
Detection of Genomic Uracil Patterns



Abstract
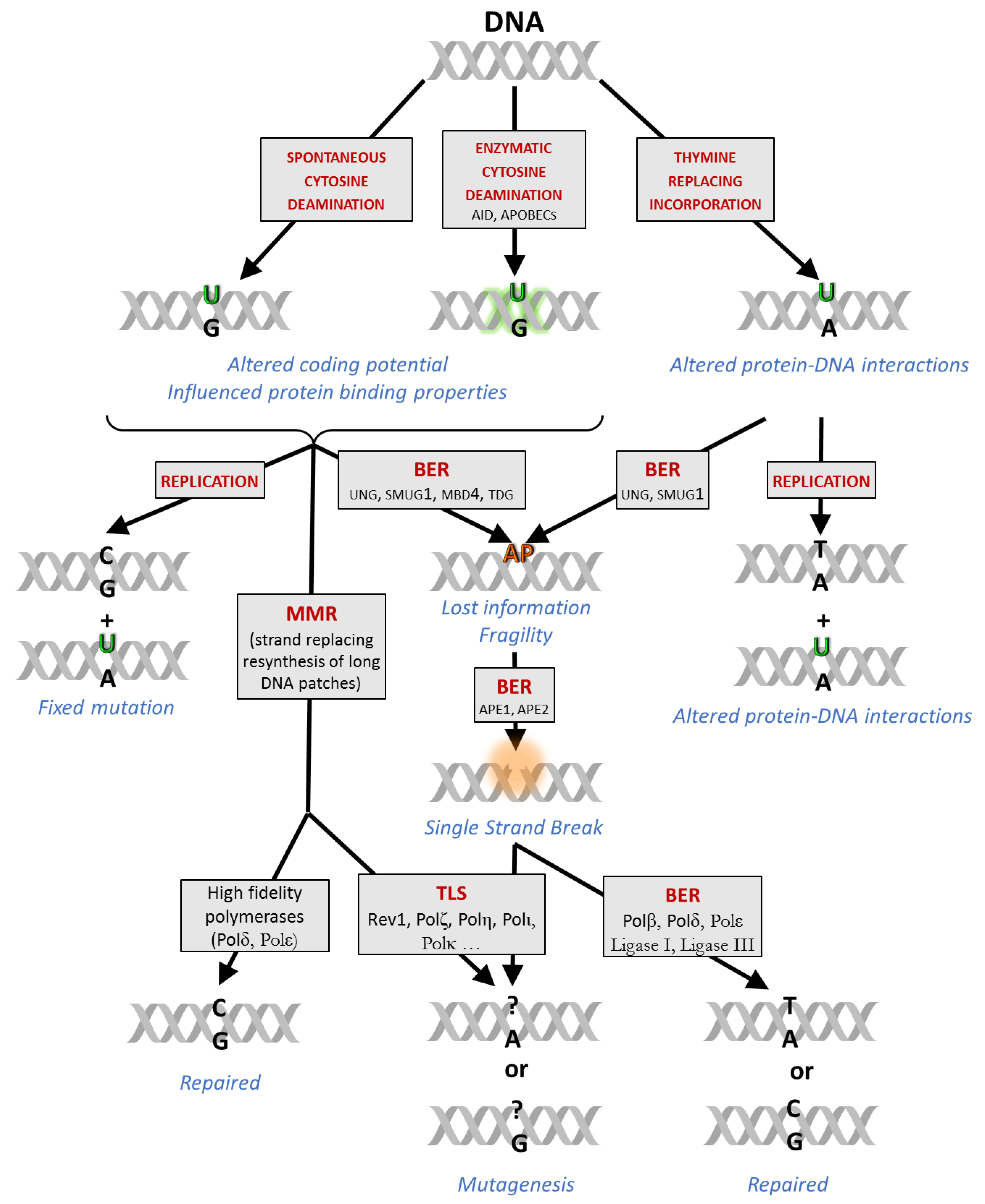
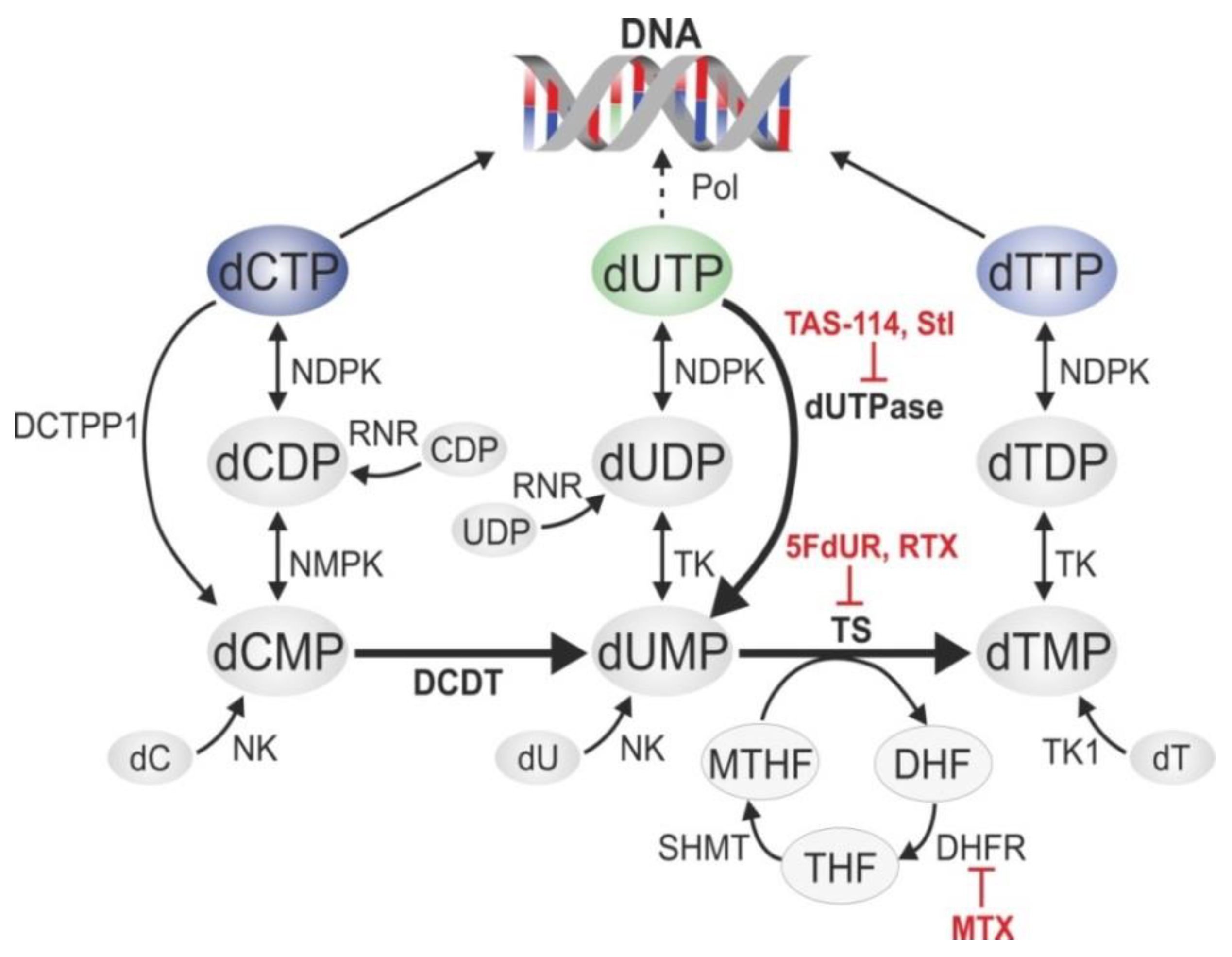
:1. Introduction
2. Uracil-DNA Detection Methods
2.1. Global Quantification of Uracil in DNA
