
Genetic Screen in Adult Drosophila Reveals That dCBP Depletion in Glial Cells Mitigates Huntington Disease Pathology through a Foxo-Dependent Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Genetic Modifiers Can Be Identified in an Adult Glial Inducible Model of HD
2.2. Known Modulators of Neuronal polyQ-Induced Toxicity Exhibit Various Effects in HD Adult Glial Cells
2.3. Impact of the Modulation of Energy Production Pathways in HD Adult Glial Cells
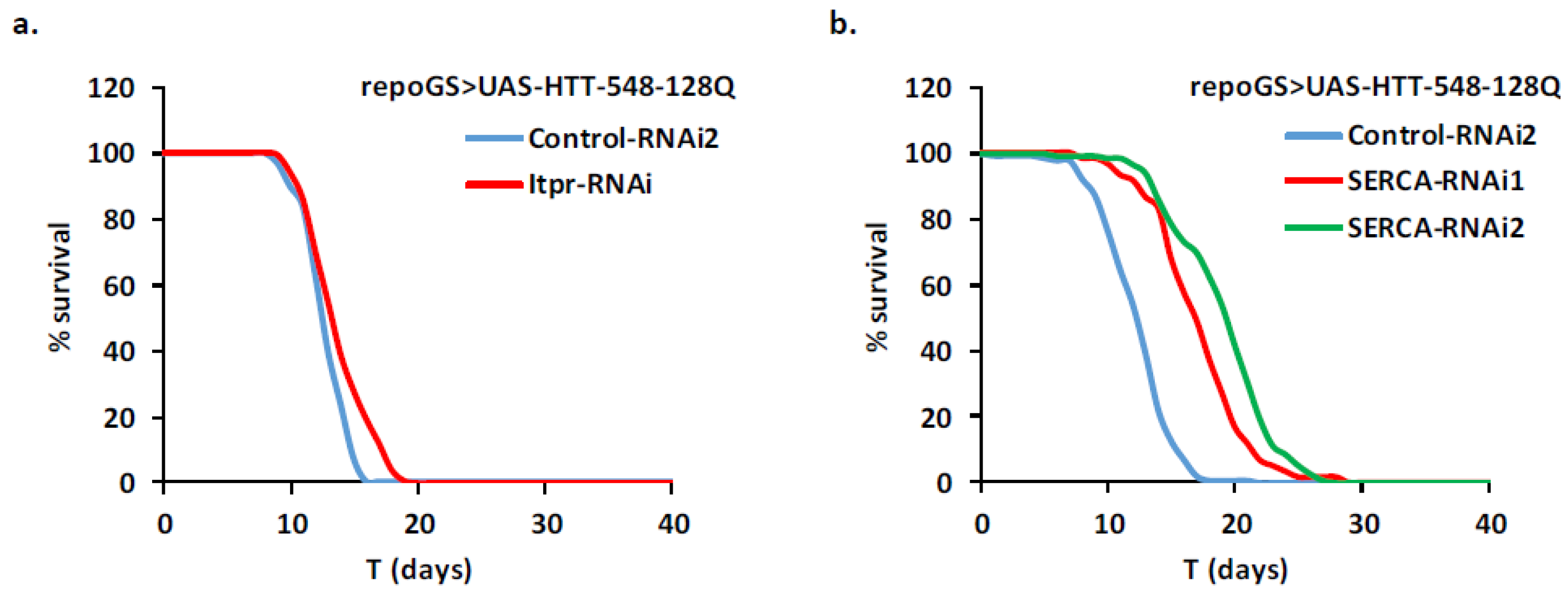
2.4. HD Modifier Genes Involved in Calcium Signaling
2.5. Depletion of dCBP in Glial Cells Mitigates HD Pathology
2.6. Depletion of dCBP in Glial Cells Improves Locomotor Defects in HD
2.7. Glia Depletion of dCBP Rescues HD in a Foxo-Dependent Manner
2.8. Identification of New HD Modifiers in the Wnt Pathway
3. Materials and Methods
3.1. Drosophila Lines and Culture Methods
3.2. Lifespan Experiments
3.3. Quantification of Transcripts by qRT-PCR
3.4. Western Blot Analysis
3.5. Negative Geotaxis Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov. Disord. 2012, 27, 1083–1091. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef]
- Carroll, J.B.; Bates, G.P.; Steffan, J.; Saft, C.; Tabrizi, S.J. Treating the whole body in Huntington’s disease. Lancet Neurol. 2015, 14, 1135–1142. [Google Scholar] [CrossRef]
- Sassone, J.; Colciago, C.; Cislaghi, G.; Silani, V.; Ciammola, A. Huntington’s disease: The current state of research with peripheral tissues. Exp. Neurol. 2009, 219, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Van Der Burg, J.M.M.; Björkqvist, M.; Brundin, P. Beyond the brain: Widespread pathology in Huntington’s disease. Lancet Neurol. 2009, 8, 765–774. [Google Scholar] [CrossRef]
- Travessa, A.M.; Rodrigues, F.B.; Mestre, T.A.; Ferreira, J.J. Fifteen years of clinical trials in Huntington’s Disease: A very low clinical drug development success rate. J. Huntingt. Dis. 2017, 6, 157–163. [Google Scholar] [CrossRef]
- Dash, D.; Mestre, T.A. Therapeutic update on Huntington’s Disease: Symptomatic treatments and emerging disease-modifying therapies. Neurotherapeutics 2020, 17, 1645–1659. [Google Scholar] [CrossRef]
- Farshim, P.P.; Bates, G.P. Mouse models of Huntington’s Disease. Methods Mol. Biol. 2018, 1780, 97–120. [Google Scholar] [CrossRef]
- Figiel, M.; Szlachcic, W.J.; Switonski, P.M.; Gabka, A.; Krzyzosiak, W.J. Mouse models of polyglutamine diseases: Review and data table. Part I. Mol. Neurobiol. 2012, 46, 393–429. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.A.; Smith, G.A. Using Drosophila models of Huntington’s disease as a translatable tool. J. Neurosci. Methods 2016, 265, 89–98. [Google Scholar] [CrossRef]
