
Palindromes in DNA—A Risk for Genome Stability and Implications in Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Recombinogenic Nature of Palindromic Sequences
2.1. DNA Palidromes Can Form Secondary Structures
2.2. Molecular Mechanisms of Palindrome Recombinogenicity
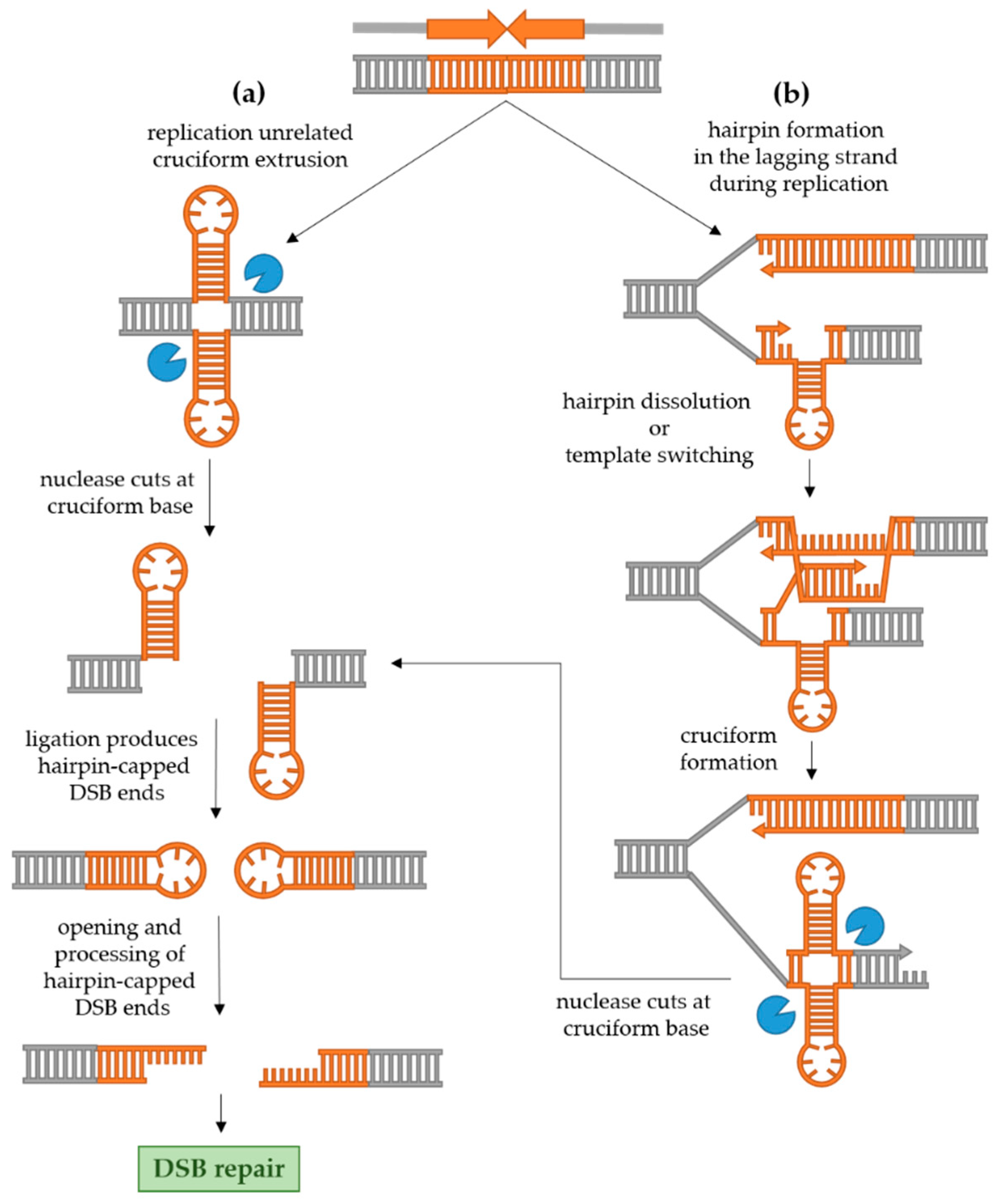
2.2.1. Replication-Independent Palindrome Recombinogenicity
2.2.2. Replication-Dependent Palindrome Recombinogenicity
3. DNA Palindromes and Genetic Instability in Cancer Cells
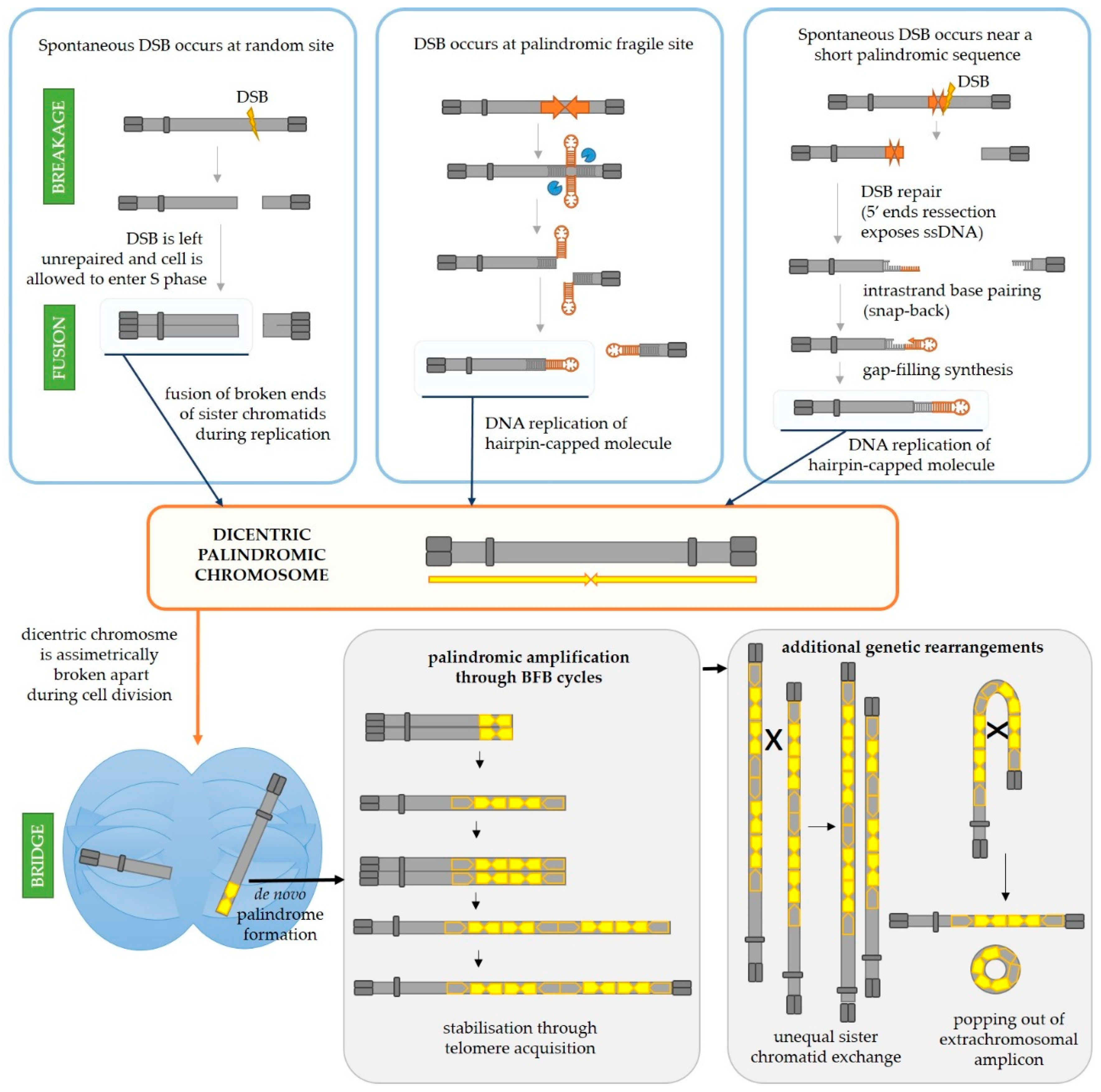
3.1. Palindromic Amplifications in Cancer
3.1.1. Palindromic Amplification in Cancer Can Arise as a Result of Iterative Break-Fusion-Bridge Cycles
3.1.2. Relatively Short Palindromes in the Genome Can Facilitate the Initiating Event of Palindromic Amplification through the Fold-Back Priming Mechanism
3.1.3. Longer Palindromes in the Genome Are Fragile Sites Which Can Lead to Palindromic Duplication
3.2. Challenges in Decyphering the Initiating Event Responsible for Palindromic Amplifications in Cancer
4. DNA Palindromes in the Human Genome
4.1. Bioinformatics in Quest for DNA Palindromes
4.2. From Short Interspersed Elements (SINEs) to Segmental Duplications—Possibilities for Palindrome Occurrence in the Human Genome
4.3. PATRRs and Other Known Palindromes in the Human Genome
5. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| BAC | bacterial artificial chromosome |
| BFB | breakage-fusion-bridge |
| DSB | double-strand break |
| GAPF | genome-wide analysis of palindrome formation |
| GWAS | genome-wide association study |
| NHEJ | non-homologous end joining |
| PATRR | palindromic AT-rich repeat |
| SINE | short interspersed element |
| SNP | single nucleotide polymorphism |
| TCGA | The Cancer Genome Atlas |
References
- Brázda, V.; Laister, R.C.; Jagelská, E.B.; Arrowsmith, C. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Murchie, A.I.; Lilley, D.M. The mechanism of cruciform formation in supercoiled DNA: Initial opening of central basepairs in salt-dependent extrusion. Nucleic Acids Res. 1987, 15, 9641–9654. [Google Scholar] [CrossRef] [Green Version]
- Courey, A.J.; Wang, J.C. Cruciform formation in a negatively supercoiled DNA may be kinetically forbidden under physiological conditions. Cell 1983, 33, 817–829. [Google Scholar] [CrossRef]