2.2. In Situ U-DNA Detection Methods
2.3. PCR-Based Methods for Uracil Localization within DNA
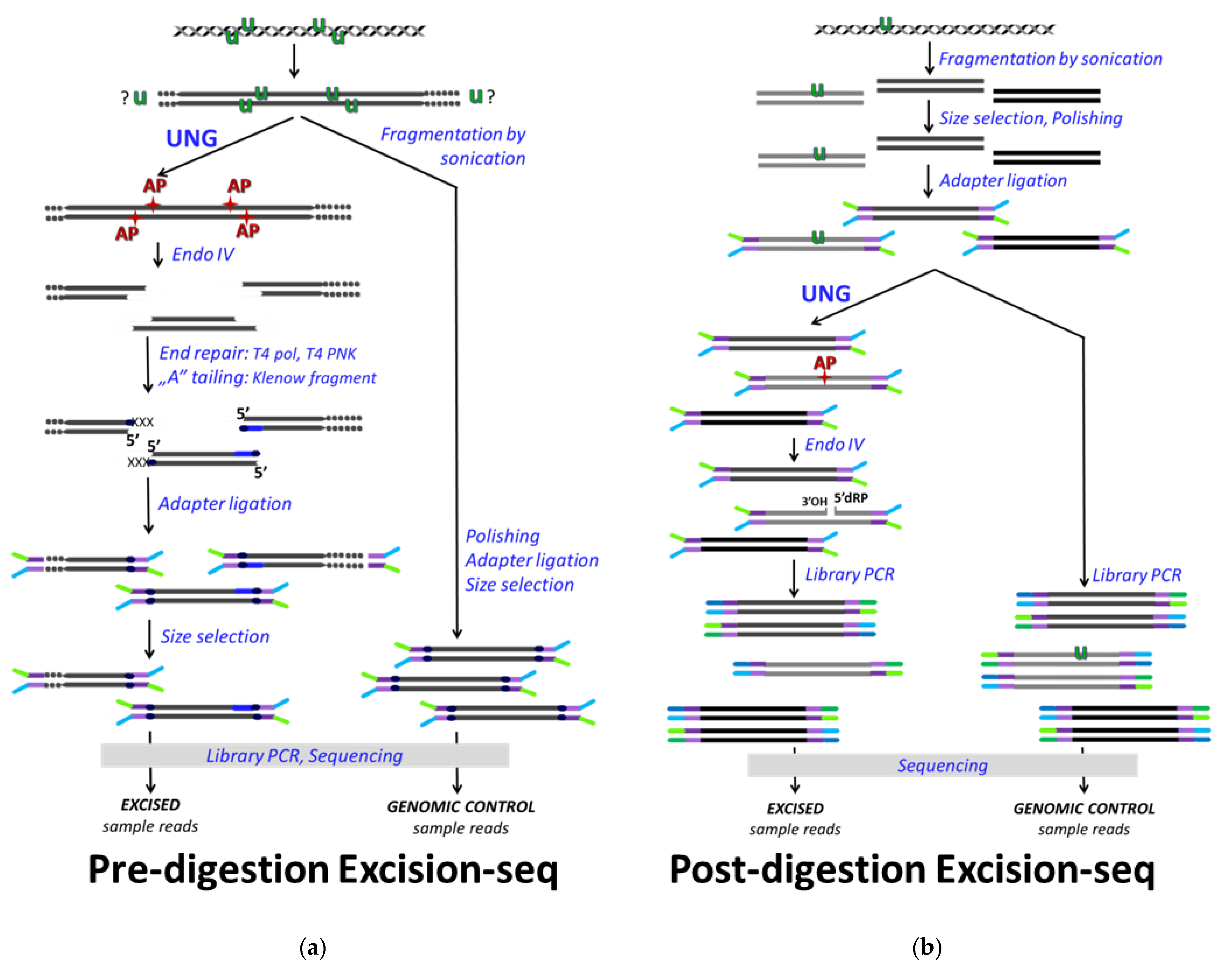
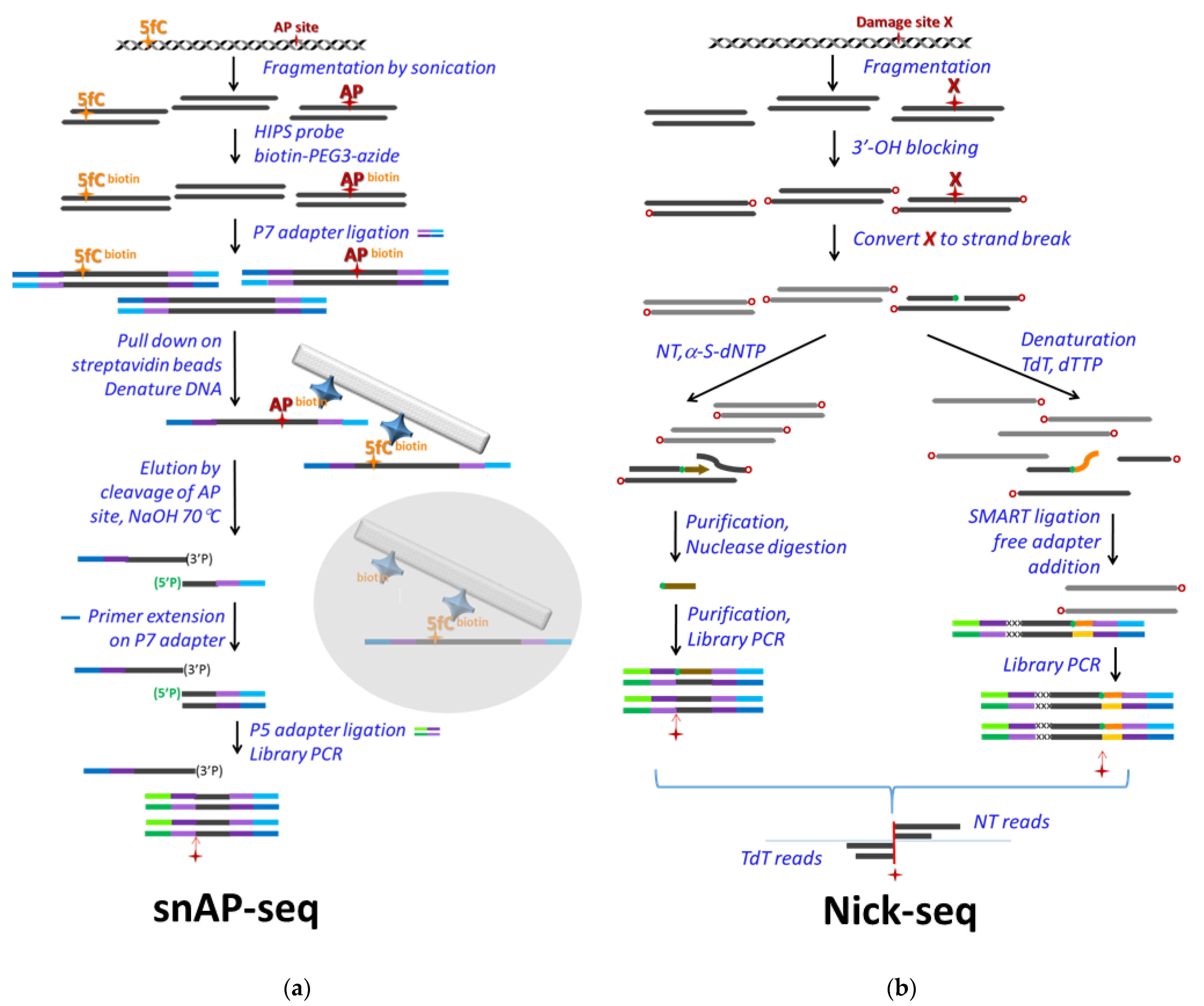