- Heidari, R.; Monnier, V.; Martin, E.; Tricoire, H. Methylene blue partially rescues heart defects in a drosophila model of Huntington’s Disease. J. Huntingt. Dis. 2015, 4, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Lakra, P.; Aditi, K.; Agrawal, N. Peripheral expression of mutant huntingtin is a critical determinant of weight loss and metabolic disturbances in Huntington’s Disease. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mielcarek, M.; Inuabasi, L.; Bondulich, M.K.; Muller, T.; Osborne, G.F.; Franklin, S.A.; Smith, D.L.; Neueder, A.; Rosinski, J.; Rattray, I.; et al. Dysfunction of the CNS-heart axis in mouse models of Huntington’s Disease. PLoS Genet. 2014, 10, e1004550. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The biology of huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s Disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef] [PubMed]
- Wilton, D.K.; Stevens, B. The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol. Dis. 2020, 143, 104963. [Google Scholar] [CrossRef]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 2019, 11, eaaw8546. [Google Scholar] [CrossRef] [Green Version]
- Gray, M. Astrocytes in Huntington’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 355–381. [Google Scholar] [CrossRef]
- Wood, T.E.; Barry, J.; Yang, Z.; Cepeda, C.; Levine, M.S.; Gray, M. Mutant huntingtin reduction in astrocytes slows disease progression in the bachd conditional huntington’s disease mouse model. Hum. Mol. Genet. 2018, 28, 487–500. [Google Scholar] [CrossRef]
- Taylor, J.P.; Taye, A.A.; Campbell, C.; Kazemi-Esfarjani, P.; Fischbeck, K.H.; Min, K.-T. Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev. 2003, 17, 1463–1468. [Google Scholar] [CrossRef] [Green Version]
- Bason, M.; Meister-Broekema, M.; Alberts, N.; Dijkers, P.; Bergink, S.; Sibon, O.C.; Kampinga, H.H. Astrocytic expression of the chaperone DNAJB6 results in non-cell autonomous protection in Huntington’s disease. Neurobiol. Dis. 2019, 124, 108–117. [Google Scholar] [CrossRef]
- Besson, M.-T.; Dupont, P.; Fridell, Y.-W.C.; Liévens, J.-C. Increased energy metabolism rescues glia-induced pathology in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3372–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, P.; Besson, M.-T.; Devaux, J.; Liévens, J.-C. Reducing canonical Wingless/Wnt signaling pathway confers protection against mutant Huntingtin toxicity in Drosophila. Neurobiol. Dis. 2012, 47, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Liévens, J.-C.; Iché, M.; Laval, M.; Faivre-Sarrailh, C.; Birman, S. AKT-sensitive or insensitive pathways of toxicity in glial cells and neurons in Drosophila models of Huntington’s disease. Hum. Mol. Genet. 2007, 17, 882–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liévens, J.-C.; Rival, T.; Iché, M.; Chneiweiss, H.; Birman, S. Expanded polyglutamine peptides disrupt EGF receptor signaling and glutamate transporter expression in Drosophila. Hum. Mol. Genet. 2005, 14, 713–724. [Google Scholar] [CrossRef]
- Pearce, M.M.P.; Spartz, E.J.; Hong, W.; Luo, L.; Kopito, R.R. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Davis, A.K.; Pratt, W.B.; Lieberman, A.P.; Osawa, Y. Targeting Hsp70 facilitated protein quality control for treatment of polyglutamine diseases. Cell. Mol. Life Sci. 2020, 77, 977–996. [Google Scholar] [CrossRef]
