
JNK Pathway in CNS Pathologies


{kind=link}
{kind=link}
Abstract
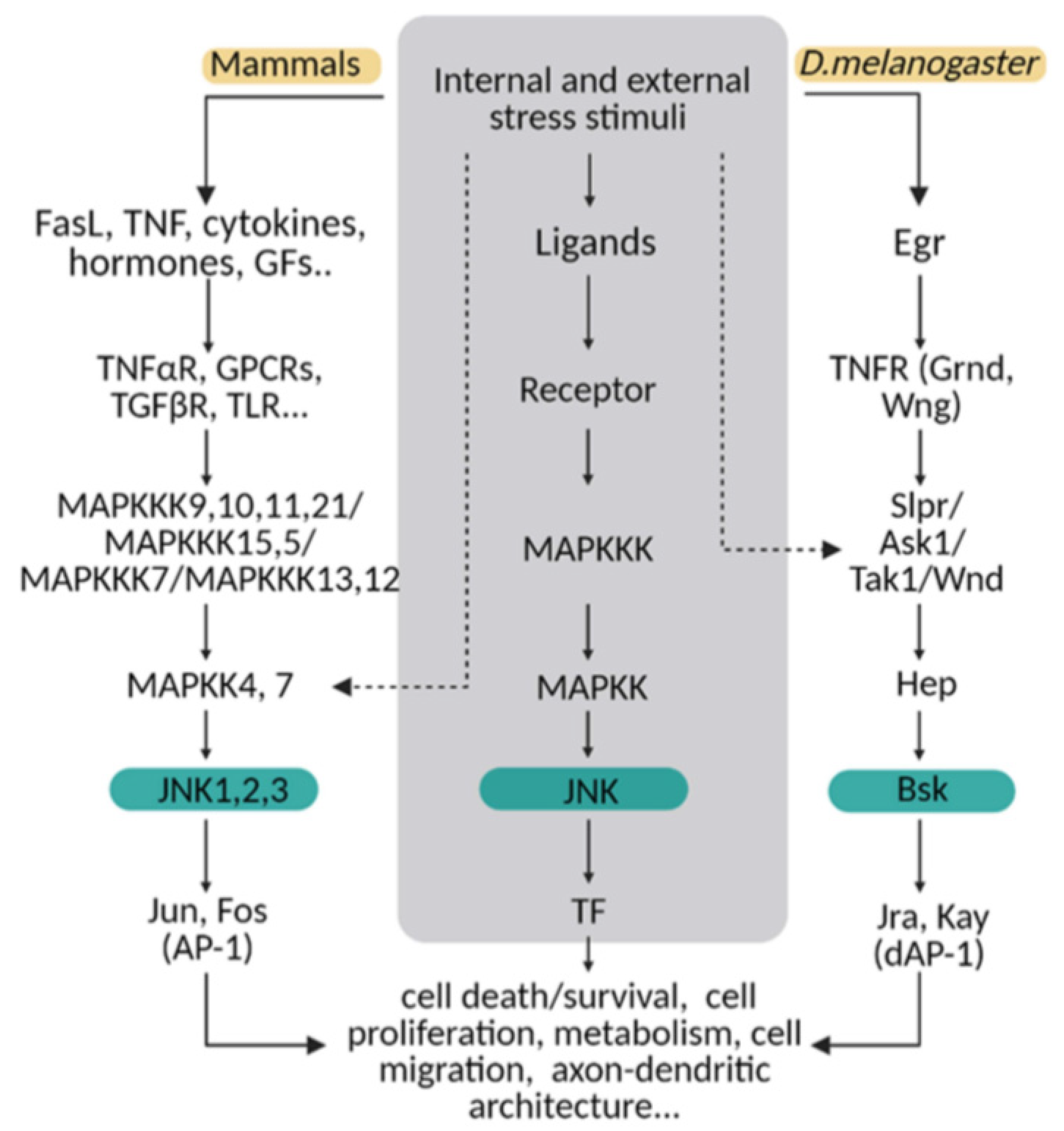
:1. Introduction
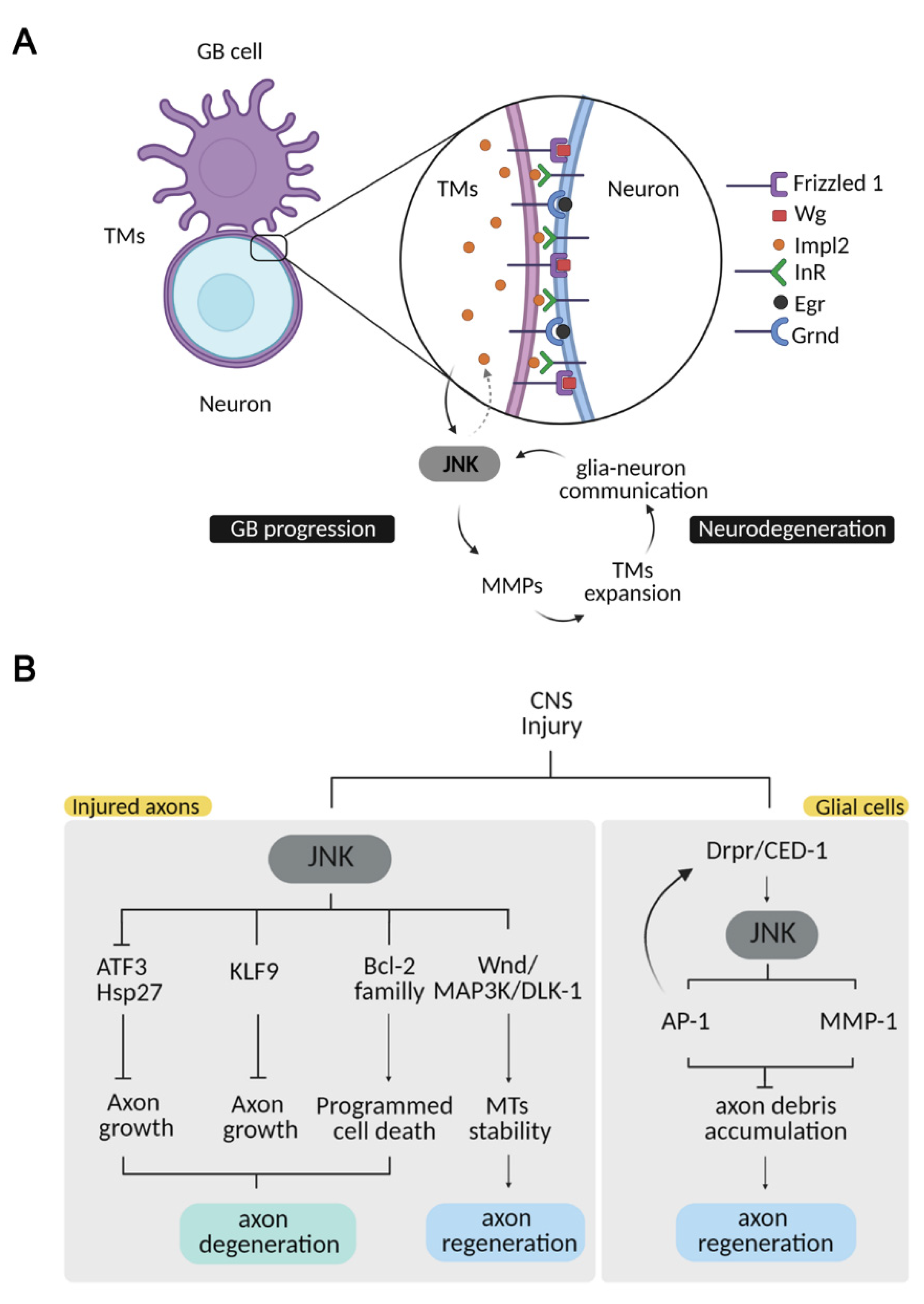
2. JNK in Glioblastoma Tumor Progression
3. JNK in Neurodegenerative Disorders
3.1. Alzheimer’s Disease (AD)
3.2. Parkinson Disease (PD)
3.3. Huntington’s Disease (HD)
4. JNK in Regeneration/Repair after an Injury to the CNS
4.1. JNK Signalling in Glial Cells upon Injury
4.2. JNK in Neurogenesis and Regeneration
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Anfinogenova, N.D.; Quinn, M.T.; Schepetkin, I.A.; Atochin, D.N. Alarmins and c-Jun N-Terminal Kinase (JNK) Signaling in Neuroinflammation. Cells 2020, 9, 2350. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, M.; Kool, J.; Cornelissen-Steijger, P.; Carlotti, F.; Popeijus, H.E.; Van Der Burgt, C.; Janssen, J.M.; Yasui, A.; Hoeben, R.C.; Terleth, C.; et al. DNA damage in transcribed genes induces apoptosis via the JNK pathway and the JNK-phosphatase MKP-1. Oncogene 2005, 24, 7135–7144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamata, H.; Honda, S.I.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Camarillo, C.; Ocampo, E.A.; Casamichana, M.L.; Pérez-Plasencia, C.; Álvarez-Sánchez, E.; Marchat, L.A. Protein kinases and transcription factors activation in response to UV-radiation of skin: Implications for carcinogenesis. Int. J. Mol. Sci. 2012, 13, 142–172. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, I.; Leiva, M.; Sabio, G. The role of stress kinases in metabolic disease. Nat. Rev. Endocrinol. 2020, 16, 697–716. [Google Scholar] [CrossRef] [PubMed]
- Rosette, C.; Karin, M. Ultraviolet light and osmotic stress: Activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 1996, 274, 1194–1197. [Google Scholar] [CrossRef]
- Song, J.J.; Lee, Y.J. Differential activation of the JNK signal pathway by UV irradiation and glucose deprivation. Cell. Signal. 2007, 19, 563–572. [Google Scholar] [CrossRef]
- Zanke, B.W.; Boudreau, K.; Rubie, E.; Winnett, E.; Tibbles, L.A.; Zon, L.; Kyriakis, J.; Liu, F.F.; Woodgett, J.R. The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr. Biol. 1996, 6, 606–613. [Google Scholar] [CrossRef] [Green Version]
- La Marca, J.E.; Richardson, H.E. Two-Faced: Roles of JNK Signalling During Tumourigenesis in the Drosophila Model. Front. Cell Dev. Biol. 2020, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Igaki, T.; Kanda, H.; Yamamoto-Goto, Y.; Kanuka, H.; Kuranaga, E.; Aigaki, T.; Miura, M. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002, 21, 3009–3018. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Yan, M.; Basler, K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 2002, 12, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.A.; Mesquita, D.; Ingaramo, M.C.; Ariel, F.; Milan, M.; Dekanty, A. Eiger/TNFα-mediated Dilp8 and ROS production coordinate intra-organ growth in drosophila. PLoS Genet. 2019, 15, e1008133. [Google Scholar] [CrossRef] [Green Version]