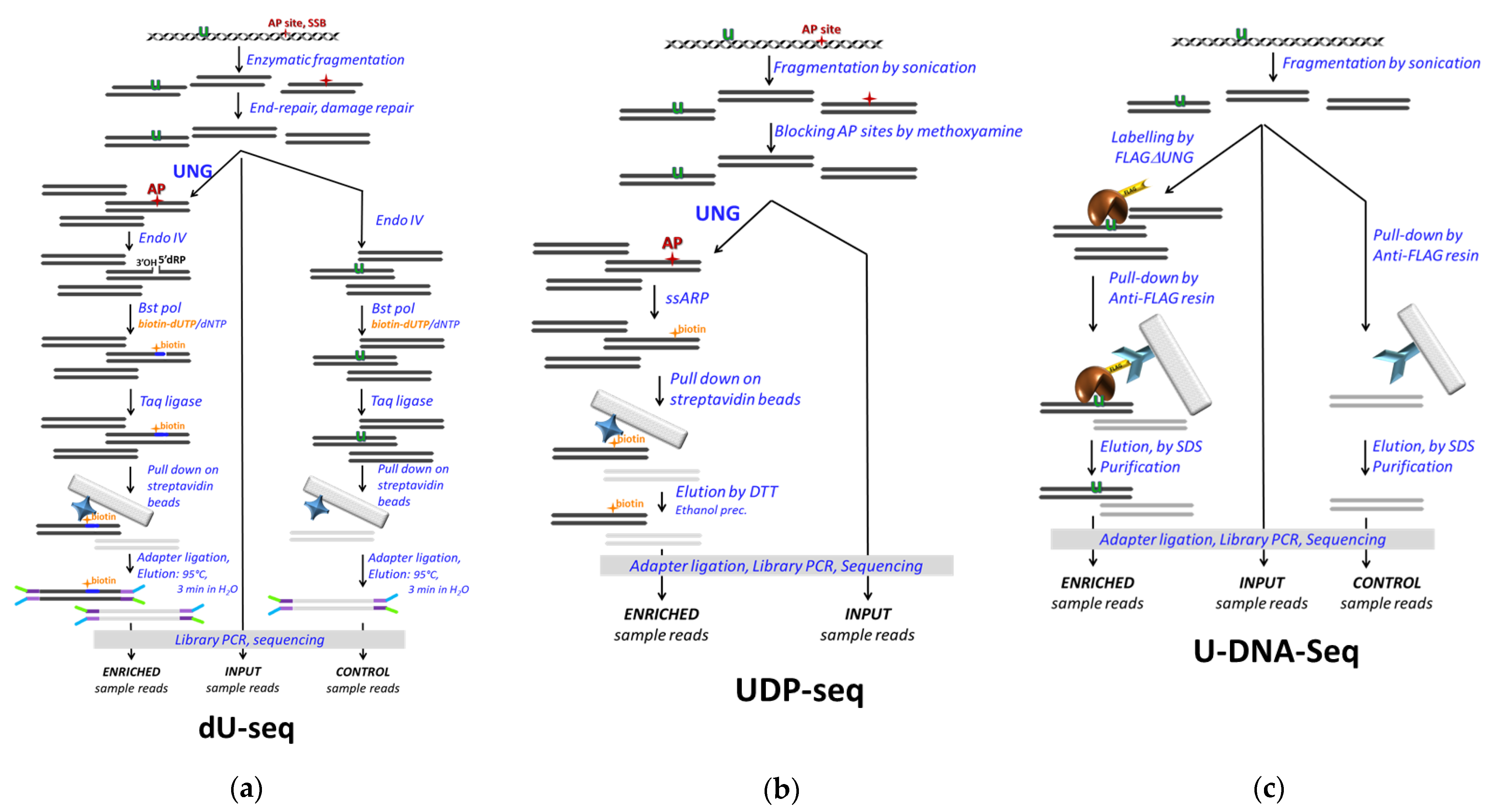
2.4. NGS Based U-DNA Detection for Genome-Wide Mapping
3. Factors to Consider in Analysis of NGS Data to Maximize Relevant Information While Avoiding Over- or Misinterpretation