- Furlong, J.C.; Sullivan, K.M.; Murchie, A.I.H.; Gough, G.W.; Lilley, D.M.J. Localized chemical hyperreactivity in supercoiled DNA. Evidence for base unpairing in sequences that induce low-salt cruciform extrusion. Biochemistry 1989, 28, 2009–2017. [Google Scholar] [CrossRef] [PubMed]
- Miklenić, M.S.; Gatalica, N.; Matanović, A.; Žunar, B.; Štafa, A.; Lisnić, B.; Svetec, I.K. Size-dependent antirecombinogenic effect of short spacers on palindrome recombinogenicity. DNA Repair 2020, 90, 102848. [Google Scholar] [CrossRef]
- Lilley, D.M. The inverted repeat as a recognizable structural feature in supercoiled DNA molecules. Proc. Natl. Acad. Sci. USA 1980, 77, 6468–6472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuuchi, K.; Kemper, B.; Hays, J.; Weisberg, R.A. T4 endonuclease VII cleaves holliday structures. Cell 1982, 29, 357–365. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Lilley, D.M. Influence of cation size and charge on the extrusion of a salt-dependent cruciform. J. Mol. Biol. 1987, 193, 397–404. [Google Scholar] [CrossRef]
- Ramreddy, T.; Sachidanandam, R.; Strick, T.R. Real-time detection of cruciform extrusion by single-molecule DNA nanomanipulation. Nucleic Acids Res. 2011, 39, 4275–4283. [Google Scholar] [CrossRef] [Green Version]
- Vologodskii, A.; Frank-Kamenetskii, M. The relaxation time for a cruciform structure in superhelical DNA. FEBS Lett. 1983, 160, 173–176. [Google Scholar] [CrossRef] [Green Version]
- Benham, C.J.; Savitt, A.G.; Bauer, W.R. Extrusion of an imperfect palindrome to a cruciform in superhelical DNA: Complete determination of energetics using a statistical mechanical model. J. Mol. Biol. 2002, 316, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzine, F.; Sanford, S.; Elisha-Feil, Z.; Levens, D. The functional response of upstream DNA to dynamic supercoiling in vivo. Nat. Struct. Mol. Biol. 2008, 15, 146–154. [Google Scholar] [CrossRef]
- Naughton, C.; Avlonitis, N.; Corless, S.; Prendergast, J.G.; Mati, I.K.; Eijk, P.P.; Cockroft, S.L.; Bradley, M.; Ylstra, B.; Gilbert, N. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 2013, 20, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, U.; Weston-Hafer, K.; Berg, D.E. Local DNA Sequence Control of Deletion Formation in Escherichia coli Plasmid pBR322. Genetics 1987, 115, 41–49. [Google Scholar] [CrossRef]