- Igaki, T.; Pastor-Pareja, J.C.; Aonuma, H.; Miura, M.; Xu, T. Intrinsic Tumor Suppression and Epithelial Maintenance by Endocytic Activation of Eiger/TNF Signaling in Drosophila. Dev. Cell 2009, 16, 458–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stronach, B.E.; Perrimon, N. Stress signaling in Drosophila. Oncogene 1999, 18, 6172–6182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Thomas, J.M.; Li, T.; Lee, Y.; Liu, Z.; Smith, W.W. The Drosophila hep pathway mediates Lrrk2-induced neurodegeneration. Biochem. Cell Biol. 2018, 96, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The Many and Varied Substrates of the c-Jun N-Terminal Kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [Green Version]
- Bogoyevitch, M.A. The isoform-specific functions of the c-Jun N-terminal kinases (JNKs): Differences revealed by gene targeting. BioEssays 2006, 28, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, X.; Borsello, T. The JNK signalling transduction pathway in the brain. Front. Biosci. 2012, 4, 2110–2120. [Google Scholar] [CrossRef]
- Yamasaki, T.; Kawasaki, H.; Nishina, H. Diverse Roles of JNK and MKK Pathways in the Brain. J. Signal Transduct. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Agnès, F.; Suzanne, M.; Noselli, S. The Drosophila JNK pathway controls the morphogenesis of imaginal discs during metamorphosis. Development 1999, 126, 5453–5462. [Google Scholar] [PubMed]
- Horiuchi, D.; Collins, C.A.; Bhat, P.; Barkus, R.V.; DiAntonio, A.; Saxton, W.M. Control of a Kinesin-Cargo Linkage Mechanism by JNK Pathway Kinases. Curr. Biol. 2007, 17, 1313–1317. [Google Scholar] [CrossRef] [Green Version]
- Igaki, T. Correcting developmental errors by apoptosis: Lessons from Drosophila JNK signaling. Apoptosis 2009, 14, 1021–1028. [Google Scholar] [CrossRef]
- Nikoloudaki, G.; Brooks, S.; Peidl, A.P.; Tinney, D.; Hamilton, D.W. JNK Signaling as a Key Modulator of Soft Connective Tissue Physiology, Pathology, and Healing. Int. J. Mol. Sci. 2020, 21, 1015. [Google Scholar] [CrossRef] [Green Version]
- Tafesh-Edwards, G.; Eleftherianos, I. JNK signaling in Drosophila immunity and homeostasis. Immunol. Lett. 2020, 226, 7–11. [Google Scholar] [CrossRef]
- Musi, C.A.; Agrò, G.; Santarella, F.; Iervasi, E.; Borsello, T. JNK3 as Therapeutic Target and Biomarker in Neurodegenerative and Neurodevelopmental Brain Diseases. Cells 2020, 9, 2190. [Google Scholar] [CrossRef]
- Portela, M.; Venkataramani, V.; Fahey-Lozano, N.; Seco, E.; Losada-Perez, M.; Winkler, F.; Casas-Tintó, S. Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol. 2019, 17, e3000545. [Google Scholar] [CrossRef]
