
The Multifaceted Role of HSF1 in Pathophysiology: Focus on Its Interplay with TG2



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HSF1: The Master Regulator of the HSR
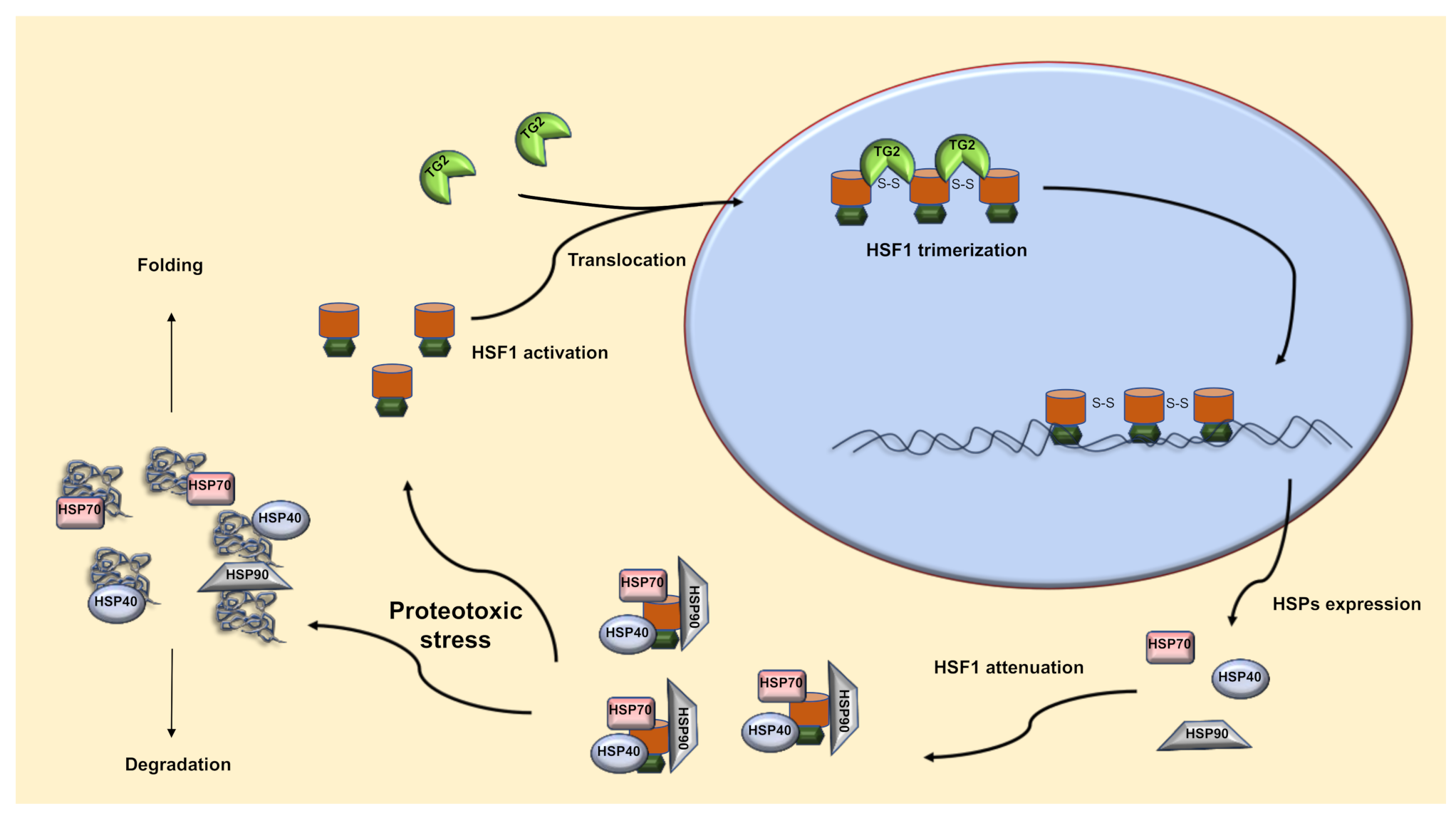
2.1. TG2-Dependent Activation of HSF1
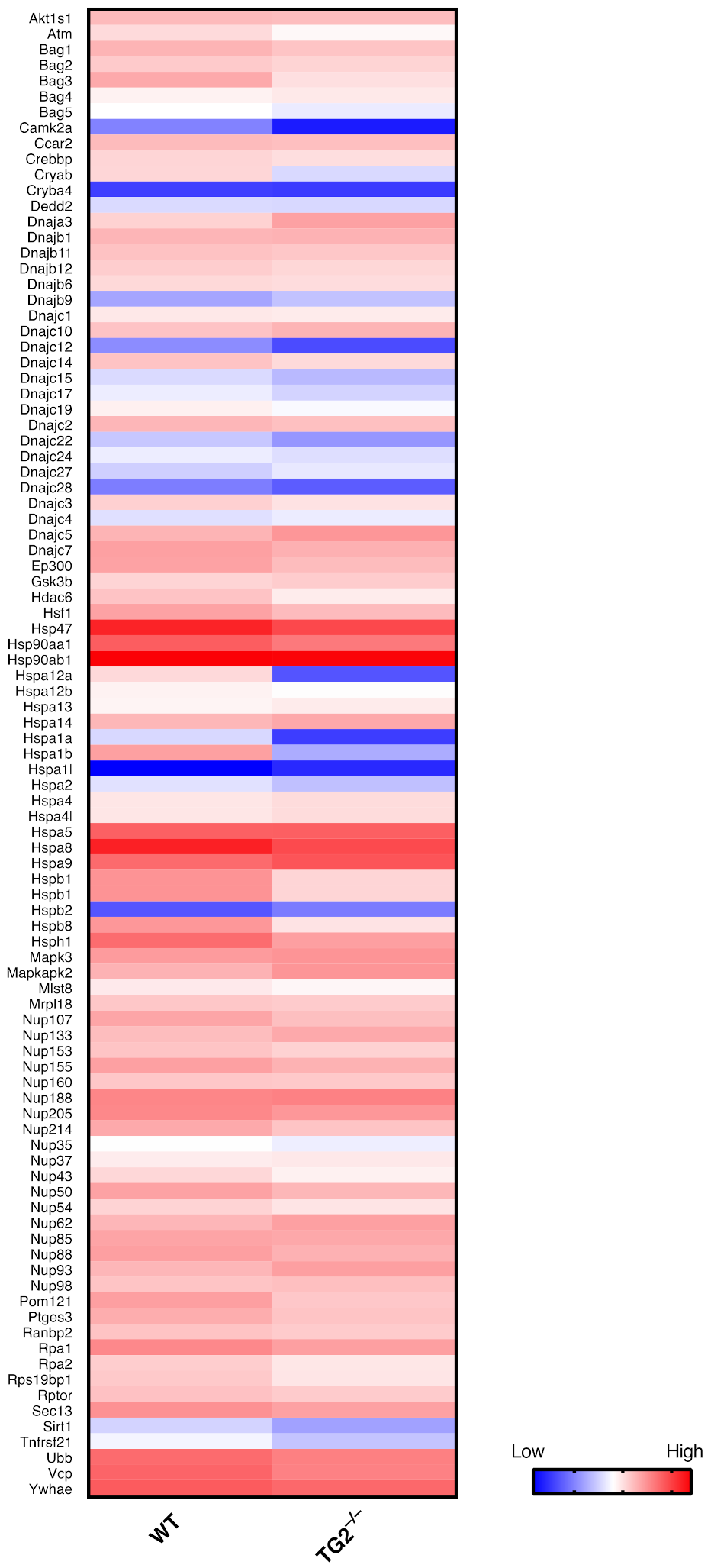
2.2. TG2 and HSF1 Axis in the Regulation of the HSR
3. HSF1 in Diseases
3.1. Cystic Fibrosis
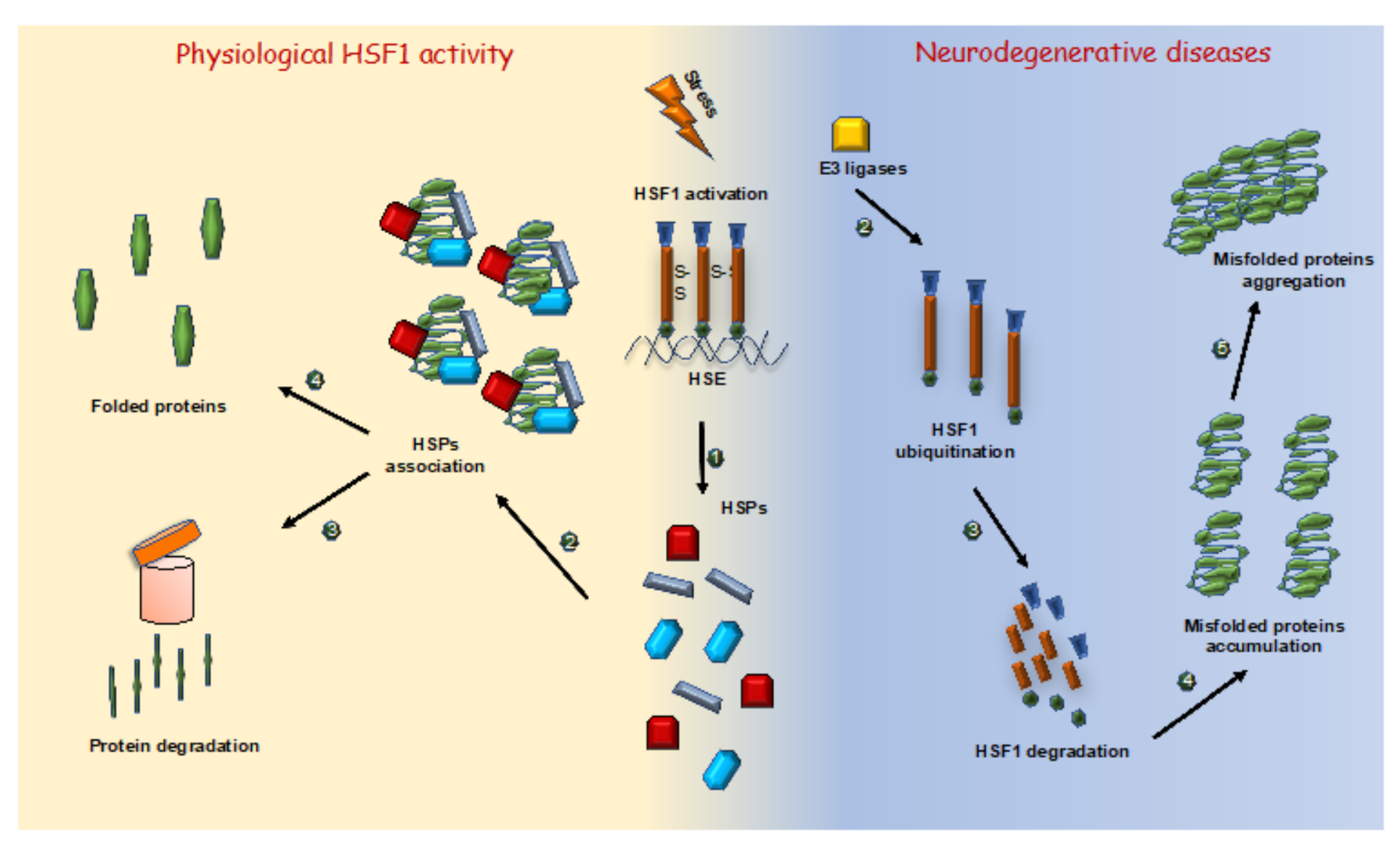
3.2. Neurodegenerative Diseases
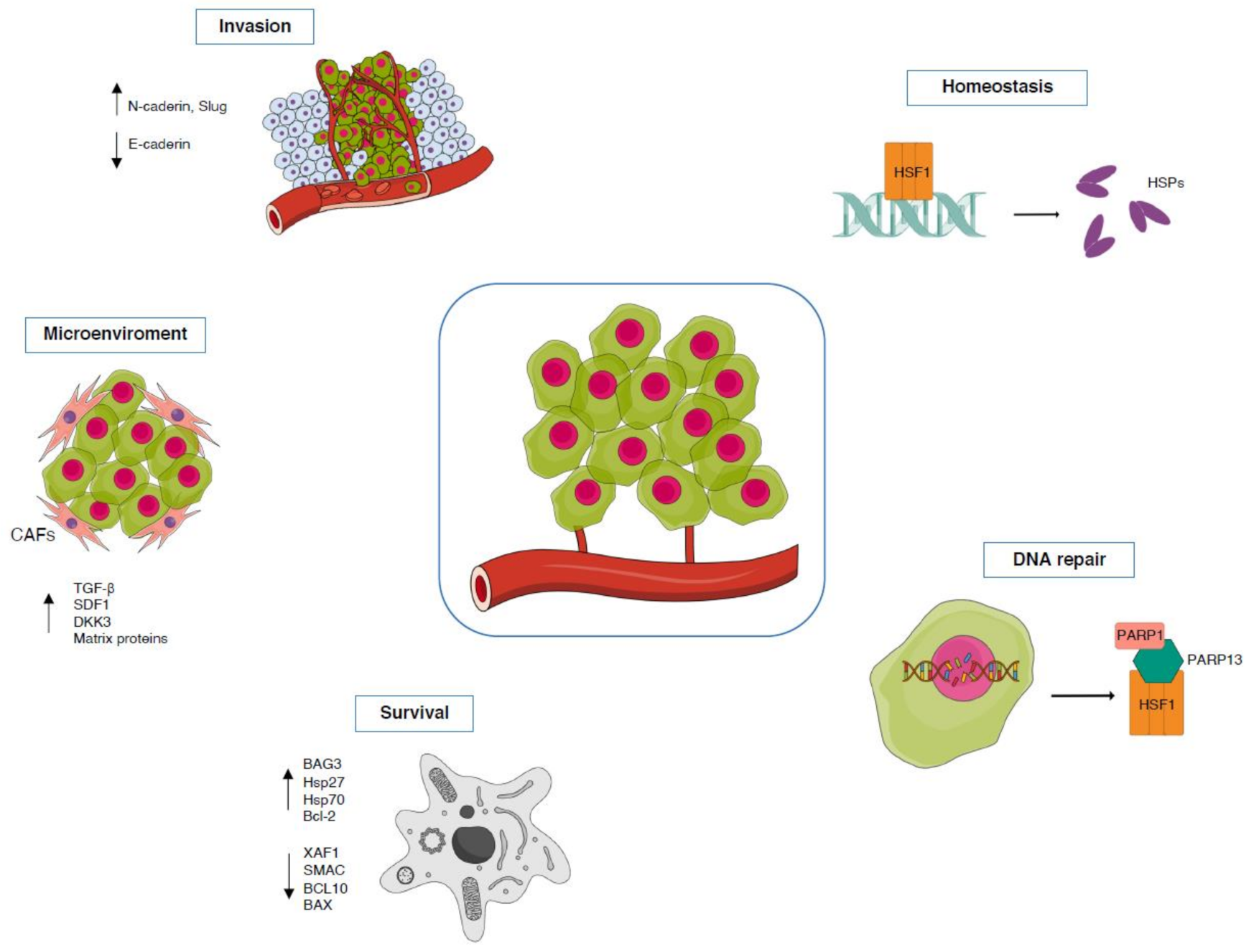
3.3. Cancer
TG2-HSF1 Axis in Cancer
4. HSF1: A Regulator of Development
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 220, 1427–1438. [Google Scholar] [CrossRef] [Green Version]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Sala, A.J.; Bott, L.C.; Morimoto, R.I. Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol. 2017, 216, 1231–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C. Heat shock transcription factors: Structure and regulation. Annu. Rev. Cell Dev. Biol. 1995, 11, 441–469. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anckar, J.; Sistonen, L. Heat shock factor 1 as a coordinator of stress and developmental pathways. Adv. Exp. Med. Biol. 2007, 594, 78–88. [Google Scholar] [PubMed]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Zorzi, E.; Bonvini, P. Inducible Hsp70 in the Regulation of Cancer Cell Survival: Analysis of Chaperone Induction, Expression and Activity. Cancers 2011, 3, 3921–3956. [Google Scholar] [CrossRef]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef]
- Kampinga, H.H. Chaperones in preventing protein denaturation in living cells and protecting against cellular stress. Handb. Exp. Pharmacol. 2006, 172, 1–42. [Google Scholar]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Kurochkin, A.V.; Yip, G.N.; Zhang, Y.; Bertelsen, E.B.; Zuiderweg, E.R. Allostery in Hsp70 chaperones is transduced by subdomain rotations. J. Mol. Biol. 2009, 388, 475–490. [Google Scholar] [CrossRef] [Green Version]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Giordanetto, F.; Kroemer, G. Targeting HSP70 for cancer therapy. Mol. Cell 2009, 36, 176–177. [Google Scholar] [CrossRef]
- Vogel, M.; Mayer, M.P.; Bukau, B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J. Biol. Chem. 2006, 281, 38705–38711. [Google Scholar] [CrossRef] [Green Version]
- Swain, J.F.; Dinler, G.; Sivendran, R.; Montgomery, D.L.; Stotz, M.; Gierasch, L.M. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell 2007, 26, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Laufen, T.; Mayer, M.P.; Beisel, C.; Klostermeier, D.; Mogk, A.; Reinstein, J.; Bukau, B. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Shevde, L.A.; Samant, R.S. Multi-faceted role of HSP40 in cancer. Clin. Exp. Metastasis 2009, 26, 559–567. [Google Scholar] [CrossRef]
- Amin, J.; Ananthan, J.; Voellmy, R. Key features of heat shock regulatory elements. Mol. Cell. Biol. 1988, 8, 3761–3769. [Google Scholar] [CrossRef]
- Fernandes, M.; Xiao, H.; Lis, J.T. Binding of heat shock factor to and transcriptional activation of heat shock genes in Drosophila. Nucleic Acids Res. 1995, 23, 4799–4804. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Perisic, O.; Lis, J.T. Cooperative binding of Drosophila heat shock factor to arrays of a conserved 5 bp unit. Cell 1991, 64, 585–593. [Google Scholar] [CrossRef]
- Zuo, J.; Rungger, D.; Voellmy, R. Multiple layers of regulation of human heat shock transcription factor 1. Mol. Cell. Biol. 1995, 15, 4319–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damberger, F.F.; Pelton, J.G.; Harrison, C.J.; Nelson, H.C.; Wemmer, D.E. Solution structure of the DNA-binding domain of the heat shock transcription factor determined by multidimensional heteronuclear magnetic resonance spectroscopy. Protein Sci. 1994, 3, 1806–1821. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.J.; Bohm, A.A.; Nelson, H.C. Crystal structure of the DNA binding domain of the heat shock transcription factor. Science 1994, 263, 224–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerfelt, M.; Trouillet, D.; Mezger, V.; Sistonen, L. Heat shock factors at a crossroad between stress and development. Ann. N. Y. Acad. Sci. 2007, 1113, 15–27. [Google Scholar] [CrossRef]
- Pirkkala, L.; Alastalo, T.P.; Nykanen, P.; Seppa, L.; Sistonen, L. Differentiation lineage-specific expression of human heat shock transcription factor 2. FASEB J. 1999, 13, 1089–1098. [Google Scholar] [CrossRef]
- Min, J.N.; Zhang, Y.; Moskophidis, D.; Mivechi, N.F. Unique contribution of heat shock transcription factor 4 in ocular lens development and fiber cell differentiation. Genesis 2004, 40, 205–217. [Google Scholar] [CrossRef]
- Izu, H.; Inouye, S.; Fujimoto, M.; Shiraishi, K.; Naito, K.; Nakai, A. Heat shock transcription factor 1 is involved in quality-control mechanisms in male germ cells. Biol. Reprod. 2004, 70, 18–24. [Google Scholar] [CrossRef]