- Sinden, R.R.; Zheng, G.X.; Brankamp, R.G.; Allen, K.N. On the deletion of inverted repeated DNA in Escherichia coli: Effects of length, thermal stability, and cruciform formation in vivo. Genetics 1991, 129, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Chalker, A.F.; Okely, E.A.; Davison, A.; Leach, D.R. The effects of central asymmetry on the propagation of palin-dromic DNA in bacteriophage lambda are consistent with cruciform extrusion in vivo. Genetics 1993, 133, 143–148. [Google Scholar] [CrossRef]
- Lobachev, K.S.; Shor, B.M.; Tran, H.T.; Taylor, W.; Keen, J.D.; Resnick, M.A.; Gordenin, D.A. Factors affecting in-verted repeat stimulation of recombination and deletion in Saccharomyces cerevisiae. Genetics 1998, 148, 1507–1524. [Google Scholar] [CrossRef]
- Lobachev, K.S.; Stenger, J.E.; Kozyreva, O.G.; Jurka, J.; Gordenin, D.A.; Resnick, M.A. InvertedAlurepeats unstable in yeast are excluded from the human genome. EMBO J. 2000, 19, 3822–3830. [Google Scholar] [CrossRef] [Green Version]
- Lisnić, B.; Svetec, I.-K.; Štafa, A.; Zgaga, Z. Size-dependent palindrome-induced intrachromosomal recombination in yeast. DNA Repair 2009, 8, 383–389. [Google Scholar] [CrossRef]
- Kato, T.; Inagaki, H.; Tong, M.; Kogo, H.; Ohye, T.; Yamada, K.; Tsutsumi, M.; Emanuel, B.S.; Kurahashi, H. DNA secondary structure is influenced by genetic variation and alters susceptibility to de novo translocation. Mol. Cytogenet. 2011, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eykelenboom, J.K.; Blackwood, J.K.; Okely, E.; Leach, D.R. SbcCD Causes a Double-Strand Break at a DNA Palindrome in the Escherichia coli Chromosome. Mol. Cell 2008, 29, 644–651. [Google Scholar] [CrossRef]
- Azeroglu, B.; Lincker, F.; White, M.A.; Jain, D.; Leach, D.R. A perfect palindrome in the Escherichia coli chromosome forms DNA hairpins on both leading- and lagging-strands. Nucleic Acids Res. 2014, 42, 13206–13213. [Google Scholar] [CrossRef] [Green Version]
- Lai, P.J.; Lim, C.T.; Le, H.P.; Katayama, T.; Leach, D.R.F.; Furukohri, A.; Maki, H. Long inverted repeat transiently stalls DNA replication by forming hairpin structures on both leading and lagging strands. Genes Cells 2016, 21, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Lobachev, K.S.; Gordenin, D.A.; Resnick, M.A. The Mre11 Complex Is Required for Repair of Hairpin-Capped Double-Strand Breaks and Prevention of Chromosome Rearrangements. Cell 2002, 108, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Voineagu, I.; Narayanan, V.; Lobachev, K.S.; Mirkin, S.M. Replication stalling at unstable inverted repeats: Interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 9936–9941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Saini, N.; Sheng, Z.; Lobachev, K.S. Genome-Wide Screen Reveals Replication Pathway for Quasi-Palindrome Fragility Dependent on Homologous Recombination. PLoS Genet. 