- Curran, B.P.; Murray, H.J.; O’Connor, J.J. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1β and long-term depression in the rat dentate gyrus in vitro. Neuroscience 2003, 118, 347–357. [Google Scholar] [CrossRef]
- Schellino, R.; Boido, M.; Vercelli, A. JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death. Cells 2019, 8, 1576. [Google Scholar] [CrossRef] [Green Version]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, e273–e281. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical Report: Primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro. Oncol. 2015, 17, iv1–iv62. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [Green Version]
- Kitanaka, C.; Sato, A.; Okada, M. JNK Signaling in the Control of the Tumor-Initiating Capacity Associated with Cancer Stem Cells. Genes Cancer 2013, 4, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, K.I.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 134, pp. 381–397. ISBN 9780128029978. [Google Scholar]
- Li, J.Y.; Wang, H.; May, S.; Song, X.; Fueyo, J.; Fuller, G.N.; Wang, H. Constitutive activation of c-Jun N-terminal kinase correlates with histologic grade and EGFR expression in diffuse gliomas. J. Neurooncol. 2008, 88, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Manole, S.; Richards, E.J.; Meyer, A.S. JNK pathway activation modulates acquired resistance to EGFR/HER2-targeted therapies. Cancer Res. 2016, 76, 5219–5228. [Google Scholar] [CrossRef] [Green Version]
- Portela, M.; Mitchell, T.; Casas-Tintó, S. Cell-to-cell communication mediates glioblastoma progression in Drosophila. Biol. Open 2020, 9, bio053405. [Google Scholar] [CrossRef]
- Casas-Tintó, S.; Portela, M. Cytonemes, Their Formation, Regulation, and Roles in Signaling and Communication in Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 5641. [Google Scholar] [CrossRef] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Jarabo, P.; de Pablo, C.; Herranz, H.; Martín, F.A.; Casas-Tintó, S. Insulin signaling mediates neurodegeneration in glioma. Life Sci. Alliance 2021, 4, e202000693. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, K.; Clavreul, A.; Lagarce, F. Toward an effective strategy in glioblastoma treatment. Part I: Resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov. Today 2015, 20, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Vo, V.A.; Lee, J.-W.; Lee, H.J.; Chun, W.; Lim, S.Y.; Kim, S.-S. Inhibition of JNK potentiates temozolomide-induced cytotoxicity in U87MG glioblastoma cells via suppression of Akt phosphorylation. Anticancer Res. 2014, 34. [Google Scholar]
- Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Sakaki, H.; Seino, S.; Seino, M.; Suzuki, S.; Kitanaka, C. The novel JNK inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo. Oncotarget 2016, 7, 27021–27032. [Google Scholar] [CrossRef] [Green Version]