4. Single Base Resolution—When Is It Truly Relevant?
Author Contributions
Funding
Conflicts of Interest
References
- Chon, J.; Field, M.S.; Stover, P.J. Deoxyuracil in DNA and disease: Genomic signal or managed situation? DNA Repair 2019, 77, 36–44. [Google Scholar] [CrossRef]
- Krokan, H.E.; Drabløs, F.; Slupphaug, G. Uracil in DNA--occurrence, consequences and repair. Oncogene 2002, 21, 8935–8948. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Petersen-Mahrt, S.K.; Harris, R.S.; Neuberger, M.S. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 2002, 418, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, M.S.; Harris, R.S.; Di Noia, J.; Petersen-Mahrt, S.K. Immunity through DNA deamination. Trends Biochem. Sci. 2003, 28, 305–312. [Google Scholar] [CrossRef]
- Beale, R.C.L.; Petersen-Mahrt, S.K.; Watt, I.N.; Harris, R.S.; Rada, C.; Neuberger, M.S. Comparison of the Differential Context-dependence of DNA Deamination by APOBEC Enzymes: Correlation with Mutation Spectra In Vivo. J. Mol. Biol. 2004, 337, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Honjo, T.; Muramatsu, M.; Fagarasan, S. Aid: How does it aid antibody diversity? Immunity 2004, 20, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siriwardena, S.U.; Chen, K.; Bhagwat, A.S. Functions and Malfunctions of Mammalian DNA-Cytosine Deaminases. Chem. Rev. 2016, 116, 12688–12710. [Google Scholar] [CrossRef] [Green Version]
- Maul, R.W.; Gearhart, P.J. Refining the Neuberger model: Uracil processing by activated B cells. Eur. J. Immunol. 2014, 44, 1913–1916. [Google Scholar] [CrossRef]
- Caradonna, S.J.; Cheng, Y.C. The role of deoxyuridine triphosphate nucleotidohydrolase, uracil-DNA glycosylase, and DNA polymerase α in the metabolism of FUdR in human tumor cells. Mol. Pharmacol. 1980, 18, 513–520. [Google Scholar]
- Tanaka, M.; Yoshida, S.; Saneyoshi, M.; Yamaguchi, T. Utilization of 5-fluoro-2′-deoxyuridine triphosphate and 5-fluoro-2′-deoxycytidine triphosphate in DNA synthesis by DNA polymerases alpha and beta from calf thymus. Cancer Res. 1981, 41, 4132–4135. [Google Scholar] [PubMed]
- Hirmondó, R.; Szabó, J.E.; Nyíri, K.; Tarjányi, S.; Dobrotka, P.; Tóth, J.; Vértessy, B.G. Cross-species inhibition of dUTPase via the Staphylococcal Stl protein perturbs dNTP pool and colony formation in Mycobacterium. DNA Repair 2015, 30. [Google Scholar] [CrossRef] [Green Version]
- Muha, V.; Horváth, A.; Békési, A.; Pukáncsik, M.; Hodoscsek, B.; Merényi, G.; Róna, G.; Batki, J.; Kiss, I.; Jankovics, F.; et al. Uracil-containing DNA in Drosophila: Stability, stage-specific accumulation, and developmental involvement. PLoS Genet 2012, 8, e1002738. [Google Scholar] [CrossRef] [Green Version]
- Andersen, S.; Heine, T.; Sneve, R.; König, I.; Krokan, H.E.; Epe, B.; Nilsen, H. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis 2005, 26, 547–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.H.; Xu, G.F.; Gu, T.P.; Chen, G.D.; Han, B.B.; Xu, Z.M.; Bjørås, M.; Krokan, H.E.; Xu, G.L.; Du, Y.R. Uracil-DNA glycosylase UNG promotes tet-mediated DNA demethylation. J. Biol. Chem. 2016, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serebrenik, A.A.; Starrett, G.J.; Leenen, S.; Jarvis, M.C.; Shaban, N.M.; Salamango, D.J.; Nilsen, H.; Brown, W.L.; Harris, R.S. The deaminase APOBEC3B triggers the death of cells lacking uracil DNA glycosylase. Proc. Natl. Acad. Sci. USA 2019, 116, 22158–22163. [Google Scholar] [CrossRef]
- Chandra, V.; Bortnick, A.; Murre, C. AID targeting: Old mysteries and new challenges. Trends Immunol. 2015, 36, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Pavri, R.; Nussenzweig, M.C. AID targeting in antibody diversity. Adv. Immunol. 2011, 110, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H. The role of activation-induced deaminase in antibody diversification and genomic instability. Immunol. Res. 2013, 55, 287–297. [Google Scholar] [CrossRef]
- Stenglein, M.D.; Burns, M.B.; Li, M.; Lengyel, J.; Harris, R.S. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010, 17, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Law, E.K.; Levin-Klein, R.; Jarvis, M.C.; Kim, H.; Argyris, P.P.; Carpenter, M.A.; Starrett, G.J.; Temiz, N.A.; Larson, L.K.; Durfee, C.; et al. APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J. Exp. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Leonard, B.; Harris, R.S. APOBEC3B: Pathological consequences of an innate immune DNA mutator. Biomed. J. 2015, 38, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Nikkilä, J.; Kumar, R.; Campbell, J.; Brandsma, I.; Pemberton, H.N.; Wallberg, F.; Nagy, K.; Scheer, I.; Vertessy, B.G.; Serebrenik, A.A.; et al. Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br. J. Cancer 2017, 117, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Periyasamy, M.; Singh, A.K.; Gemma, C.; Kranjec, C.; Farzan, R.; Leach, D.A.; Navaratnam, N.; Pálinkás, H.L.; Vértessy, B.G.; Fenton, T.R.; et al. p53 controls expression of the DNA deaminase APOBEC3B to limit its potential mutagenic activity in cancer cells. Nucleic Acids Res. 2017, 45, 11056–11069. [Google Scholar] [CrossRef] [Green Version]
- Horváth, A.; Vértessy, B.G. A one-step method for quantitative determination of uracil in DNA by real-time PCR. Nucleic Acids Res. 2010, 38, e196. [Google Scholar] [CrossRef] [Green Version]
- Róna, G.; Scheer, I.; Nagy, K.; Pálinkás, H.L.; Tihanyi, G.; Borsos, M.; Békési, A.; Vértessy, B.G. Detection of uracil within DNA using a sensitive labeling method for in vitro and cellular applications. Nucleic Acids Res. 2016, 44, e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, M.D.; Wilson, D.M. Participation of DNA repair in the response to 5-fluorouracil. Cell. Mol. Life Sci. 2009, 66, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Visnes, T.; Doseth, B.; Pettersen, H.S.; Hagen, L.; Sousa, M.M.L.; Akbari, M.; Otterlei, M.; Kavli, B.; Slupphaug, G.; Krokan, H.E. Uracil in DNA and its processing by different DNA glycosylases. Philos. Trans. R Soc. Lond. B Biol. Sci. 2009, 364, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Krokan, H.E.; Otterlei, M.; Nilsen, H.; Kavli, B.; Skorpen, F.; Andersen, S.; Skjelbred, C.; Akbari, M.; Aas, P.A.; Slupphaug, G. Properties and functions of human uracil-DNA glycosylase from the UNG gene. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 365–386. [Google Scholar]