2013, 9, e1003979. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, H.; Takahagi, M.; Shiba, T.; Nakata, A.; Shinagawa, H. Escherichia coli RuvC protein is an endonuclease that resolves the Holliday structure. EMBO J. 1991, 10, 4381–4389. [Google Scholar] [CrossRef]
- Inagaki, H.; Ohye, T.; Kogo, H.; Tsutsumi, M.; Kato, T.; Tong, M.; Emanuel, B.S.; Kurahashi, H. Two sequential cleavage reactions on cruciform DNA structures cause palindrome-mediated chromosomal translocations. Nat. Commun. 2013, 4, 1592. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, V.; Mieczkowski, P.A.; Kim, H.-M.; Petes, T.D.; Lobachev, K.S. The Pattern of Gene Amplification Is Determined by the Chromosomal Location of Hairpin-Capped Breaks. Cell 2006, 125, 1283–1296. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- DePamphilis, M.L. Eukaryotic DNA replication forks. ChemTracts-Biochem. Mol. Biol. 2002, 15, 313–325. [Google Scholar]
- Casper, A.M.; Greenwell, P.W.; Tang, W.; Petes, T.D. Chromosome Aberrations Resulting From Double-Strand DNA Breaks at a Naturally Occurring Yeast Fragile Site Composed of Inverted Ty Elements Are Independent of Mre11p and Sae2p. Genetics 2009, 183, 423–439. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef]
- Wang, T.-L.; Diaz, L.A.; Iacobuzio-Donahue, C.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C.; Velculescu, V.E.; Romans, K.; Bardelli, A.; Saha, S.; et al. Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3089–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, K.; DeVries, S.; Fridlyand, J.; Spellman, P.T.; Roydasgupta, R.; Kuo, W.-L.; Lapuk, A.; Neve, R.M.; Qian, Z.; Ryder, T.; et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006, 10, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, J.; Krasnitz, A.; Lakshmi, B.; Navin, N.E.; Riggs, M.; Leibu, E.; Esposito, D.; Alexander, J.; Troge, J.; Grubor, V.; et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006, 16, 1465–1479. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas (TCGA) Program. Available online: https://www.cancer.gov/tcga (accessed on 15 December 2020).