- Trinklein, N.D.; Chen, W.C.; Kingston, R.E.; Myers, R.M. Transcriptional regulation and binding of heat shock factor 1 and heat shock factor 2 to 32 human heat shock genes during thermal stress and differentiation. Cell Stress Chaperones 2004, 9, 21–28. [Google Scholar] [CrossRef]
- Hahn, J.S.; Hu, Z.; Thiele, D.J.; Iyer, V.R. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 2004, 24, 5249–5256. [Google Scholar] [CrossRef] [Green Version]
- Christians, E.S.; Benjamin, I.J. Heat shock response: Lessons from mouse knockouts. Mol. Chaperones Health Dis. 2006, 172, 139–152. [Google Scholar]
- Zhang, Y.; Huang, L.; Zhang, J.; Moskophidis, D.; Mivechi, N.F. Targeted disruption of hsf1 leads to lack of thermotolerance and defines tissue-specific regulation for stress-inducible Hsp molecular chaperones. J. Cell. Biochem. 2002, 86, 376–393. [Google Scholar] [CrossRef]
- Zou, J.; Guo, Y.; Guettouche, T.; Smith, D.F.; Voellmy, R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stresssensitive complex with HSF1. Cell 1998, 94, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, S.; Ali, A.; Ovsenek, N. Multiple components of the HSP90 chaperone complex function in regulation of heat shock factor 1 In vivo. Mol. Cell. Biol. 1999, 19, 8033–8041. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Guettouche, T.; Fenna, M.; Boellmann, F.; Pratt, W.B.; Toft, D.O.; Smith, D.F.; Voellmy, R. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J. Biol. Chem. 2001, 276, 45791–45799. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Munson, K.M.; Webb, W.W.; Lis, J.T. Dynamics of heat shock factor association with native gene loci in living cells. Nature 2006, 442, 1050–1053. [Google Scholar] [CrossRef]
- Holmberg, C.I.; Hietakangas, V.; Mikhailov, A.; Rantanen, J.O.; Kallio, M.; Meinander, A.; Hellman, J.; Morrice, N.; MacKintosh, C.; Morimoto, R.I.; et al. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J. 2001, 20, 3800–3810. [Google Scholar] [CrossRef] [Green Version]
- Guettouche, T.; Boellmann, F.; Lane, W.S.; Voellmy, R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Chu, B.; Zhong, R.; Soncin, F.; Stevenson, M.A.; Calderwood, S.K. Transcriptional activity of heat shock factor 1 at 37 degrees C is repressed through phosphorylation on two distinct serine residues by glycogen synthase kinase 3 and protein kinases Calpha and Czeta. J. Biol. Chem. 1998, 273, 18640–18646. [Google Scholar] [CrossRef] [Green Version]
- Kline, M.P.; Morimoto, R.I. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol. Cell. Biol. 1997, 17, 2107–2115. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.G.; Thiele, D.J. Redox regulation of mammalian heat shock factor 1 is essential for HSP gene activation and protection from stress. Genes Dev. 2002, 17, 516–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossin, F.; Villella, V.R.; D’Eletto, M.; Farrace, M.G.; Esposito, S.; Ferrari, E.; Monzani, R.; Occhigrossi, L.; Pagliarini, V.; Sette, C.; et al. TG2 regulates the heat-shock response by the post-translational modification of HSF1. EMBO Rep. 2018, 19, e45067. [Google Scholar] [CrossRef] [PubMed]
- Baler, R.; Welch, W.J.; Voellmy, R. Heat shock gene regulation by nascent polypeptides and denatured proteins: hsp70 as a potential autoregulatory factor. J. Cell Biol. 1992, 117, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Mosser, D.D.; Morimoto, R.I. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998, 12, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Eletto, M.; Rossin, F.; Fedorova, O.; Farrace, M.G.; Piacentini, M. Transglutaminase type 2 in the regulation of proteostasis. Biol. Chem. 2019, 400, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Fesus, L.; Piacentini, M. Transglutaminase 2: An enigmatic enzyme with diverse functions. Trends Biochem. Sci. 2002, 10, 534–539. [Google Scholar] [CrossRef]
- Rossin, F.; D’Eletto, M.; Macdonald, D.; Farrace, M.G.; Piacentini, M. TG2 transamidating activity acts as a reostat controlling the interplay between apoptosis and autophagy. Amino Acids 2011, 42, 1793–1802. [Google Scholar] [CrossRef]
- D’Eletto, M.; Farrace, M.G.; Falasca, L.; Reali, V.; Oliverio, S.; Melino, G.; Griffin, M.; Fimia, G.M.; Piacentini, M. Transglutaminase 2 is involved in autophagosome maturation. Autophagy 2009, 5, 1145–1154. [Google Scholar] [CrossRef]
- D’Eletto, M.; Farrace, M.G.; Rossin, F.; Strappazzon, F.; Giacomo, G.D.; Cecconi, F.; Melino, G.; Sepe, S.; Moreno, S.; Fimia, G.M.; et al. Type 2 transglutaminase is involved in the autophagy-dependent clearance of ubiquitinated proteins. Cell Death Differ. 2012, 19, 1228–1238. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Hidalgo, L.; Altuntas, S.; Rossin, F.; D’Eletto, M.; Marsella, C.; Farrace, M.G.; Falasca, L.; Antonioli, M.; Fimia, G.M.; Piacentini, M. Transglutaminase type 2-dependent selective recruitment of proteins into exosomes under stressful cellular conditions. Biochim. Biophys. Acta 2016, 1863, 2084–2092. [Google Scholar] [CrossRef]