- Messoussi, A.; Feneyrolles, C.; Bros, A.; Deroide, A.; Daydé-Cazals, B.; Chevé, G.; Van Hijfte, N.; Fauvel, B.; Bougrin, K.; Yasri, A. Recent progress in the design, study, and development of c-Jun N-terminal kinase inhibitors as anticancer agents. Chem. Biol. 2014, 21, 1433–1443. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Nakano, R.; Nakayama, T.; Sugiya, H. Biological Properties of JNK3 and Its Function in Neurons, Astrocytes, Pancreatic β-Cells and Cardiovascular Cells. Cells 2020, 9, 1802. [Google Scholar] [CrossRef] [PubMed]
- Sclip, A.; Tozzi, A.M.A.; Abaza, A.; Cardinetti, D.; Colombo, I.; Calabresi, P.A.; Salmona, M.; Welker, E.; Borsello, T. c-Jun N-terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. Cell Death Dis. 2014, 5, e1019. [Google Scholar] [CrossRef] [PubMed]
- Hepp Rehfeldt, S.C.; Majolo, F.; Goettert, M.I.; Laufer, S. c-Jun N-Terminal Kinase Inhibitors as Potential Leads for New Therapeutics for Alzheimer’s Diseases. Int. J. Mol. Sci. 2020, 21, 9677. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Nguyen, T.V.V.; Pike, C.J. β-Amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. 2005, 25, 1149–1158. [Google Scholar] [CrossRef]
- Yoshida, H.; Hastie, J.; Mclauchlan, H.; Cohen, P.; Goedert, M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J. Neurochem. 2004, 90, 352–358. [Google Scholar] [CrossRef]
- Bozyczko-Coyne, D.; Saporito, M.S.; Hudkins, R.L. Targeting the JNK pathway for therapeutic benefit in CNS disease. Curr. Drug Targets. CNS Neurol. Disord. 2002, 1, 31–49. [Google Scholar] [CrossRef]
- Gourmaud, S.; Paquet, C.; Dumurgier, J.; Pace, C.; Bouras, C.; Gray, F.; Laplanche, J.L.; Meurs, E.F.; Mouton-Liger, F.; Hugon, J. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: Links to cognitive decline. J. Psychiatry Neurosci. 2015, 40, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Borsello, T.; Forloni, G. JNK Signalling: A Possible Target to Prevent Neurodegeneration. Curr. Pharm. Des. 2007, 13, 1875–1886. [Google Scholar] [CrossRef]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.S.; Abel, G.; Klintworth, H.; Flavell, R.A.; Xia, Z. JNK3 mediates paraquat-and rotenone-induced dopaminergic neuron death. J. Neuropathol. Exp. Neurol. 2010, 69, 511–520. [Google Scholar] [CrossRef]
- Bekker, M.; Abrahams, S.; Loos, B.; Bardien, S. Can the interplay between autophagy and apoptosis be targeted as a novel therapy for Parkinson’s disease? Neurobiol. Aging 2021, 100, 91–105. [Google Scholar] [CrossRef]
- Peng, J.; Andersen, J.K. The role of c-Jun N-terminal kinase (JNK) in Parkinson’s disease. IUBMB Life 2003, 55, 267–271. [Google Scholar] [CrossRef]
- Wang, W.; Ma, C.; Mao, Z.; Li, M. JNK inhibition as a potential strategy in treating Parkinson’s disease. Drug News Perspect. 2004, 17, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, H.; Wu, B. RIPK1 inhibitor ameliorates the MPP+/MPTP-induced Parkinson’s disease through the ASK1/JNK signalling pathway. Brain Res. 2021, 1757, 147310. [Google Scholar] [CrossRef] [PubMed]