- Tanaka, H.; Watanabe, T. Mechanisms Underlying Recurrent Genomic Amplification in Human Cancers. Trends Cancer 2020, 6, 462–477. [Google Scholar] [CrossRef]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar] [PubMed]
- Tanaka, H.; Yao, M.-C. Palindromic gene amplification—An evolutionarily conserved role for DNA inverted repeats in the genome. Nat. Rev. Cancer 2009, 9, 216–224. [Google Scholar] [CrossRef]
- Marotta, M.; Chen, X.; Watanabe, T.; Faber, P.W.; Diede, S.J.; Tapscott, S.; Tubbs, R.; Kondratova, A.; Stephens, R.; Tanaka, H. Homology-mediated end-capping as a primary step of sister chromatid fusion in the breakage-fusion-bridge cycles. Nucleic Acids Res. 2013, 41, 9732–9740. [Google Scholar] [CrossRef] [Green Version]
- Feijoo, P.; Dominguez, D.; Tusell, L.; Genesca, A. Telomere-Dependent Genomic Integrity: Evolution of the Fusion-Bridge-Breakage Cycle Concept. Curr. Pharm. Des. 2014, 20, 6375–6385. [Google Scholar] [CrossRef]
- Kiwerska, K.; Szyfter, K. DNA repair in cancer initiation, progression, and therapy—A double-edged sword. J. Appl. Genet. 2019, 60, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gisselsson, D.; Pettersson, L.; Höglund, M.; Heidenblad, M.; Gorunova, L.; Wiegant, J.; Mertens, F.; Cin, P.D.; Mitelman, F.; Mandahl, N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc. Natl. Acad. Sci. USA 2000, 97, 5357–5362. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.W.I.; Sabatier, L.; Fouladi, B.; Pottier, G.; Ricoul, M.; Mumane, J.P. DNA Amplification by Breakage/Fusion/Bridge Cycles Initiated by Spontaneous Telomere Loss in a Human Cancer Cell Line. Neoplasia 2002, 4, 531–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellman, A.; Zlotorynski, E.; Scherer, S.W.; Cheung, J.; Vincent, J.B.; Smith, D.I.; Trakhtenbrot, L.; Kerem, B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 2002, 1, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Shuster, M.I.; Han, L.; Le Beau, M.M.; Davis, E.; Sawicki, M.; Lese, C.M.; Park, N.H.; Colicelli, J.; Gollin, S.M. A consistent pattern of RIN1 rearrangements in oral squamous cell carcinoma cell lines supports a breakage-fusion-bridge cycle model for 11q13 amplification. Genes Chromosom. Cancer 2002, 28, 153–163. [Google Scholar] [CrossRef]
- Coquelle, A.; Pipiras, E.; Toledo, F.; Buttin, G.; Debatisse, M. Expression of Fragile Sites Triggers Intrachromosomal Mammalian Gene Amplification and Sets Boundaries to Early Amplicons. Cell 1997, 89, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Shao, Z.-Y.; Wen, X.; Zhang, L.-Z. New insights of extrachromosomal DNA in tumorigenesis and therapeutic resistance of cancer. Am. J. Cancer Res. 2020, 10, 4056–4065. [Google Scholar] [PubMed]
- Kim, H.; Nguyen, N.-P.; Turner, K.; Wu, S.; Gujar, A.D.; Luebeck, J.; Liu, J.; Deshpande, V.; Rajkumar, U.; Namburi, S.; et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet. 2020, 52, 1–7. [Google Scholar] [CrossRef]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nat. Cell Biol. 2017, 543, 122–125. [Google Scholar] [CrossRef]
- Xu, K.; Ding, L.; Chang, T.-C.; Shao, Y.; Chiang, J.; Mulder, H.; Wang, S.; Shaw, T.I.; Wen, J.; Hover, L.; et al. Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol. 2019, 137, 123–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symington, L.S. End Resection at Double-Strand Breaks: Mechanism and Regulation. Cold Spring Harb. Perspect. Biol. 2014, 6, a016436. [Google Scholar] [CrossRef] [Green Version]
- Butler, D.K.; Yasuda, L.; Yao, M.-C. Induction of Large DNA Palindrome Formation in Yeast: Implications for Gene Amplification and Genome Stability in Eukaryotes. Cell 1996, 87, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Butler, D.K.; Gillespie, D.; Steele, B. Formation of large palindromic DNA by homologous recombination of short in-verted repeat sequences in Saccharomyces cerevisiae. Genetics 2002, 161, 1065–1075. [Google Scholar] [PubMed]