- Altuntas, S.; Rossin, F.; Marsella, C.; D’Eletto, M.; Diaz-Hidalgo, L.; Farrace, M.G.; Campanella, M.; Antonioli, M.; Fimia, G.M.; Piacentini, M. The transglutaminase type 2 and pyruvate kinase isoenzyme M2 interplay in autophagy regulation. Oncotarget 2015, 6, 44941–44954. [Google Scholar] [CrossRef]
- McMillan, D.R.; Xiao, X.; Shao, L.; Graves, K.; Benjamin, I.J. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem. 1998, 273, 7523–7528. [Google Scholar] [CrossRef] [Green Version]
- Rossin, F.; Costa, R.; Bordi, M.; D’Eletto, M.; Occhigrossi, L.; Farrace, M.G.; Barlev, N.; Ciccosanti, F.; Muccioli, S.; Chieregato, L.; et al. Transglutaminase Type 2 regulates the Wnt/β-catenin pathway in vertebrates. Cell Death Dis. 2021, 12, 249. [Google Scholar] [CrossRef]
- Farrelly, L.A.; Thompson, R.E.; Zhao, S.; Lepack, A.E.; Lyu, Y.; Bhanu, N.V.; Zhang, B.; Loh, Y.E.; Ramakrishnan, A.; Vadodaria, K.C.; et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 2019, 567, 535–539. [Google Scholar] [CrossRef]
- Oliverio, S.; Amendola, A.; Di Sano, F.; Farrace, M.G.; Fesus, L.; Nemes, Z.; Piredda, L.; Spinedi, A.; Piacentini, M. Tissue transglutaminase-dependent posttranslational modification of the retinoblastoma gene product in promonocytic cells undergoing apoptosis. Mol. Cell. Biol. 1997, 17, 6040–6048. [Google Scholar] [CrossRef] [Green Version]
- Tatsukawa, H.; Fukaya, Y.; Frampton, G.; Martinez-Fuentes, A.; Suzuki, K.; Kuo, T.F.; Nagatsuma, K.; Shimokado, K.; Okuno, M.; Wu, J. Role of transglutaminase 2 in liver injury via cross-linking and silencing of transcription factor Sp1. Gastroenterology 2009, 136, 1783–1795. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Zhang, Y.; Zhang, H.; Graner, S.; Williams, J.F.; Levitt, M.L.; Lokshin, A. Interaction of tissue transglutaminase with nuclear transport protein importin-alpha3. FEBS Lett. 1999, 446, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, D.C.; Sarge, K.D. RNA polymerase II interacts with the Hspa1b promoter in mouse epididymal spermatozoa. Reproduction 2009, 137, 923–929. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, J.; Liu, S.; Yan, Q. HSF1 and Sp1 regulate FUT4 gene expression and cell proliferation in breast cancer cells. J. Cell. Biochem. 2014, 115, 168–178. [Google Scholar] [CrossRef]
- Caccamo, D.; Condello, S.; Ferlazzo, N.; Currò, M.; Griffin, M.; Ientile, R. Transglutaminase 2 interaction with small heat shock proteins mediate cell survival upon excitotoxic stress. Amino Acids 2013, 44, 151–159. [Google Scholar] [CrossRef]
- Sileno, S.; D’Oria, V.; Stucchi, R.; Alessio, M.; Petrini, S.; Bonetto, V.; Maechler, P.; Bertuzzi, F.; Grasso, V.; Paolella, K.; et al. A possible role of transglutaminase 2 in the nucleus of INS-1E and of cells of human pancreatic islets. J. Proteom. 2014, 96, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ergülen, E.; Bécsi, B.; Csomós, I.; Fésüs, L.; Kanchan, K. Identification of DNAJA1 as a novel interacting partner and a substrate of human transglutaminase 2. Biochem. J. 2016, 473, 3889–3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, B.; Park, H.; Lee, S.; Li, Y.; Choi, J.M.; Lee, J.Y.; Kim, J.; Choi, Y.D.; Kwon, Y.G.; Lee, H.W.; et al. CHIP-mediated degradation of transglutaminase 2 negatively regulates tumor growth and angiogenesis in renal cancer. Oncogene 2016, 35, 3718–3728. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Brunsting, J.F.; Lambert, H.; Landry, J.; Kampinga, H.H. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J. Biol. Chem. 2009, 284, 5523–5532. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Narem, A.P. CFTR Chloride Channel in the Apical Compartments: Spatiotemporal Coupling to its Interacting Partners. Integr. Biol. 2010, 2, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Lubamba, B.; Dhooghe, B.; Noel, S.; Leal, T. Cystic fibrosis: Insight into CFTR pathophysiology and pharmacotherapy. Clin. Biochem. 2012, 45, 1132–1144. [Google Scholar] [CrossRef]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, E.; Monzani, R.; Villella, V.R.; Esposito, S.; Saluzzo, F.; Rossin, F.; D’Eletto, M.; Tosco, A.; De Gregorio, F.; Izzo, V.; et al. Cysteamine re-establishes the clearance of Pseudomonas aeruginosa by macrophages bearing the cystic fibrosis-relevant F508del-CFTR mutation. Cell Death Dis. 2017, 8, e2544. [Google Scholar] [CrossRef] [Green Version]
- Roth, D.M.; Hutt, D.M.; Tong, J.; Bouchecareilh, M.; Wang, N.; Seeley, T.; Dekkers, J.F.; Beekman, J.M.; Garza, D.; Drew, L.; et al. Modulation of the maladaptive stress response to manage diseases of protein folding. PLoS Biol. 2014, 11, e1001998. [Google Scholar] [CrossRef] [Green Version]