- Slupphaug, G.; Eftedal, I.; Kavli, B.; Bharati, S.; Helle, N.M.; Haug, T.; Levine, D.W.; Krokan, H.E. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry 1995, 34, 128–138. [Google Scholar] [CrossRef]
- Kavli, B.; Sundheim, O.; Akbari, M.; Otterlei, M.; Nilsen, H.; Skorpen, F.; Aas, P.A.; Hagen, L.; Krokan, H.E.; Slupphaug, G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 2002, 277, 39926–39936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caradonna, S.; Muller-weeks, S. The Nature of Enzymes Involved in Uracil-DNA Repair: Isoform Characteristics of Proteins Responsible for Nuclear and Mitochondrial Genomic Integrity. Curr. Protein Pept. Sci. 2005, 2. [Google Scholar] [CrossRef] [PubMed]
- Wibley, J.E.A.; Waters, T.R.; Haushalter, K.; Verdine, G.L.; Pearl, L.H. Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol. Cell 2003, 11. [Google Scholar] [CrossRef]
- Masaoka, A.; Matsubara, M.; Hasegawa, R.; Tanaka, T.; Kurisu, S.; Terato, H.; Ohyama, Y.; Karino, N.; Matsuda, A.; Ide, H. Mammalian 5-formyluracil-DNA glycosylase. 2. Role of SMUG1 uracil-DNA glycosylase in repair of 5-formyluracil and other oxidized and deaminated base lesions. Biochemistry 2003, 42. [Google Scholar] [CrossRef]
- Boorstein, R.J.; Cummings, A.; Marenstein, D.R.; Chan, M.K.; Ma, Y.; Neubert, T.A.; Brown, S.M.; Teebor, G.W. Definitive Identification of Mammalian 5-Hydroxymethyluracil DNA N-Glycosylase Activity as SMUG1. J. Biol. Chem. 2001, 276. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, H. Structural and mutation studies of two DNA demethylation related glycosylases: MBD4 and TDG. Biophysics 2014, 10, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Burdzy, A.; Sowers, L.C. Substrate recognition by a family of uracil-DNA glycosylases: UNG, MUG, and TDG. Chem. Res. Toxicol. 2002, 15, 1001–1009. [Google Scholar] [CrossRef]
- Gallinari, P.; Jiricny, J. A new class of uracil-DNA glycosylases related to human thymine-DNA glycosylase. Nature 1996, 383. [Google Scholar] [CrossRef]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Cortázar, D.; Kunz, C.; Selfridge, J.; Lettieri, T.; Saito, Y.; MacDougall, E.; Wirz, A.; Schuermann, D.; Jacobs, A.L.; Siegrist, F.; et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011, 470, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.M.; Kolendowski, B.; Isovic, M.; Bose, K.; Dranse, H.J.; Sampaio, A.V.; Underhill, T.M.; Torchia, J. Regulation of Active DNA Demethylation through RAR-Mediated Recruitment of a TET/TDG Complex. Cell Rep. 2017, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellacosa, A.; Drohat, A.C. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair 2015, 32. [Google Scholar] [CrossRef] [Green Version]
- Krokan, H.E.; Sætrom, P.; Aas, P.A.; Pettersen, H.S.; Kavli, B.; Slupphaug, G. Error-free versus mutagenic processing of genomic uracil-Relevance to cancer. DNA Repair 2014, 19. [Google Scholar] [CrossRef] [Green Version]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Wilson, D.M., III. The Base Excision Repair Pathway; World Scientific: Singapore, 2017. [Google Scholar]
- Schanz, S.; Castor, D.; Fischer, F.; Jiricny, J. Interference of mismatch and base excision repair during the processing of adjacent U/G mispairs may play a key role in somatic hypermutation. Proc. Natl. Acad. Sci. USA 2009, 106, 5593–5598. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ. 2001, 8, 1076–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Li, W.; Adebali, O.; Yang, Y.; Oztas, O.; Selby, C.P.; Sancar, A. Genome-wide mapping of nucleotide excision repair with XR-seq. Nat. Protoc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Weiner, K.X.B.; Weiner, R.S.; Maley, F.; Maley, G.F. Primary structure of human deoxycytidylate deaminase and overexpression of its functional protein in Escherichia coli. J. Biol. Chem. 1993, 268. [Google Scholar] [CrossRef]
- Mancini, W.R.; Cheng, Y.C. Human deoxycytidylate deaminase: Substrate and regulator specificities and their chemotherapeutic implications. Mol. Pharmacol. 1983, 23, 159–164. [Google Scholar] [PubMed]
- Vértessy, B.G.; Tóth, J. Keeping uracil out of DNA: Physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 2009, 42, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Islam, Z.; Gurevic, I.; Strutzenberg, T.S.; Ghosh, A.K.; Iqbal, T.; Kohen, A. Bacterial versus human thymidylate synthase: Kinetics and functionality. PLoS ONE 2018, 13, e0196506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, D.B.; Boyer, J.; Allen, W.L.; Latif, T.; Ferguson, P.R.; Maxwell, P.J.; McDermott, U.; Lynch, M.; Harkin, D.P.; Johnston, P.G. The role of thymidylate synthase induction in modulating p53-regulated gene expression in response to 5-fluorouracil and antifolates. Cancer Res. 2002, 62, 2644–2649. [Google Scholar] [PubMed]
- Chu, E.; Voeller, D.M.; Jones, K.L.; Takechi, T.; Maley, G.F.; Maley, F.; Segal, S.; Allegra, C.J. Identification of a thymidylate synthase ribonucleoprotein complex in human colon cancer cells. Mol. Cell. Biol. 1994, 14. [Google Scholar] [CrossRef] [PubMed]
- Garg, D.; Beribisky, A.V.; Ponterini, G.; Ligabue, A.; Marverti, G.; Martello, A.; Paola Costi, M.; Sattler, M.; Wade, R.C. Translational repression of thymidylate synthase by targeting its mRNA. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [Green Version]