- Rattray, A.J.; Shafer, B.K.; Neelam, B.; Strathern, J.N. A mechanism of palindromic gene amplification in Saccharomyces cerevisiae. Genes Dev. 2005, 19, 1390–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Tapscott, S.J.; Trask, B.J.; Yao, M.-C. Short inverted repeats initiate gene amplification through the formation of a large DNA palindrome in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 8772–8777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, S.K.; Yin, Y.; Petes, T.D.; Symington, L.S. Mre11-Sae2 and RPA Collaborate to Prevent Palindromic Gene Amplification. Mol. Cell 2015, 60, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.K.; Gibb, B.; De Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Li, B.-Z.; Putnam, C.D.; Kolodner, R.D. Mechanisms underlying genome instability mediated by formation of foldback inversions in Saccharomyces cerevisiae. eLife 2020, 9, 58223. [Google Scholar] [CrossRef]
- Patel, D.R.; Weiss, R.S. A tough row to hoe: When replication forks encounter DNA damage. Biochem. Soc. Trans. 2018, 46, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.A.; Lambert, S.A.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Pilzecker, B.; Buoninfante, O.A.; Jacobs, H. DNA damage tolerance in stem cells, ageing, mutagenesis, disease and cancer therapy. Nucleic Acids Res. 2019, 47, 7163–7181. [Google Scholar] [CrossRef] [Green Version]
- Shastri, N.; Tsai, Y.-C.; Hile, S.; Jordan, D.; Powell, B.; Chen, J.; Maloney, D.; Dose, M.; Lo, Y.; Anastassiadis, T.; et al. Genome-wide Identification of Structure-Forming Repeats as Principal Sites of Fork Collapse upon ATR Inhibition. Mol. Cell 2018, 72, 222–238.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Maciejowski, J.; De Lange, J.M.T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Bergstrom, A.D.; Yao, M.-C.; Tapscott, S.J. Widespread and nonrandom distribution of DNA palindromes in cancer cells provides a structural platform for subsequent gene amplification. Nat. Genet. 2005, 37, 320–327. [Google Scholar] [CrossRef]
- Yang, H.; Volfovsky, N.; Rattray, A.; Chen, X.; Tanaka, H.; Strathern, J. GAP-Seq: A method for identification of DNA palindromes. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Bergstrom, D.A.; Yao, M.-C.; Tapscott, S.J. Large DNA palindromes as a common form of structural chromosome aberrations in human cancers. Hum. Cell 2006, 19, 17–23. [Google Scholar] [CrossRef]
- Guenthoer, J.; Diede, S.J.; Tanaka, H.; Chai, X.; Hsu, L.; Tapscott, S.J.; Porter, P.L. Assessment of palindromes as platforms for DNA amplification in breast cancer. Genome Res. 2011, 22, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Ganapathiraju, M.K.; Subramanian, S.; Chaparala, S.; Karunakaran, K.B. A reference catalog of DNA palindromes in the human genome and their variations in 1000 Genomes. Hum. Genome Var. 2020, 7, 1–12. [Google Scholar] [CrossRef]
- Lu, L.; Jia, H.; Dröge, P.; Li, J. The human genome-wide distribution of DNA palindromes. Funct. Integr. Genom. 2007, 7, 221–227. [Google Scholar] [CrossRef]
- Lisnić, B.; Svetec, I.-K.; Šarić, H.; Nikolić, I.; Zgaga, Z. Palindrome content of the yeast Saccharomyces cerevisiae genome. Curr. Genet. 2005, 47, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, E.M.; Benson, G.; Gelfand, Y.; Benham, C.J. The distribution of inverted repeat sequences in the Saccharomyces cerevisiae genome. Curr. Genet. 2010, 56, 321–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey-Dixon, E.L.; Sharp, R.; Schuckers, M.; Lock, R. Comparative genome analysis suggests characteristics of yeast inverted repeats that are important for transcriptional activity. Genome 2011, 54, 934–942. [Google Scholar] [CrossRef]