- Ong, T.; Ramsey, B.W. New Therapeutic Approaches to Modulate and Correct Cystic Fibrosis Transmembrane Conductance Regulator. Pediatric Clin. N. Am. 2016, 63, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef]
- Villella, V.R.; Esposito, S.; Ferrari, E.; Monzani, R.; Tosco, A.; Rossin, F.; Castaldo, A.; Silano, M.; Marseglia, G.L.; Romani, L.; et al. Autophagy suppresses the pathogenic immune response to dietary antigens in cystic fibrosis. Cell Death Dis. 2019, 10, 258. [Google Scholar] [CrossRef] [Green Version]
- De Stefano, D.; Villella, V.R.; Esposito, S.; Tosco, A.; Sepe, A.; De Gregorio, F.; Salvadori, L.; Grassia, R.; Leone, C.A.; De Rosa, G.; et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del- CFTR mutation. Autophagy 2014, 10, 2053–2074. [Google Scholar] [CrossRef] [Green Version]
- Tosco, A.; De Gregorio, F.; Esposito, S.; De Stefano, D.; Sana, I.; Ferrari, E.; Sepe, A.; Salvadori, L.; Buonpensiero, P.; Di Pasqua, A.; et al. A novel treatment of cystic fibrosis acting on-target: Cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2016, 23, 1380–1393. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Kuo, T.F.; Tatsukawa, H.; Kojima, S. New insights into the functions and localization of nuclear transglutaminase 2. FEBS J. 2011, 278, 4756–4767. [Google Scholar] [CrossRef]
- Ruan, Q.; Johnson, G.V. Transglutaminase 2 in neurodegenerative disorders. Front Biosci. 2007, 12, 891–904. [Google Scholar] [CrossRef] [Green Version]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar]
- Meriin, A.B.; Sherman, M.Y. Role of molecular chaperones in neurodegenerative disorders. Int. J. Hyperth. 2005, 21, 403–419. [Google Scholar] [CrossRef]
- Webster, J.M.; Darling, A.L.; Uversky, V.N.; Blair, L.J. Small Heat Shock Proteins, Big Impact on Protein Aggregation in Neurodegenerative Disease. Front. Pharmacol. 2019, 10, 1047. [Google Scholar] [CrossRef]
- Kim, E.; Wang, B.; Sastry, N.; Masliah, E.; Nelson, P.T.; Cai, H.; Liao, F.F. NEDD4-mediated HSF1 degradation underlies α-synucleinopathy. Hum. Mol. Genet. 2016, 25, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Pastor, R.; Burchfiel, E.T.; Neef, D.W.; Jaeger, A.M.; Cabiscol, E.; McKinstry, S.U.; Doss, A.; Aballay, A.; Lo, D.C.; Akimov, S.S.; et al. Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington’s disease. Nat. Commun. 2017, 8, 14405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.Q.; Wang, X.L.; Cao, X.H.; Ye, Z.Y.; Li, L.; Cai, W.Q. Increased heat shock transcription factor 1 in the cerebellum reverses the deficiency of Purkinje cells in Alzheimer’s disease. Brain Res. 2013, 1519, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Khalsa, D.S. Stress, Meditation, and Alzheimer’s Disease Prevention: Where The Evidence Stands. J. Alzheimer’s Dis. 2015, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Shin, M.S.; Ji, E.S.; Kim, T.W.; Cho, H.S.; Kim, C.J.; Jang, M.S.; Kim, T.W.; Kim, B.K.; Kim, D.H. Treadmill exercise improves motor coordination through ameliorating Purkinje cell loss in amyloid beta23-35-induced Alzheimer’s disease rats. J. Exerc. Rehabil. 2014, 10, 258–264. [Google Scholar] [CrossRef]
- Kozuki, M.; Kurata, T.; Miyazaki, K.; Morimoto, N.; Ohta, Y.; Ikeda, Y.; Abe, K. Atorvastatin and pitavastatin protect cerebellar Purkinje cells in AD model mice and preserve the cytokines MCP-1 and TNF-α. Brain Res. 2011, 1388, 32–38. [Google Scholar] [CrossRef]
- Adachi, H.; Katsuno, M.; Waza, M.; Minamiyama, M.; Tanaka, F.; Sobue, G. Heat shock proteins in neurodegenerative diseases: Pathogenic roles and therapeutic implications. Int. J. Hyperth. 2009, 25, 647–654. [Google Scholar] [CrossRef]
- Papp, E.; Csermely, P. Chemical chaperones: Mechanisms of action and potential use. Handb. Exp. Pharmacol. 2006, 172, 405–416. [Google Scholar]
- Pujols, J.; Peña-Díaz, S.; Pallarès, I.; Ventura, S. Chemical Chaperones as Novel Drugs for Parkinson’s Disease. Trends Mol. Med. 2020, 26, 408–421. [Google Scholar] [CrossRef]
- Santagata, S.; Hu, R.; Lin, N.U.; Mendillo, M.L.; Collins, L.C.; Hankinson, S.E.; Schnitt, S.J.; Whitesell, L.; Tamimi, R.M.; Lindquist, S. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 18378–18383. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Tian, H.; Chen, G. Upregulation of Nuclear Heat Shock Factor 1 Contributes to Tumor Angiogenesis and Poor Survival in Patients With Non-Small Cell Lung Cancer. Ann. Thorac. Surg. 2015, 100, 465–472. [Google Scholar] [CrossRef]
- Björk, J.K.; Ahonen, I.; Mirtti, T.; Erickson, A.; Rannikko, A.; Bützow, A.; Nordling, S.; Lundin, J.; Lundin, M.; Sistonen, L. Increased HSF1 expression predicts shorter disease-specific survival of prostate cancer patients following radical prostatectomy. Oncotarget 2018, 9, 31200–31213. [Google Scholar] [CrossRef] [Green Version]