- Ravikumar, B.; Imarisio, S.; Sarkar, S.; O’Kane, C.J.; Rubinsztein, D.C. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J. Cell Sci. 2008, 121, 1649–1660. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Pallos, J.; Jacques, V.; Lau, A.; Tang, B.; Cooper, A.; Syed, A.; Purcell, J.; Chen, Y.; Sharma, S.; et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol. Dis. 2012, 46, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Pallos, J.; Bodai, L.; Lukacsovich, T.; Purcell, J.M.; Steffan, J.S.; Thompson, L.M.; Marsh, J.L. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2008, 17, 3767–3775. [Google Scholar] [CrossRef] [Green Version]
- Thomas, E.A. Involvement of HDAC1 and HDAC3 in the pathology of polyglutamine disorders: Therapeutic implications for selective HDAC1/HDAC3 inhibitors. Pharmaceuticals 2014, 7, 634–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The kynurenine pathway modulates neurodegeneration in a drosophila model of Huntington’s Disease. Curr. Biol. 2011, 21, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Rajamani, K.; Liu, J.-W.; Wu, C.-H.; Chiang, I.-T.; You, D.-H.; Lin, S.-Y.; Hsieh, D.-K.; Lin, S.-Z.; Harn, H.-J.; Chiou, T.-W. n-Butylidenephthalide exhibits protection against neurotoxicity through regulation of tryptophan 2, 3 dioxygenase in spinocerebellar ataxia type 3. Neuropharmacology 2017, 117, 434–446. [Google Scholar] [CrossRef]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007, 3, e82. [Google Scholar] [CrossRef] [Green Version]
- Volkenhoff, A.; Weiler, A.; Letzel, M.; Stehling, M.; Klämbt, C.; Schirmeier, S. Glial glycolysis is essential for neuronal survival in drosophila. Cell Metab. 2015, 22, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [Green Version]
- Benchoua, A.; Trioulier, Y.; Zala, D.; Gaillard, M.-C.; Lefort, N.; Dufour, N.; Saudou, F.; Elalouf, J.-M.; Hirsch, E.; Hantraye, P.; et al. involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol. Biol. Cell 2006, 17, 1652–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s disease. Biochim. Biophys. Acta 2010, 1802, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruetenik, A.L.; Ocampo, A.; Ruan, K.; Zhu, Y.; Li, C.; Zhai, R.G.; Barrientos, A. Attenuation of polyglutamine-induced toxicity by enhancement of mitochondrial OXPHOS in yeast and fly models of aging. Microb. Cell 2016, 3, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Di Cristo, F.; Finicelli, M.; Digilio, F.A.; Paladino, S.; Valentino, A.; Scialò, F.; D’Apolito, M.; Saturnino, C.; Galderisi, U.; Giordano, A.; et al. Meldonium improves Huntington’s disease mitochondrial dysfunction by restoring peroxisome proliferator-activated receptor γ coactivator 1α expression. J. Cell. Physiol. 2019, 234, 9233–9246. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I. Role of inositol 1,4,5-trishosphate receptors in pathogenesis of Huntington’s Disease and spinocerebellar ataxias. Neurochem. Res. 2011, 36, 1186–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, T.