- Subramanian, S.; Chaparala, S.; Avali, V.; Ganapathiraju, M.K. A pilot study on the prevalence of DNA palindromes in breast cancer genomes. BMC Med. Genom. 2016, 9, 73–259. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Han, Y.-S. OMPPM: Online multiple palindrome pattern matching. Bioinformatics 2015, 32, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, J.-M. Lirex: A Package for Identification of Long Inverted Repeats in Genomes. Genom. Proteom. Bioinform. 2017, 15, 141–146. [Google Scholar] [CrossRef]
- Lewis, S.M. New approaches to the analysis of palindromic sequences from the human genome: Evolution and polymorphism of an intronic site at the NF1 locus. Nucleic Acids Res. 2005, 33, e186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houck, C.M.; Rinehart, F.P.; Schmid, C.W. A ubiquitous family of repeated DNA sequences in the human genome. J. Mol. Biol. 1979, 132, 289–306. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nat. Cell Biol. 2001, 409, 860–921. [CrossRef] [Green Version]
- Batzer, M.A.; Deininger, P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef]
- Peixoto, A.; Pinheiro, M.; Massena, L.; Santos, C.; Pinto, P.; Rocha, P.; Pinto, C.; Teixeira, M.R. Genomic characterization of two large Alu-mediated rearrangements of the BRCA1 gene. J. Hum. Genet. 2012, 58, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.; Bhatt, S.S.; Janssen, I.M.; Xia, Z.; Lalani, S.R.; Pfundt, R.; Derwinska, K.; De Vries, B.B.; Gilissen, C.; Hoischen, A.; et al. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009, 18, 3579–3593. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Beck, C.R.; Du, R.; Campbell, I.M.; Coban-Akdemir, Z.; Gu, S.; Breman, A.M.; Stankiewicz, P.; Ira, G.; Shaw, C.A.; et al. Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res. 2018, 28, 1228–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deininger, P.L.; Batzer, M.A. Alu Repeats and Human Disease. Mol. Genet. Metab. 1999, 67, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lu, H.H.S.; Chung, W.-Y.; Yang, J.; Li, W.-H. Patterns of Segmental Duplication in the Human Genome. Mol. Biol. Evol. 2004, 22, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Trombetta, B.; Cruciani, F. Y chromosome palindromes and gene conversion. Qual. Life Res. 2017, 136, 605–619. [Google Scholar] [CrossRef]
- Lange, J.; Skaletsky, H.; Van Daalen, S.K.; Embry, S.L.; Korver, C.M.; Brown, L.G.; Oates, R.D.; Silber, S.J.; Repping, S.; Page, D.C. Isodicentric Y Chromosomes and Sex Disorders as Byproducts of Homologous Recombination that Maintains Palindromes. Cell 2009, 138, 855–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repping, S.; Skaletsky, H.; Lange, J.; Silber, S.; Van Der Veen, F.; Oates, R.D.; Page, D.C.; Rozen, S. Recombination between Palindromes P5 and P1 on the Human Y Chromosome Causes Massive Deletions and Spermatogenic Failure. Am. J. Hum. Genet. 2002, 71, 906–922. [Google Scholar] [CrossRef] [Green Version]
- Rozen, S.G.; Marszalek, J.D.; Irenze, K.; Skaletsky, H.; Brown, L.G.; Oates, R.D.; Silber, S.J.; Ardlie, K.; Page, D.C. AZFc Deletions and Spermatogenic Failure: A Population-Based Survey of 20,000 Y Chromosomes. Am. J. Hum. Genet. 2012, 91, 890–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannarella, R.; Condorelli, R.A.; Duca, Y.; La Vignera, S.; Calogero, A.E. New insights into the genetics of spermatogenic failure: A review of the literature. Qual. Life Res. 2019, 138, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Murata, M.M.; Manguso, N.; Watanabe, T.; Mouakkad-Montoya, L.; Igari, F.; Rahman, M.; Qu, Y.; Cui, X.; Giuliano, E.A.; et al. The fragility of a structurally diverse duplication block triggers recurrent genomic amplification. Nucleic Acids Res. 2021, 49, 244–256. [Google Scholar] [CrossRef]
- Inagaki, H.; Kato, T.; Tsutsumi, M.; Ouchi, Y.; Ohye, T.; Kurahashi, H. Palindrome-Mediated Translocations in Humans: A New Mechanistic Model for Gross Chromosomal Rearrangements. Front. Genet. 2016, 7, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
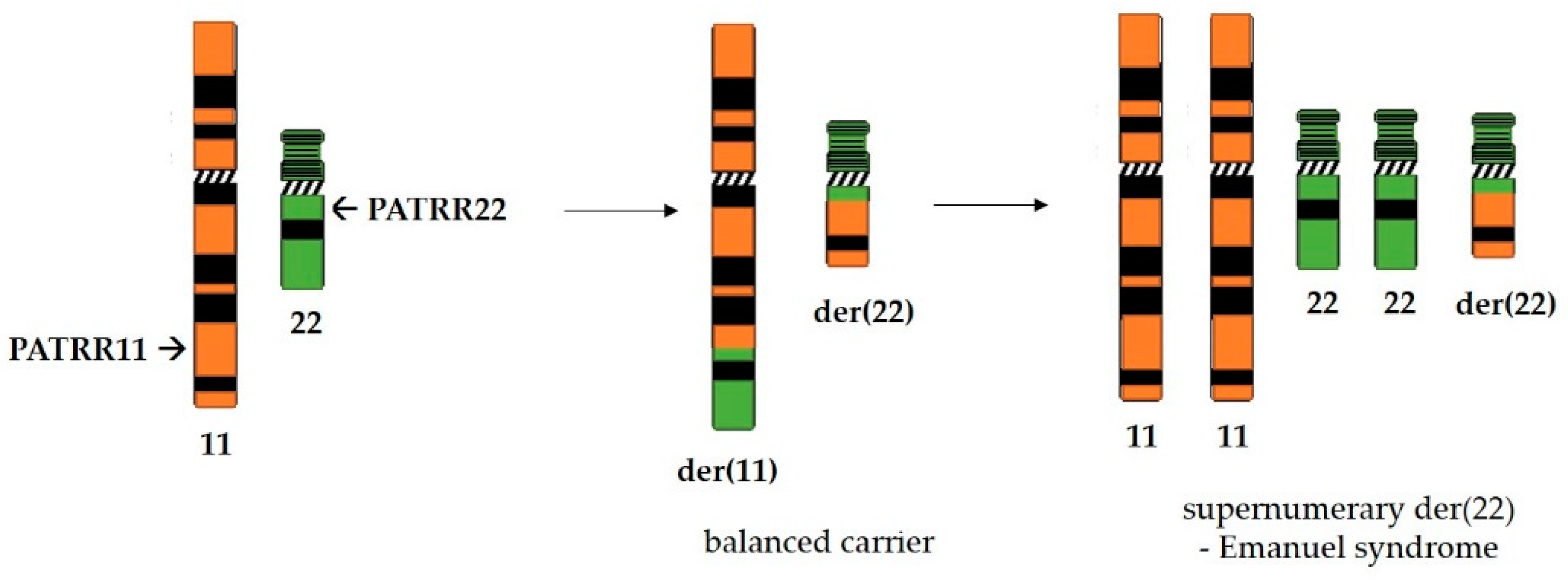