- Levi-Galibov, O.; Lavon, H.; Wassermann-Dozorets, R.; Pevsner-Fischer, M.; Mayer, S.; Wershof, E.; Stein, Y.; Brown, L.E.; Zhang, W.; Friedman, G.; et al. Heat Shock Factor 1-dependent extracellular matrix remodeling mediates the transition from chronic intestinal inflammation to colon cancer. Nat. Commun. 2020, 11, 6245. [Google Scholar] [CrossRef]
- Fok, J.; Hedayat, S.; Zhang, L.; Aronson, L.I.; Mirabella, F.; Pawlyn, C.; Bright, M.D.; Wardell, C.P.; Keats, J.J.; De Billy, E.; et al. HSF1 Is Essential for Myeloma Cell Survival and A Promising Therapeutic Target. Clin. Cancer Res. 2018, 24, 2395–2407. [Google Scholar] [CrossRef] [Green Version]
- Dudeja, V.; Chugh, R.K.; Sangwan, V.; Skube, S.J.; Mujumdar, N.R.; Antonoff, M.B.; Dawra, R.K.; Vickers, S.M.; Saluja, A.K. Prosurvival role of heat shock factor 1 in the pathogenesis of pancreatobiliary tumors. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G948–G955. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Moskophidis, D.; Mivechi, N.F. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011, 14, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, S.K.; Xie, Y.; Wang, X.; Khaleque, M.A.; Chou, S.D.; Murshid, A.; Prince, T.; Zhang, Y. Signal Transduction Pathways Leading to Heat Shock Transcription. Signal Transduct. Insights 2010, 2, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Antonietti, P.; Linder, B.; Hehlgans, S.; Mildenberger, I.C.; Burger, M.C.; Fulda, S.; Steinbach, J.P.; Gessler, F.; Rödel, F.; Mittelbronn, M.; et al. Interference with the HSF1/HSP70/BAG3 Pathway Primes Glioma Cells to Matrix Detachment and BH3 Mimetic-Induced Apoptosis. Mol. Cancer Ther. 2017, 16, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Gabai, V.L.; Sherman, M.Y. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 2010, 29, 5204–5213. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, A.T.; Marnett, L.J. HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J. Biol. Chem. 2009, 284, 9176–9183. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; He, H.; Yu, L.; Xia, H.H.; Lin, M.C.; Gu, Q.; Li, M.; Zou, B.; An, X.; Jiang, B.; et al. HSF1 down-regulates XAF1 through transcriptional regulation. J. Biol. Chem. 2006, 281, 2451–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.; Liao, Y.; Zhang, J.; Huang, Q.; Luo, W.; Yu, J.; Gong, J.; Zhou, Y.; Li, X.; Tang, B.; et al. Heat shock factor 1 inhibits the mitochondrial apoptosis pathway by regulating second mitochondria-derived activator of caspase to promote pancreatic tumorigenesis. J. Exp. Clin. Cancer Res. 2017, 36, 64. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, D.; Cao, M.; Ba, J.; Wu, B.; Liu, T.; Nie, C. A study on the biological function of heat shock factor 1 proteins in breast cancer. Oncol. Lett. 2018, 16, 3145–3149. [Google Scholar]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, K.H.; Cao, J.; Tang, Z.; Dai, S.; He, Y.; Sampson, S.B.; Benjamin, I.J.; Dai, C. HSF1 critically attunes proteotoxic stress sensing by mTORC1 to combat stress and promote growth. Nat. Cell Biol. 2016, 18, 527–539. [Google Scholar] [CrossRef]
- Kumar, S.; Tomar, M.S.; Acharya, A. HSF1-mediated regulation of tumor cell apoptosis: A novel target for cancer therapeutics. Future Oncol. 2013, 9, 1573–1586. [Google Scholar] [CrossRef]
- Dietlein, F.; Thelen, L.; Reinhardt, H.C. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Gen. 2014, 30, 326–339. [Google Scholar] [CrossRef]
- Fujimoto, M.; Takii, R.; Takaki, E.; Katiyar, A.; Nakato, R.; Shirahige, K.; Nakai, A. The HSF1-PARP13-PARP1 complex facilitates DNA repair and promotes mammary tumorigenesis. Nat. Commun. 2017, 8, 1638. [Google Scholar] [CrossRef]
- Li, Q.; Martinez, J.D. Loss of HSF1 results in defective radiation-induced G(2) arrest and DNA repair. Radiat. Res. 2011, 176, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.D.; Paullin, T.R.; Aoisa, C.; Menzie, C.J.; Ubaldini, A.; Westerheide, S.D. The Heat Shock Transcription Factor HSF1 Induces Ovarian Cancer Epithelial-Mesenchymal Transition in a 3D Spheroid Growth Model. PLoS ONE 2016, 11, e0168389. [Google Scholar] [CrossRef] [Green Version]
- Xi, C.; Hu, Y.; Buckhaults, P.; Moskophidis, D.; Mivechi, N.F. Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J. Biol. Chem. 2012, 87, 35646–35657. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Chang, R.; Yang, L. Heat shock factor 1 promotes invasion and metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer 2012, 118, 1782–1794. [Google Scholar] [CrossRef]
- Kourtis, N.; Moubarak, R.S.; Aranda-Orgilles, B.; Lui, K.; Aydin, I.T.; Trimarchi, T.; Darvishian, F.; Salvaggio, C.; Zhong, J.; Bhatt, K.; et al. FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat. Cell Biol. 2015, 17, 322–332. [Google Scholar] [CrossRef]