-S.; Tu, H.; Chan, E.Y.; Maximov, A.; Wang, Z.; Wellington, C.L.; Hayden, M.R.; Bezprozvanny, I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type. Neuron 2003, 39, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Chorna, T.; Hasan, G. The genetics of calcium signaling in Drosophila melanogaster. Biochim. Biophys. Acta 2012, 1820, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011, 12, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaywitz, A.J.; Greenberg, M.E. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 1999, 68, 821–861. [Google Scholar] [CrossRef] [PubMed]
- Dansen, T.B.; Smits, L.M.M.; Van Triest, M.H.; De Keizer, P.L.J.; Van Leenen, D.; Koerkamp, M.G.; Szypowska, A.; Meppelink, A.; Brenkman, A.B.; Yodoi, J.; et al. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat. Chem. Biol. 2009, 5, 664–672. [Google Scholar] [CrossRef]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef]
- Li, J.; Sutter, C.; Parker, D.S.; Blauwkamp, T.; Fang, M.; Cadigan, K.M. CBP/p300 are bimodal regulators of Wnt signaling. EMBO J. 2007, 26, 2284–2294. [Google Scholar] [CrossRef] [Green Version]
- Godin, J.D.; Poizat, G.; Hickey, M.; Maschat, F.; Humbert, S. Mutant huntingtin-impaired degradation of β-catenin causes neurotoxicity in Huntington’s disease. EMBO J. 2010, 29, 2433–2445. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.A.; Vazquez-Manrique, R.P.; Tourette, C.; Farina, F.; Offner, N.; Mukhopadhyay, A.; Orfila, A.-M.; Darbois, A.; Menet, S.; Tissenbaum, H.A.; et al. Integration of β -catenin, sirtuin, and FOXO signaling protects from mutant huntingtin toxicity. J. Neurosci. 2012, 32, 12630–12640. [Google Scholar] [CrossRef] [Green Version]
- Reim, G.; Hruzova, M.; Goetze, S.; Basler, K. Protection of armadillo/β-catenin by armless, a novel positive regulator of wingless signaling. PLoS Biol. 2014, 12, e1001988. [Google Scholar] [CrossRef] [Green Version]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [Green Version]
- Artiushin, G.; Zhang, S.L.; Tricoire, H.; Sehgal, A. Endocytosis at the Drosophila blood–brain barrier as a function for sleep. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Barnat, M.; Capizzi, M.; Aparicio, E.; Boluda, S.; Wennagel, D.; Kacher, R.; Kassem, R.; Lenoir, S.; Agasse, F.; Braz, B.Y.; et al. Huntington’s disease alters human neurodevelopment. Science 2020, 369, 787–793. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Almeida, A.; Moncada, S. Glycolysis: A bioenergetic or a survival pathway? Trends Biochem. Sci. 2010, 35, 145–149. [Google Scholar] [CrossRef]