- Brunn, N.D.; Dibrov, S.M.; Kao, M.B.; Ghassemian, M.; Hermann, T. Analysis of mRNA recognition by human thymidylate synthase. Biosci. Rep. 2014, 34. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, A.; Takahari, D.; Keisho, C.; Nakamura, Y.; Ikeno, T.; Wakabayashi, M.; Nomura, S.; Tamura, H.; Fukutani, M.; Hirano, N.; et al. A multicenter phase II study of TAS-114 in combination with S-1 in patients with pretreated advanced gastric cancer (EPOC1604). Gastric Cancer 2020, 1, 3. [Google Scholar] [CrossRef]
- Yano, W.; Yokogawa, T.; Wakasa, T.; Yamamura, K.; Fujioka, A.; Yoshisue, K.; Matsushima, E.; Miyahara, S.; Miyakoshi, H.; Taguchi, J.; et al. TAS-114, a first-in-class dual dUTPase/DPD inhibitor, demonstrates potential to improve therapeutic efficacy of fluoropyrimidine-based chemotherapy. Mol. Cancer Ther. 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Yokogawa, T.; Yano, W.; Tsukioka, S.; Osada, A.; Wakasa, T.; Ueno, H.; Hoshino, T.; Yamamura, K.; Fujioka, A.; Fukuoka, M.; et al. dUTPase inhibition confers susceptibility to a thymidylate synthase inhibitor in DNA-repair-defective human cancer cells. Cancer Sci. 2021, 112, 422–432. [Google Scholar] [CrossRef]
- Nyíri, K.; Harris, M.J.; Matejka, J.; Ozohanics, O.; Vékey, K.; Borysik, A.J.; Vértessy, B.G. HDX and native mass spectrometry reveals the different structural basis for interaction of the staphylococcal pathogenicity island repressor stl with dimeric and trimeric phage dUTPases. Biomolecules 2019, 9, 488. [Google Scholar] [CrossRef] [Green Version]
- Surányi, É.V.; Hírmondó, R.; Nyíri, K.; Tarjányi, S.; Kőhegyi, B.; Tóth, J.; Vértessy, B.G. Exploiting a phage-bacterium interaction system as a molecular switch to decipher macromolecular interactions in the living cell. Viruses 2018, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Nyíri, K.; Mertens, H.D.T.; Tihanyi, B.; Nagy, G.N.; Kohegyi, B.; Matejka, J.; Harris, M.J.; Szabó, J.E.; Papp-Kádár, V.; Németh-Pongrácz, V.; et al. Structural model of human dUTPase in complex with a novel proteinaceous inhibitor. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Benedek, A.; Pölöskei, I.; Ozohanics, O.; Vékey, K.; Vértessy, B.G. The Stl repressor from Staphylococcus aureus is an efficient inhibitor of the eukaryotic fruitfly dUTPase. FEBS Open Bio 2018, 8. [Google Scholar] [CrossRef]
- Papp-Kádár, V.; Szabó, J.E.; Nyíri, K.; Vertessy, B.G. In Vitro analysis of predicted DNA-binding sites for the Stl repressor of the Staphylococcus aureus SaPIBov1 pathogenicity island. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyíri, K.; Kohegyi, B.; Micsonai, A.; Kardos, J.; Vertessy, B.G. Evidence-based structural model of the staphylococcal repressor protein: Separation of functions into different domains. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, J.E.; Németh, V.; Papp-Kádár, V.; Nyíri, K.; Leveles, I.; Bendes, Á.; Zagyva, I.; Róna, G.; Pálinkás, H.L.; Besztercei, B.; et al. Highly potent dUTPase inhibition by a bacterial repressor protein reveals a novel mechanism for gene expression control. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merényi, G.; Kovári, J.; Tóth, J.; Takács, E.; Zagyva, I.; Erdei, A.; Vértessy, B.G. Cellular response to efficient dUTPase RNAi silencing in stable hela cell lines perturbs expression levels of genes involved in thymidylate metabolism. Nucleosides Nucleotides Nucleic Acids 2011, 30. [Google Scholar] [CrossRef]
- Koehler, S.E.; Ladner, R.D. Small interfering RNA-mediated suppression of dUTPase sensitizes cancer cell lines to thymidylate synthase inhibition. Mol. Pharmacol. 2004, 66. [Google Scholar] [CrossRef]
- Wilson, P.M.; Danenberg, P.V.; Johnston, P.G.; Lenz, H.-J.; Ladner, R.D. Standing the test of time: Targeting thymidylate biosynthesis in cancer therapy. Nat. Rev. Clin. Oncol. 2014, 11, 282–298. [Google Scholar] [CrossRef]
- Li, W.; Sancar, A. Methodologies for detecting environmentally induced DNA damage and repair. Environ. Mol. Mutagen. 2020, 61, 664–679. [Google Scholar] [CrossRef]
- Mingard, C.; Wu, J.; McKeague, M.; Sturla, S.J. Next-generation DNA damage sequencing. Chem. Soc. Rev. 2020, 49, 7354–7377. [Google Scholar] [CrossRef]
- Galashevskaya, A.; Sarno, A.; Vågbø, C.B.; Aas, P.A.; Hagen, L.; Slupphaug, G.; Krokan, H.E. A robust, sensitive assay for genomic uracil determination by LC/MS/MS reveals lower levels than previously reported. DNA Repair 2013, 12, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Blount, B.C.; Ames, B.N. Analysis of uracil in DNA by gas chromatography-mass spectrometry. Anal. Biochem. 1994, 219, 195–200. [Google Scholar] [CrossRef]
- Mashiyama, S.T.; Courtemanche, C.; Elson-Schwab, I.; Crott, J.; Lee, B.L.; Ong, C.N.; Fenech, M.; Ames, B.N. Uracil in DNA, determined by an improved assay, is increased when deoxynucleosides are added to folate-deficient cultured human lymphocytes. Anal. Biochem. 2004, 330, 58–69. [Google Scholar] [CrossRef]
- Atamna, H.; Cheung, I.; Ames, B.N. A method for detecting abasic sites in living cells: Age-dependent changes in base excision repair. Proc. Natl. Acad. Sci. USA 2000, 97, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Ulvik, A.; Refsum, H.; Ueland, P.M. Uracil in human DNA from subjects with normal and impaired folate status as determined by high-performance liquid chromatography-tandem mass spectrometry. Anal. Chem. 2002, 74. [Google Scholar] [CrossRef]
- Kubo, K.; Ide, H.; Wallace, S.S.; Kow, Y.W. A Novel, Sensitive, and Specific Assay for Abasic Sites, the Most Commonly Produced DNA Lesion. Biochemistry 1992, 31. [Google Scholar] [CrossRef] [PubMed]
- Kow, Y.W.; Dare, A. Detection of abasic sites and oxidative DNA base damage using an ELISA-like assay. Methods 2000, 22. [Google Scholar] [CrossRef] [PubMed]