- Nakamura, Y.; Fujimoto, M.; Fukushima, S.; Nakamura, A.; Hayashida, N.; Takii, R.; Takaki, E.; Nakai, A.; Muto, M. Heat shock factor 1 is required for migration and invasion of human melanoma in vitro and in vivo. Cancer Lett. 2014, 354, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Qian, W.; Li, J.; Jiang, Z.; Cheng, L.; Yan, B.; Cao, J.; Sun, L.; Zhou, C.; Lei, M.; et al. Loss of AMPK activation promotes the invasion and metastasis of pancreatic cancer through an HSF1-dependent pathway. Mol. Oncol. 2017, 11, 1475–1492. [Google Scholar] [CrossRef]
- Tchénio, T.; Havard, M.; Martinez, L.A.; Dautry, F. Heat shock-independent induction of multidrug resistance by heat shock factor 1. Mol. Cell. Biol. 2006, 26, 580–591. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.L.; Paw, I.; Dewhirst, M.W.; Lo, H.W. Akt phosphorylates and activates HSF-1 independent of heat shock, leading to Slug overexpression and epithelial-mesenchymal transition (EMT) of HER2-overexpressing breast cancer cells. Oncogene 2015, 34, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Feng, B.; Meng, Y.; Wang, J.; Geng, B.; Cui, Q.; Zhang, H.; Yang, Y.; Yang, J. FAM3C-YY1 axis is essential for TGFβ-promoted proliferation and migration of human breast cancer MDA-MB-231 cells via the activation of HSF1. J. Cell. Mol. Med. 2019, 23, 3464–3475. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Santagata, S.; Mendillo, M.L.; Sholl, L.M.; Ben-Aharon, I.; Beck, A.H.; Dias-Santagata, D.; Koeva, M.; Stemmer, S.M.; Whitesell, L.; et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 2014, 158, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, N.; Ranftl, R.; Chicherova, I.; Slaven, N.D.; Moeendarbary, E.; Farrugia, A.J.; Lam, M.; Semiannikova, M.; Westergaard, M.; Tchou, J.; et al. Dickkopf-3 links HSF1 and YAP/TAZ signalling to control aggressive behaviours in cancerassociated fibroblasts. Nat. Commun. 2019, 10, 130. [Google Scholar] [CrossRef]
- Huang, L.; Xu, A.M.; Liu, W. Transglutaminase 2 in cancer. Am. J. Cancer Res. 2015, 5, 2756–2776. [Google Scholar] [PubMed]
- Tabolacci, C.; De Martino, A.; Mischiati, C.; Feriotto, G.; Beninati, S. The Role of Tissue Transglutaminase in Cancer Cell Initiation, Survival and Progression. Med. Sci. 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.; Qin, X.Y.; Furutani, Y. Transglutaminase 2 as a Marker for Inflammation and Therapeutic Target in Sepsis. Int. J. Mol. Sci. 2021, 22, 1897. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Korponay-Szabó, I.; Király, R.; Sarang, Z.; Tsay, G.J. Transglutaminase 2 in human diseases. BioMedicine 2017, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Chrobok, N.L.; Sestito, C.; Wilhelmus, M.M.; Drukarch, B.; van Dam, A.M. Is monocyte- and macrophage-derived tissue transglutaminase involved in inflammatory processes? Amino Acids 2017, 49, 441–452. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Mehta, K. Tissue transglutaminase, inflammation, and cancer: How intimate is the relationship? Amino Acids 2013, 44, 81–88. [Google Scholar] [CrossRef]
- Brown, K.D. Transglutaminase 2 and NF-κB: An odd couple that shapes breast cancer phenotype. Breast Cancer Res. Treat. 2013, 137, 329–336. [Google Scholar] [CrossRef]
- Bayardo, M.; Punzi, F.; Bondar, C.; Chopita, N.; Chirdo, F. Transglutaminase 2 expression is enhanced synergistically by interferon-γ and tumour necrosis factor-α in human small intestine. Clin. Exp. Immunol. 2012, 168, 95–104. [Google Scholar] [CrossRef]
- Nunes, I.; Gleizes, P.E.; Metz, C.N.; Rifkin, D.B. Latent transforming growth factor-beta binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-beta. J. Cell. Bio. 1997, 136, 1151–1163. [Google Scholar] [CrossRef]
- Cao, L.; Shao, M.; Schilder, J.; Guise, T.; Mohammad, K.S.; Matei, D. Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. [Google Scholar] [CrossRef] [Green Version]
- Karicheva, O.; Rodriguez-Vargas, J.M.; Wadier, N.; Martin-Hernandez, K.; Vauchelles, R.; Magroun, N.; Tissier, A.; Schreiber, V.; Dantzer, F. PARP3 controls TGFβ and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget 2016, 7, 64109–64123. [Google Scholar] [CrossRef] [Green Version]
- Ayinde, O.; Wang, Z.; Griffin, M. Tissue transglutaminase induces Epithelial-Mesenchymal-Transition and the acquisition of stem cell like characteristics in colorectal cancer cells. Oncotarget 2017, 8, 20025–20041. [Google Scholar] [CrossRef] [Green Version]
- Tatsukawa, H.; Furutani, Y.; Hitomi, K.; Kojima, S. Transglutaminase 2 has opposing roles in the regulation of cellular functions as well as cell growth and death. Cell Death Dis. 2016, 7, e2244. [Google Scholar] [CrossRef] [Green Version]