- Spigolon, G.; Cavaccini, A.; Trusel, M.; Tonini, R.; Fisone, G. cJun N-terminal kinase (JNK) mediates cortico-striatal signaling in a model of Parkinson’s disease. Neurobiol. Dis. 2018, 110, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Sieradzan, K.A.; Mann, D.M.A. The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.A.; You, Y.M.; Pollema, S.L.; Kaminska, A.; Liu, K.; Yoshioka, K.; Björkblom, B.; Coffey, E.T.; Bagnato, C.; Han, D.; et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 2009, 12, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Perrin, V.; Dufour, N.; Raoul, C.; Hassig, R.; Brouillet, E.; Aebischer, P.; Luthi-Carter, R.; Déglon, N. Implication of the JNK pathway in a rat model of Huntington’s disease. Exp. Neurol. 2009, 215, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, C.; Kanje, M. Retrograde axonal transport of JNK signaling molecules influence injury induced nuclear changes in p-c-Jun and ATF3 in adult rat sensory neurons. Mol. Cell. Neurosci. 2005, 29, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Clark, S.; Bronfman, F.C.; Deppmann, C.D.; Carter, B.D. Long-distance regressive signaling in neural development and disease. Wiley Interdiscip. Rev. Dev. Biol. 2021, 10, e382. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.M.; Doherty, J.; Hackett, R.; Freeman, M.R. The c-Jun kinase signaling cascade promotes glial engulfment activity through activation of draper and phagocytic function. Cell Death Differ. 2013, 20, 1140–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syc-Mazurek, S.B.; Libby, R.T. Axon injury signaling and compartmentalized injury response in glaucoma. Prog. Retin. Eye Res. 2019, 73, 100769. [Google Scholar] [CrossRef]
- Apara, A.; Galvao, J.; Wang, Y.; Blackmore, M.; Trillo, A.; Iwao, K.; Brown, D.P.; Fernandes, K.A.; Huang, A.; Nguyen, T.; et al. KLF9 and JNK3 Interact to Suppress Axon Regeneration in the Adult CNS. J. Neurosci. 2017, 37, 9632–9644. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zheng, B. Multitasking: Dual Leucine Zipper-Bearing Kinases in Neuronal Development and Stress Management Shall we add “MAPK” or “MAP3K”? Or both? Annu. Rev. Cell Dev. Biol. 2019, 35, 501–521. [Google Scholar] [CrossRef]
- Valakh, V.; Frey, E.; Babetto, E.; Walker, L.J.; DiAntonio, A. Cytoskeletal disruption activates the DLK/JNK pathway, which promotes axonal regeneration and mimics a preconditioning injury. Neurobiol. Dis. 2015, 77, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Wang, X.; Ewanek, R.; Bhat, P.; DiAntonio, A.; Collins, C.A. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J. Cell Biol. 2010, 191, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Collins, C.A. A Conditioning Lesion Protects Axons from Degeneration via the Wallenda/DLK MAP Kinase Signaling Cascade. J. Neurosci. 2012, 32, 610–615. [Google Scholar] [CrossRef]