- Lari, S.-U.; Chen, C.-Y.; Vértessy, B.G.; Morré, J.; Bennett, S.E. Quantitative determination of uracil residues in Escherichia coli DNA: Contribution of ung, dug, and dut genes to uracil avoidance. DNA Repair 2006, 5, 1407–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Shalhout, S.; Ahn, Y.H.; Bhagwat, A.S. A versatile new tool to quantify abasic sites in DNA and inhibit base excision repair. DNA Repair 2015, 27. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Perera, M.L.W.; Sakhtemani, R.; Bhagwat, A.S. A novel class of chemicals that react with abasic sites in DNA and specifically kill B cell cancers. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Siriwardena, S.U.; Perera, M.L.W.; Senevirathne, V.; Stewart, J.; Bhagwat, A.S. A Tumor-Promoting Phorbol Ester Causes a Large Increase in APOBEC3A Expression and a Moderate Increase in APOBEC3B Expression in a Normal Human Keratinocyte Cell Line without Increasing Genomic Uracils. Mol. Cell. Biol. 2018, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.A.; Schauer, G.; Bhagwat, A.S. Visualization of uracils created by APOBEC3A using UdgX shows colocalization with RPA at stalled replication forks. Nucleic Acids Res. 2020, 48, 118. [Google Scholar] [CrossRef] [PubMed]
- Sakhtemani, R.; Senevirathne, V.; Stewart, J.; Perera, M.L.W.; Pique-Regi, R.; Lawrence, M.S.; Bhagwat, A.S. Genome-wide mapping of regions preferentially targeted by the human DNA-cytosine deaminase APOBEC3A using uracil-DNA pulldown and sequencing. J. Biol. Chem. 2019, 294, 15037–15051. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, T.A.; Haug, T.; Singh, K.K.; Keshav, K.F.; Skorpen, F.; Otterlei, M.; Bharati, S.; Lindmo, T.; Benichou, S.; Benarous, R.; et al. A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J. Biol. Chem. 1997, 272, 6561–6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torseth, K.; Doseth, B.; Hagen, L.; Olaisen, C.; Liabakk, N.-B.; Græsmann, H.; Durandy, A.; Otterlei, M.; Krokan, H.E.; Kavli, B.; et al. The UNG2 Arg88Cys variant abrogates RPA-mediated recruitment of UNG2 to single-stranded DNA. DNA Repair 2012, 11, 559–569. [Google Scholar] [CrossRef]
- Green, D.A.; Deutsch, W.A. Direct determination of uracil in [32P,uracil-3H]poly(dA·dT) and bisulfite-treated phage PM2 DNA. Anal. Biochem. 1984, 142. [Google Scholar] [CrossRef]
- Duthie, S.J.; McMillan, P. Uracil misincorporation in human DNA detected using single cell gel electrophoresis. Carcinogenesis 1997, 18. [Google Scholar] [CrossRef] [Green Version]
- Palinkas, H.L.; Bekesi, A.; Rona, G.; Pongor, L.; Papp, G.; Tihanyi, G.; Holub, E.; Poti, A.; Gemma, C.; Ali, S.; et al. Genome-wide alterations of uracil distribution patterns in human DNA upon chemotherapeutic treatments. eLife 2020, 9, e60498. [Google Scholar] [CrossRef]
- Sang, P.B.; Srinath, T.; Patil, A.G.; Woo, E.-J.; Varshney, U. A unique uracil-DNA binding protein of the uracil DNA glycosylase superfamily. Nucleic Acids Res. 2015, 43, 8452–8463. [Google Scholar] [CrossRef] [Green Version]
- Tu, J.; Chen, R.; Yang, Y.; Cao, W.; Xie, W. Suicide inactivation of the uracil DNA glycosylase UdgX by covalent complex formation. Nat. Chem. Biol. 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Ahn, W.C.; Aroli, S.; Kim, J.H.; Moon, J.H.; Lee, G.S.; Lee, M.H.; Sang, P.B.; Oh, B.H.; Varshney, U.; Woo, E.J. Covalent binding of uracil DNA glycosylase UdgX to abasic DNA upon uracil excision. Nat. Chem. Biol. 2019, 15, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Zeng, H.; Tu, J.; Sun, L.; Cao, W.; Xie, W. Structural insights into an MsmUdgX mutant capable of both crosslinking and uracil excision capability. DNA Repair 2021, 97. [Google Scholar] [CrossRef]
- Datta, M.; Aroli, S.; Karmakar, K.; Dutta, S.; Chakravortty, D.; Varshney, U. Development of mCherry tagged UdgX as a highly sensitive molecular probe for specific detection of uracils in DNA. Biochem. Biophys. Res. Commun. 2019, 518, 38–43. [Google Scholar] [CrossRef]
- Suspene, R.; Henry, M.; Guillot, S.; Wain-Hobson, S.; Vartanian, J.-P. Recovery of APOBEC3-edited human immunodeficiency virus G→A hypermutants by differential DNA denaturation PCR. J. Gen. Virol. 2005, 86, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Martomo, S.A.; Fu, D.; Yang, W.W.; Joshi, N.S.; Gearhart, P.J. Deoxyuridine Is Generated Preferentially in the Nontranscribed Strand of DNA from Cells Expressing Activation-Induced Cytidine Deaminase. J. Immunol. 2005, 174, 7787–7791. [Google Scholar] [CrossRef] [Green Version]
- Maul, R.W.; Saribasak, H.; Martomo, S.A.; McClure, R.L.; Yang, W.; Vaisman, A.; Gramlich, H.S.; Schatz, D.G.; Woodgate, R.; Wilson, D.M.; et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat. Immunol. 2011, 12, 70–76. [Google Scholar] [CrossRef]
- Hansen, E.C.; Ransom, M.; Hesselberth, J.R.; Hosmane, N.N.; Capoferri, A.A.; Bruner, K.M.; Pollack, R.A.; Zhang, H.; Drummond, M.B.; Siliciano, J.M.; et al. Diverse fates of uracilated HIV-1 DNA during infection of myeloid lineage cells. Elife 2016, 5. [Google Scholar] [CrossRef]
- Connolly, B.A. Recognition of deaminated bases by archaeal family-B DNA polymerases. Biochem. Soc. Trans. 2009, 37, 65–68. [Google Scholar] [CrossRef]
- Fogg, M.J.; Pearl, L.H.; Connolly, B.A. Structural basis for uracil recognition by archaeal family B DNA polymerases. Nat. Struct. Biol. 2002. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Martínez Cuesta, S.; van Delft, P.; Balasubramanian, S. Sequencing abasic sites in DNA at single-nucleotide resolution. Nat. Chem. 2019. [Google Scholar] [CrossRef]
- Keszthelyi, A.; Daigaku, Y.; Ptasińska, K.; Miyabe, I.; Carr, A.M. Mapping ribonucleotides in genomic DNA and exploring replication dynamics by polymerase usage sequencing (Pu-seq). Nat. Protoc. 2015. [Google Scholar] [CrossRef]
- Rauluseviciute, I.; Drabløs, F.; Rye, M.B. DNA methylation data by sequencing: Experimental approaches and recommendations for tools and pipelines for data analysis. Clin. Epigenetics 2019, 11, 193. [Google Scholar] [CrossRef] [Green Version]