- Autuori, F.; Farrace, M.G.; Oliverio, S.; Piredda, L.; Piacentini, M. “Tissue” transglutaminase and apoptosis. Adv. Biochem. Eng. Biotechnol. 1998, 62, 129–136. [Google Scholar]
- Rodolfo, C.; Mormone, E.; Matarrese, P.; Ciccosanti, F.; Farrace, M.G.; Garofano, E.; Piredda, L.; Fimia, G.M.; Malorni, W.; Piacentini, M. Tissue transglutaminase is a multifunctional BH3-only protein. J. Biol. Chem. 2004, 279, 54783–54792. [Google Scholar] [CrossRef] [Green Version]
- Jang, G.Y.; Jeon, J.H.; Cho, S.Y.; Shin, D.M.; Kim, C.W.; Jeong, E.M.; Bae, H.C.; Kim, T.W.; Lee, S.H.; Choi, Y.; et al. Transglutaminase 2 suppresses apoptosis by modulating caspase 3 and NF-kappaB activity in hypoxic tumor cells. Oncogene 2010, 29, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Boehm, J.E.; Singh, U.; Combs, C.; Antonyak, M.A.; Cerione, R.A. Tissue transglutaminase protects against apoptosis by modifying the tumor suppressor protein p110 Rb. J. Biol. Chem. 2002, 277, 20127–20130. [Google Scholar] [CrossRef] [Green Version]
- Erdem, S.; Yegen, G.; Telci, D.; Yildiz, I.; Tefik, T.; Issever, H.; Kilicaslan, I.; Sanli, O. The increased transglutaminase 2 expression levels during initial tumorigenesis predict increased risk of metastasis and decreased disease-free and cancer-specific survivals in renal cell carcinoma. World J. Urol. 2015, 33, 1553–1560. [Google Scholar] [CrossRef]
- Satpathy, M.; Cao, L.; Pincheira, R.; Emerson, R.; Bigsby, R.; Nakshatri, H.; Matei, D. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res. 2007, 67, 7194–7202. [Google Scholar] [CrossRef] [Green Version]
- Mangala, L.S.; Fok, J.Y.; Zorrilla-Calancha, I.R.; Verma, A.; Mehta, K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene 2007, 26, 2459–2470. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.; Guha, S.; Diagaradjane, P.; Kunnumakkara, A.B.; Sanguino, A.M.; Lopez-Berestein, G.; Sood, A.K.; Aggarwal, B.B.; Krishnan, S.; Gelovani, J.G.; et al. Therapeutic significance of elevated tissue transglutaminase expression in pancreatic cancer. Clin. Cancer Res. 2008, 14, 2476–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimov, S.S.; Belkin, A.M. Cell-surface transglutaminase promotes fibronectin assembly via interaction with the gelatin-binding domain of fibronectin: A role in TGFbeta-dependent matrix deposition. J. Cell Sci. 2001, 114, 2989–3000. [Google Scholar] [CrossRef] [PubMed]
- Chau, D.Y.; Collighan, R.J.; Verderio, E.A.; Addy, V.L.; Griffin, M. The cellular response to transglutaminase-cross-linked collagen. Biomaterials 2005, 26, 6518–6529. [Google Scholar] [CrossRef] [PubMed]
- Bagatur, Y.; Ilter Akulke, A.Z.; Bihorac, A.; Erdem, M.; Telci, D. Tissue transglutaminase expression is necessary for adhesion, metastatic potential and cancer stemness of renal cell carcinoma. Cell Adh. Migr. 2018, 12, 138–151. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Perez, M.; Caja, S.; Melino, G.; Johnson, T.S.; Lindfors, K.; Griffin, M. A novel extracellular role for tissue transglutaminase in matrix-bound VEGF-mediated angiogenesis. Cell Death Dis. 2013, 4, e808. [Google Scholar] [CrossRef] [Green Version]
- Jedlicka, P.; Mortin, M.A.; Wu, C. Multiple functions of Drosophila heat shock transcription factor in vivo. EMBO J. 1997, 16, 2452–2462. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Zuo, X.; Davis, A.A.; McMillan, D.R.; Curry, B.B.; Richardson, J.A.; Benjamin, I.J. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 1999, 8, 5943–5952. [Google Scholar] [CrossRef] [Green Version]
- Trinklein, N.D.; Murray, J.I.; Hartman, S.J.; Botstein, D.; Myers, R.M. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol. Biol. Cell. 2004, 15, 1254–1261. [Google Scholar] [CrossRef]
- Li, J.; Chauve, L.; Phelps, G.; Brielmann, R.M.; Morimoto, R.I. E2F coregulates an essential HSF developmental program that is distinct from the heat-shock response. Genes Dev. 2016, 30, 2062–2075. [Google Scholar] [CrossRef] [Green Version]
- Metchat, A.; Akerfelt, M.; Bierkamp, C.; Delsinne, V.; Sistonen, L.; Alexandre, H.; Christians, E.S. Mammalian heat shock factor 1 is essential for oocyte meiosis and directly regulates Hsp90alpha expression. J. Biol. Chem. 2009, 284, 9521–9528. [Google Scholar] [CrossRef] [Green Version]
- Christians, E.; Davis, A.A.; Thomas, S.D.; Benjamin, I.J. Maternal effect of Hsf1 on reproductive success. Nature 2000, 407, 693–694. [Google Scholar] [CrossRef]
- Le Masson, F.; Razak, Z.; Kaigo, M.; Audouard, C.; Charry, C.; Cooke, H.; Westwood, J.T.; Christians, E.S. Identification of heat shock factor 1 molecular and cellular targets during embryonic and adult female meiosis. Mol. Cell. Biol. 2011, 3116, 3410–3423. [Google Scholar] [CrossRef] [Green Version]