- Varma, H.; Cheng, R.; Voisine, C.; Hart, A.C.; Stockwell, B.R. Inhibitors of metabolism rescue cell death in Huntington’s disease models. Proc. Natl. Acad. Sci. USA 2007, 104, 14525–14530. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.D.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, G.; Bao, G.; Xu, G.; Sun, Y.; Li, W.; Wang, L.; Chen, J.; Jin, H.; Cui, Z. Nuclear translocation of PKM2 modulates astrocyte proliferation via p27 and β-catenin pathway after spinal cord injury. Cell Cycle 2015, 14, 2609–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Ryskamp, D.; Birnbaumer, L.; Bezprozvanny, I. Inhibition of TRPC1-dependent store-operated calcium entry improves synaptic stability and motor performance in a mouse model of Huntington’s Disease. J. Huntingt. Dis. 2018, 7, 35–50. [Google Scholar] [CrossRef]
- Wu, J.; Ryskamp, D.A.; Liang, X.; Egorova, P.; Zakharova, O.; Hung, G.; Bezprozvanny, I. Enhanced store-operated calcium entry leads to striatal synaptic loss in a huntington’s disease mouse model. J. Neurosci. 2016, 36, 125–141. [Google Scholar] [CrossRef]
- Kumar, J.P.; Jamal, T.; Doetsch, A.; Turner, F.R.; Duffy, J.B. CREB binding protein functions during successive stages of eye development in drosophila. Genetics 2004, 168, 877–893. [Google Scholar] [CrossRef] [Green Version]
- Van Tienen, L.M.; Mieszczanek, J.; Fiedler, M.; Rutherford, T.J.; Bienz, M. Constitutive scaffolding of multiple Wnt enhanceosome components by Legless/BCL9. eLife 2017, 6, e20882. [Google Scholar] [CrossRef]
- Carrera, I.; Janody, F.; Leeds, N.; Duveau, F.; Treisman, J.E. Pygopus activates Wingless target gene transcription through the mediator complex subunits Med12 and Med13. Proc. Natl. Acad. Sci. USA 2008, 105, 6644–6649. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Guo, X.; Chen, C.; Chen, Y.; Li, J.; Sun, Y.; Wu, C.; Yang, Y.; Jiang, C.; Li, W.; et al. dFoxO promotes wingless signaling in drosophila. Sci. Rep. 2016, 6, 22348. [Google Scholar] [CrossRef] [Green Version]
- Essers, M.A.G.; De Vries-Smits, L.M.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.T.; Korswagen, H.C. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, E.; Heidari, R.; Monnier, V.; Tricoire, H. Genetic Screen in Adult Drosophila Reveals That dCBP Depletion in Glial Cells Mitigates Huntington Disease Pathology through a Foxo-Dependent Pathway. Int. J. Mol. Sci. 2021, 22, 3884. https://doi.org/10.3390/ijms22083884
Martin E, Heidari R, Monnier V, Tricoire H. Genetic Screen in Adult Drosophila Reveals That dCBP Depletion in Glial Cells Mitigates Huntington Disease Pathology through a Foxo-Dependent Pathway. International Journal of Molecular Sciences. 2021; 22(8):3884. https://doi.org/10.3390/ijms22083884
Chicago/Turabian StyleMartin, Elodie, Raheleh Heidari, Véronique Monnier, and Hervé Tricoire. 2021. "Genetic Screen in Adult Drosophila Reveals That dCBP Depletion in Glial Cells Mitigates Huntington Disease Pathology through a Foxo-Dependent Pathway" International Journal of Molecular Sciences 22, no. 8: 3884. https://doi.org/10.3390/ijms22083884
APA StyleMartin, E., Heidari, R., Monnier, V., & Tricoire, H. (2021). Genetic Screen in Adult Drosophila Reveals That dCBP Depletion in Glial Cells Mitigates Huntington Disease Pathology through a Foxo-Dependent Pathway. International Journal of Molecular Sciences, 22(8), 3884. https://doi.org/10.3390/ijms22083884