- Huntwork-Rodriguez, S.; Wang, B.; Watkins, T.; Ghosh, A.S.; Pozniak, C.D.; Bustos, D.; Newton, K.; Kirkpatrick, D.S.; Lewcock, J.W. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J. Cell Biol. 2013, 202, 747–763. [Google Scholar] [CrossRef] [Green Version]
- Hirai, S.-I.; Banba, Y.; Satake, T.; Ohno, S. Axon Formation in Neocortical Neurons Depends on Stage-Specific Regulation of Microtubule Stability by the Dual Leucine Zipper Kinase-c-Jun N-Terminal Kinase Pathway. J. Neurosci. 2011, 31, 6468–6480. [Google Scholar] [CrossRef]
- Simard-Bisson, C.; Bidoggia, J.; Larouche, D.; Guérin, S.L.; Blouin, R.; Hirai, S.-I.; Germain, L. A Role for DLK in Microtubule Reorganization to the Cell Periphery and in the Maintenance of Desmosomal and Tight Junction Integrity. J. Investig. Dermatol. 2017, 137, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Hellal, F.; Hurtado, A.; Ruschel, J.; Flynn, K.C.; Laskowski, C.J.; Umlauf, M.; Kapitein, L.C.; Strikis, D.; Lemmon, V.; Bixby, J.; et al. Microtubule Stabilization Reduces Scarring and Causes Axon Regeneration after Spinal Cord Injury. Science 2011, 331, 928–931. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.E.; Cho, Y.; Beirowski, B.; Milbrandt, J.; Cavalli, V.; DiAntonio, A. Dual Leucine Zipper Kinase is Required for Retrograde Injury Signaling and Axonal Regeneration. Neuron 2012, 74, 1015–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, S.-H.; Apple, E.C.; Liu, Z.; Chen, L. Age-dependent autophagy induction after injury promotes axon regeneration by limiting NOTCH. Autophagy 2020, 16, 2052–2068. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.; Sheehan, A.E.; Bradshaw, R.; Fox, A.N.; Lu, T.-Y.; Freeman, M.R. PI3K Signaling and Stat92E Converge to Modulate Glial Responsiveness to Axonal Injury. PLoS Biol. 2014, 12, e1001985. [Google Scholar] [CrossRef]
- Casas-Tintó, S.; Lolo, F.-N.; Moreno, E. Active JNK-dependent secretion of Drosophila Tyrosyl-tRNA synthetase by loser cells recruits haemocytes during cell competition. Nat. Commun. 2015, 6, 10022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.I.; Timmons, A.K.; Klein, A.P.; Pritchett, T.L.; Welch, E.; Meehan, T.L.; Li, C.; McCall, K. Draper acts through the JNK pathway to control synchronous engulfment of dying germline cells by follicular epithelial cells. Development 2012, 139, 4029–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 8, 28. [Google Scholar] [CrossRef]
- Purice, M.D.; Ray, A.; Münzel, E.J.; Pope, B.J.; Park, D.J.; Speese, S.D.; Logan, M.A. A novel Drosophila injury model reveals severed axons are cleared through a Draper/MMP-1 signaling cascade. eLife 2017, 6, e23611. [Google Scholar] [CrossRef]