- Adar, S.; Hu, J.; Lieb, J.D.; Sancar, A. Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Adar, S.; Selby, C.P.; Lieb, J.D.; Sancar, A. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 2015. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Sequencing DNA for the Oxidatively Modified Base 8-Oxo-7,8-Dihydroguanine. Methods Enzymol. 2017, 591, 187–210. [Google Scholar]
- Wu, J.; McKeague, M.; Sturla, S.J. Nucleotide-Resolution Genome-Wide Mapping of Oxidative DNA Damage by Click-Code-Seq. J. Am. Chem. Soc. 2018, 140, 9783–9787. [Google Scholar] [CrossRef] [PubMed]
- Bryan, D.S.; Ransom, M.; Adane, B.; York, K.; Hesselberth, J.R. High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res. 2014, 24, 1534–1542. [Google Scholar] [CrossRef] [Green Version]
- Ransom, M.; Bryan, D.S.; Hesselberth, J.R. High-resolution mapping of modified DNA nucleobases using excision repair enzymes. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2018; Volume 1672, pp. 63–76. [Google Scholar]
- Shu, X.; Liu, M.; Lu, Z.; Zhu, C.; Meng, H.; Huang, S.; Zhang, X.; Yi, C. Genome-wide mapping reveals that deoxyuridine is enriched in the human centromeric DNA. Nat. Chem. Biol. 2018, 14, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Riedl, J.; Ding, Y.; Fleming, A.M.; Burrows, C.J. Identification of DNA lesions using a third base pair for amplification and nanopore sequencing. Nat. Commun. 2015. [Google Scholar] [CrossRef] [Green Version]
- Riedl, J.; Fleming, A.M.; Burrows, C.J. Sequencing of DNA Lesions Facilitated by Site-Specific Excision via Base Excision Repair DNA Glycosylases Yielding Ligatable Gaps. J. Am. Chem. Soc. 2016, 138. [Google Scholar] [CrossRef] [Green Version]
- Craig, J.M.; Laszlo, A.H.; Derrington, I.M.; Ross, B.C.; Brinkerhoff, H.; Nova, I.C.; Doering, K.; Tickman, B.I.; Svet, M.T.; Gundlach, J.H. Direct detection of unnatural DNA nucleotides dNaM and d5SICS using the MspA nanopore. PLoS ONE 2015, 10, e0143253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, P.; Kudirka, R.; Albers, A.E.; Barfield, R.M.; De Hart, G.W.; Drake, P.M.; Jones, L.C.; Rabuka, D. Hydrazino-pictet-spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjug. Chem. 2013, 24. [Google Scholar] [CrossRef] [PubMed]
- Van Luenen, H.G.A.M.; Farris, C.; Jan, S.; Genest, P.A.; Tripathi, P.; Velds, A.; Kerkhoven, R.M.; Nieuwland, M.; Haydock, A.; Ramasamy, G.; et al. Glucosylated hydroxymethyluracil, DNA base J, prevents transcriptional readthrough in Leishmania. Cell 2012, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, D.; Cliffe, L.; Förstner, K.U.; Hon, C.C.; Siegel, T.N.; Sabatini, R. Regulation of transcription termination by glucosylated hydroxymethyluracil, base J, in Leishmania major and Trypanosoma brucei. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef]
- Bullard, W.; Lopes Da Rosa-Spiegler, J.; Liu, S.; Wang, Y.; Sabatini, R. Identification of the glucosyltransferase that converts hydroxymethyluracil to base J in the trypanosomatid genome. J. Biol. Chem. 2014, 289. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.; Wu, X.; Zhou, J.; Wu, H.; Liu, L.; Zhang, Q.; Demott, M.S.; Gu, C.; Wang, L.; You, D.; et al. Nick-seq for single-nucleotide resolution genomic maps of DNA modifications and damage. Nucleic Acids Res. 2020, 48, 6715–6725. [Google Scholar] [CrossRef]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef] [Green Version]
- McNulty, S.M.; Sullivan, B.A. Alpha satellite DNA biology: Finding function in the recesses of the genome. Chromosom. Res. 2018, 26, 115–138. [Google Scholar] [CrossRef]
- Miga, K.H.; Newton, Y.; Jain, M.; Altemose, N.; Willard, H.F.; Kent, W.J. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 2014, 24, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, F.; Beraldi, D.; Hardisty, R.E.; McInroy, G.R.; van Delft, P.; Balasubramanian, S. Genome-wide mapping of 5-hydroxymethyluracil in the eukaryote parasite Leishmania. Genome Biol. 2017, 18, 23. [Google Scholar] [CrossRef] [Green Version]
- Nabel, C.S.; Schutsky, E.K.; Kohli, R.M. Molecular targeting of mutagenic AID and APOBEC deaminases. Cell Cycle 2014, 13, 171–172. [Google Scholar] [CrossRef] [Green Version]
- Bhagwat, A.S.; Hao, W.; Townes, J.P.; Lee, H.; Tang, H.; Foster, P.L. Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in Escherichia coli. Proc. Natl. Acad. Sci. USA 2016, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dU-Seq | UDP-Seq | U-DNA-Seq | |
|---|---|---|---|
| Genome | human | E. coli | human |
| Gene deficiency | ung−/− wt | ung−/dut− vs. wt, ung−/mug− | hmlh1−/− Restored MMR |
| Transgene | - ung UDGs | - A3A, A3A* EV | ugi - |
| Treatment | - 5FdUR | - - | 5FdUR, RTX - |
| Data pre-processing | Trim, Align | Trim, Align, Filter | Trim, Align, Blacklist, Filter |
| Enrichment analysis | Peak calling | Peak calling, NDC, UI | Coverage, log2 ratio, broad regions |
| Conclusion | Centromeric enrichment | ung−/dut−: replication origin A3A in ung−/mug−: lagging strand, hairpin loops, tRNA genes | Heterochromatin, upon treatments: shifted towards early replicating and active euchromatin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Békési, A.; Holub, E.; Pálinkás, H.L.; Vértessy, B.G. Detection of Genomic Uracil Patterns. Int. J. Mol. Sci. 2021, 22, 3902. https://doi.org/10.3390/ijms22083902
Békési A, Holub E, Pálinkás HL, Vértessy BG. Detection of Genomic Uracil Patterns. International Journal of Molecular Sciences. 2021; 22(8):3902. https://doi.org/10.3390/ijms22083902
Chicago/Turabian StyleBékési, Angéla, Eszter Holub, Hajnalka Laura Pálinkás, and Beáta G. Vértessy. 2021. "Detection of Genomic Uracil Patterns" International Journal of Molecular Sciences 22, no. 8: 3902. https://doi.org/10.3390/ijms22083902
APA StyleBékési, A., Holub, E., Pálinkás, H. L., & Vértessy, B. G. (2021). Detection of Genomic Uracil Patterns. International Journal of Molecular Sciences, 22(8), 3902. https://doi.org/10.3390/ijms22083902