 prevents dopamine neurodegeneration in a rat alpha-synuclein model of Parkinson’s disease. Front. Neurol. 2018, 9, 222. [Google Scholar] [CrossRef] [Green Version]
- Hauser, D.N.; Dukes, A.A.; Mortimer, A.D.; Hastings, T.G. Dopamine quinone modifies and decreases the abundance of the mitochondrial selenoprotein glutathione peroxidase 4. Free Radic. Biol. Med. 2013, 65, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulig, M.; Ecroyd, H. The small heat-shock protein αB-crystallin uses different mechanisms of chaperone action to prevent the amorphous versus fibrillar aggregation of α-lactalbumin. Biochem. J. 2012, 448, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, J.; Huang, Q.-L.; Ding, L.; Bova, M.P. Lens α-crystallin: Chaperone-like properties. Methods Enzymol. 1998, 290, 365–383. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Final % of Soluble A53T αSyn Retained | ||
|---|---|---|
| αBc ± SEM | Hsp27 ± SEM | |
| A53T αSyn only | 27.0 ± 4.9 | |
| αSyn + sHsps:DAox (1:0) | 49.6 ± 4.9 | 37.2 ± 7.6 |
| αSyn + sHsps:DAox (1:1) | 46.2 ± 4.1 | 42.3 ± 3.7 |
| αSyn + sHsps:DAox (1:5) | 46.9 ± 2.6 | 39.3 ± 4.9 |
| Final % of Soluble αLA Retained | ||
| αBc ± SEM | Hsp27 ± SEM | |
| αLA only | 39.5 ± 3.1 | |
| αLA + sHsps:DAox (1:0) | 83.6 ± 3.1 | 65.9 ± 4.1 |
| αLA + sHsps:DAox (1:1) | 81.0 ± 3.9 | 65.8 ± 1.0 |
| αLA + sHsps:DAox (1:5) | 64.0 ± 2.4 | 53.6 ± 1.7 |
| αBc | Hsp27 | |||
|---|---|---|---|---|
| Estimated mass ± STDEV (kDa) | Actual mass (kDa) | Estimated mass ± STDEV (kDa) | Actual mass (kDa) | |
| Monomer | 21 ± 0.6 | 20.2 | 26 ± 0.1 | 22.8 |
| Dimer | 43 ± 0.6 | 40.4 | 50 ± 0.4 | 45.4 |
| Trimer | 64 ± 1.0 | 60.6 | 75 ± 0.6 | 68.1 |
| Tetramer | 82 ± 2.2 | 80.8 | 97 ± 2.6 | 90.8 |
| Pentamer | 94 ± 5.7 | 101.0 | 111 ± 0.6 | 113.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayashi, J.; Ton, J.; Negi, S.; Stephens, D.E.K.M.; Pountney, D.L.; Preiss, T.; Carver, J.A. The Effect of Oxidized Dopamine on the Structure and Molecular Chaperone Function of the Small Heat-Shock Proteins, αB-Crystallin and Hsp27. Int. J. Mol. Sci. 2021, 22, 3700. https://doi.org/10.3390/ijms22073700
Hayashi J, Ton J, Negi S, Stephens DEKM, Pountney DL, Preiss T, Carver JA. The Effect of Oxidized Dopamine on the Structure and Molecular Chaperone Function of the Small Heat-Shock Proteins, αB-Crystallin and Hsp27. International Journal of Molecular Sciences. 2021; 22(7):3700. https://doi.org/10.3390/ijms22073700
Chicago/Turabian StyleHayashi, Junna, Jennifer Ton, Sparsh Negi, Daniel E. K. M. Stephens, Dean L. Pountney, Thomas Preiss, and John A. Carver. 2021. "The Effect of Oxidized Dopamine on the Structure and Molecular Chaperone Function of the Small Heat-Shock Proteins, αB-Crystallin and Hsp27" International Journal of Molecular Sciences 22, no. 7: 3700. https://doi.org/10.3390/ijms22073700
APA StyleHayashi, J., Ton, J., Negi, S., Stephens, D. E. K. M., Pountney, D. L., Preiss, T., & Carver, J. A. (2021). The Effect of Oxidized Dopamine on the Structure and Molecular Chaperone Function of the Small Heat-Shock Proteins, αB-Crystallin and Hsp27. International Journal of Molecular Sciences, 22(7), 3700. https://doi.org/10.3390/ijms22073700