- Chiu, H.; Zou, Y.; Suzuki, N.; Hsieh, Y.-W.; Chuang, C.-F.; Wu, Y.-C.; Chang, C. Engulfing cells promote neuronal regeneration and remove neuronal debris through distinct biochemical functions of CED-1. Nat. Commun. 2018, 9, 4842. [Google Scholar] [CrossRef]
- Li, X.; Peng, Z.; Long, L.; Lu, X.; Zhu, K.; Tuo, Y.; Chen, N.; Zhao, X.; Wang, L.; Wan, Y. Transplantation of Wnt5a-modified NSCs promotes tissue repair and locomotor functional recovery after spinal cord injury. Exp. Mol. Med. 2020, 52, 2020–2033. [Google Scholar] [CrossRef]
- Blakely, B.D.; Bye, C.R.; Fernando, C.V.; Prasad, A.A.; Pasterkamp, R.J.; Macheda, M.L.; Stacker, S.A.; Parish, C.L. Ryk, a Receptor Regulating Wnt5a-Mediated Neurogenesis and Axon Morphogenesis of Ventral Midbrain Dopaminergic Neurons. Stem Cells Dev. 2013, 22, 2132–2144. [Google Scholar] [CrossRef]
- Jang, S.; Park, J.-S.; Jeong, H.-S. Neural Differentiation of Human Adipose Tissue-Derived Stem Cells Involves Activation of the Wnt5a/JNK Signalling. Stem Cells Int. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kang, M.-J.; Han, J.-S. Interleukin-1 beta promotes neuronal differentiation through the Wnt5a/RhoA/JNK pathway in cortical neural precursor cells. Mol. Brain 2018, 11, 39. [Google Scholar] [CrossRef]
- Castro-Torres, R.D.; Landa, J.; Rabaza, M.; Busquets, O.; Olloquequi, J.; Ettcheto, M.; Beas-Zarate, C.; Folch, J.; Camins, A.; Auladell, C.; et al. JNK Isoforms Are Involved in the Control of Adult Hippocampal Neurogenesis in Mice, Both in Physiological Conditions and in an Experimental Model of Temporal Lobe Epilepsy. Mol. Neurobiol. 2019, 56, 5856–5865. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.; Marchisella, F.; Ortega-Martinez, S.; Hollos, P.; Eerola, K.; Komulainen, E.; Kulesskaya, N.; Freemantle, E.; Fagerholm, V.; Savontous, E.; et al. JNK1 controls adult hippocampal neurogenesis and imposes cell-autonomous control of anxiety behaviour from the neurogenic niche. Mol. Psychiatry 2018, 23, 362–374. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de los Reyes Corrales, T.; Losada-Pérez, M.; Casas-Tintó, S. JNK Pathway in CNS Pathologies. Int. J. Mol. Sci. 2021, 22, 3883. https://doi.org/10.3390/ijms22083883
de los Reyes Corrales T, Losada-Pérez M, Casas-Tintó S. JNK Pathway in CNS Pathologies. International Journal of Molecular Sciences. 2021; 22(8):3883. https://doi.org/10.3390/ijms22083883
Chicago/Turabian Stylede los Reyes Corrales, Teresa, María Losada-Pérez, and Sergio Casas-Tintó. 2021. "JNK Pathway in CNS Pathologies" International Journal of Molecular Sciences 22, no. 8: 3883. https://doi.org/10.3390/ijms22083883
APA Stylede los Reyes Corrales, T., Losada-Pérez, M., & Casas-Tintó, S. (2021). JNK Pathway in CNS Pathologies. International Journal of Molecular Sciences, 22(8), 3883. https://doi.org/10.3390/ijms22083883