- Bierkamp, C.; Luxey, M.; Metchat, A.; Audouard, C.; Dumollard, R.; Christians, E. Lack of maternal Heat Shock Factor 1 results in multiple cellular and developmental defects, including mitochondrial damage and altered redox homeostasis, and leads to reduced survival of mammalian oocytes and embryos. Dev. Biol. 2010, 339, 338–353. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.D.; Saraiva, M.J. Enlarged ventricles, astrogliosis and neurodegeneration in heat shock factor 1 null mouse brain. Neuroscience 2004, 126, 657–663. [Google Scholar] [CrossRef]
- Homma, S.; Jin, X.; Wang, G.; Tu, N.; Min, J.; Yanasak, N.; Mivechi, N.F. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J. Neurosci. 2007, 27, 7974–7986. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Hara, K.; Kobayashi, A.; Fujimoto, M.; Otsuki, K.; Yamagata, H.; Hobara, T.; Abe, N.; Higuchi, F.; Shibata, T.; et al. Impaired hippocampal spinogenesis and neurogenesis and altered affective behavior in mice lacking heat shock factor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 1681–1686. [Google Scholar] [CrossRef] [Green Version]
- Takaki, E.; Fujimoto, M.; Sugahara, K.; Nakahari, T.; Yonemura, S.; Tanaka, Y.; Hayashida, N.; Inouye, S.; Takemoto, T.; Yamashita, H.; et al. Maintenance of olfactory neurogenesis requires HSF1, a major heat shock transcription factor in mice. J. Biol. Chem. 2006, 281, 4931–4937. [Google Scholar] [CrossRef] [Green Version]
- Varodayan, F.P.; Pignataro, L.; Harrison, N.L. Alcohol induces synaptotagmin 1 expression in neurons via activation of heat shock factor 1. Neuroscience 2011, 193, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Varodayan, F.P.; Harrison, N.L. HSF1 transcriptional activity mediates alcohol induction of Vamp2 expression and GABA release. Front. Integ. Neurosci. 2013, 7, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Tan, S.; Zheng, H.; Liu, M.; Chen, G.; Zhang, H.; Wang, K.; Tan, S.; Zhou, J.; Xiao, X.Z. HSF1 functions as a transcription regulator for Dp71 expression. Cell Stress Chaperones 2015, 20, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Kovács, D.; Sigmond, T.; Hotzi, B.; Bohár, B.; Fazekas, D.; Deák, V.V.; Vellai, T.; Barna, J. HSF1Base: A Comprehensive Database of HSF1 (Heat Shock Factor 1) Target Genes. Int. J. Mol. Sci. 2019, 20, 5815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Hu, Z.; Zhang, Y.; Huang, J.; Yang, X.; Wang, J. Proteomics Analysis of Proteins Interacting with Heat Shock Factor 1 in Squamous Cell Carcinoma of the Cervix. Oncol. Lett. 2019, 18, 2568–2575. [Google Scholar] [CrossRef] [Green Version]
- Min, B.; Chung, K.C. New insight into transglutaminase 2 and link to neurodegenerative diseases. BMB Rep. 2018, 51, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrieri, M.L.; Dicitore, A.; Benevento, R.; Di Maio, M.; Santoriello, A.; Canonico, S.; Giordano, A.; Stiuso, P. Interplay between membrane lipid peroxidation, transglutaminase activity, and cyclooxygenase 2 expression in the tissue adjoining to breast cancer. J. Cell. Physiol. 2012, 227, 1577–1582. [Google Scholar] [CrossRef]
- Lewis, T.E.; Milam, T.D.; Klingler, D.W.; Rao, P.S.; Jaggi, M.; Smith, D.J.; Hemstreet, G.P.; Balaji, K.C. Tissue transglutaminase interacts with protein kinase A anchor protein 13 in prostate cancer. Urol. Oncol. 2005, 23, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Mi, W.; Cai, J.; Ying, W.; Liu, F.; Lu, H.; Qiao, Y.; Jia, W.; Bi, X.; Lu, N.; et al. Quantitative proteomic signature of liver cancer cells: Tissue transglutaminase 2 could be a novel protein candidate of human hepatocellular carcinoma. J. Proteome Res. 2008, 7, 3847–3859. [Google Scholar] [CrossRef]
- Ashour, A.A.; Gurbuz, N.; Alpay, S.N.; Abdel-Aziz, A.A.; Mansour, A.M.; Huo, L.; Ozpolat, B. Elongation factor-2 kinase regulates TG2/beta1 integrin/Src/uPAR pathway and epithelial-mesenchymal transition mediating pancreatic cancer cells invasion. J. Cell. Mol. Med. 2014, 18, 2235–2251. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Occhigrossi, L.; D’Eletto, M.; Barlev, N.; Rossin, F. The Multifaceted Role of HSF1 in Pathophysiology: Focus on Its Interplay with TG2. Int. J. Mol. Sci. 2021, 22, 6366. https://doi.org/10.3390/ijms22126366
Occhigrossi L, D’Eletto M, Barlev N, Rossin F. The Multifaceted Role of HSF1 in Pathophysiology: Focus on Its Interplay with TG2. International Journal of Molecular Sciences. 2021; 22(12):6366. https://doi.org/10.3390/ijms22126366
Chicago/Turabian StyleOcchigrossi, Luca, Manuela D’Eletto, Nickolai Barlev, and Federica Rossin. 2021. "The Multifaceted Role of HSF1 in Pathophysiology: Focus on Its Interplay with TG2" International Journal of Molecular Sciences 22, no. 12: 6366. https://doi.org/10.3390/ijms22126366
APA StyleOcchigrossi, L., D’Eletto, M., Barlev, N., & Rossin, F. (2021). The Multifaceted Role of HSF1 in Pathophysiology: Focus on Its Interplay with TG2. International Journal of Molecular Sciences, 22(12), 6366. https://doi.org/10.3390/ijms22126366