- Kurahashi, H.; Emanuel, B.S. Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum. Mol. Genet. 2001, 10, 2605–2617. [Google Scholar] [CrossRef] [Green Version]
- Schoemaker, M.J.; Jones, E.M.; Higgins, C.D.; Wright, A.F.; Swerdlow, A.J. the UK Clinical Cytogenetics Group Mortality and cancer incidence in carriers of constitutional t(11;22)(q23;q11) translocations: A prospective study. Int. J. Cancer 2018, 145, 1493–1498. [Google Scholar] [CrossRef]
- Carter, M.T.; Pierre, S.A.S.; Zackai, E.H.; Emanuel, B.S.; Boycott, K.M. Phenotypic delineation of Emanuel syndrome (supernumerary derivative 22 syndrome): Clinical features of 63 individuals. Am. J. Med. Genet. Part A 2009, 149A, 1712–1721. [Google Scholar] [CrossRef] [Green Version]
- Kurahashi, H.; Shaikh, T.H.; Zackai, E.H.; Celle, L.; Driscoll, D.A.; Budarf, M.L.; Emanuel, B.S. Tightly Clustered 11q23 and 22q11 Breakpoints Permit PCR-Based Detection of the Recurrent Constitutional t(11;22). Am. J. Hum. Genet. 2000, 67, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Kurahashi, H.; Emanuel, B.S. Unexpectedly high rate of de novo constitutional t(11;22) translocations in sperm from normal males. Nat. Genet. 2001, 29, 139–140. [Google Scholar] [CrossRef]
- Ohye, T.; Inagaki, H.; Kogo, H.; Tsutsumi, M.; Kato, T.; Tong, M.; Macville, M.V.E.; Medne, L.; Zackai, E.H.; Emanuel, B.S.; et al. Paternal origin of the de novo constitutional t(11;22)(q23;q11). Eur. J. Hum. Genet. 2010, 18, 783–787. [Google Scholar] [CrossRef]
- Crow, J.F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 2000, 1, 40–47. [Google Scholar] [CrossRef]
- Thomas, N.S.; Morris, J.K.; Baptista, J.; Ng, B.L.; Crolla, A.J.; Jacobs, A.P. De novo apparently balanced translocations in man are predominantly paternal in origin and associated with a significant increase in paternal age. J. Med. Genet. 2009, 47, 112–115. [Google Scholar] [CrossRef] [Green Version]
- Templado, C.; Donate, A.; Giraldo, J.; Bosch, M.; Estop, A. Advanced age increases chromosome structural abnormalities in human spermatozoa. Eur. J. Hum. Genet. 2010, 19, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Yamada, K.; Inagaki, H.; Kogo, H.; Ohye, T.; Emanuel, B.S.; Kurahashi, H. Age has no effect on de novo constitutional t(11;22) translocation frequency in sperm. Fertil. Steril. 2007, 88, 1446–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurahashi, H.; Inagaki, H.; Yamada, K.; Ohye, T.; Taniguchi, M.; Emanuel, B.S.; Toda, T. Cruciform DNA Structure Underlies the Etiology for Palindrome-mediated Human Chromosomal Translocations. J. Biol. Chem. 2004, 279, 35377–35383. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, H.; Ohye, T.; Kogo, H.; Kato, T.; Bolor, H.; Taniguchi, M.; Shaikh, T.H.; Emanuel, B.S.; Kurahashi, H. Chromosomal instability mediated by non-B DNA: Cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res. 2008, 19, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correll-Tash, S.; Lilley, B.; Iv, H.S.; Mlynarski, E.; Franconi, C.P.; McNamara, M.; Woodbury, C.; Easley, A.C.; Emanuel, B.S. Double strand breaks (DSBs) as indicators of genomic instability in PATRR-mediated translocations. Hum. Mol. Genet. 2021, 29, 3872–3881. [Google Scholar] [CrossRef] [PubMed]
- Deen, D.F.; Morgan, W.F.; Tofilon, P.J.; Barcellos-Hoff, M.H. Measurement of sister chromatid exchanges and their relationship to DNA damage, repair and cell killing. Pharmacol. Ther. 1989, 42, 349–360. [Google Scholar] [CrossRef]
- Johnson, R.D. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000, 19, 3398–3407. [Google Scholar] [CrossRef]
- Correll-Tash, S.; Conlin, L.; Mininger, B.A.; Lilley, B.; Mennuti, M.T.; Emanuel, B.S. The Recurrent t(11;22)(q23;q11.2) Can Occur as a Post-Zygotic Event. Cytogenet. Genome Res. 2018, 156, 185–190. [Google Scholar] [CrossRef]
- Feng, X.; Xie, F.-Y.; Ou, X.-H.; Ma, J.-Y. Cruciform DNA in mouse growing oocytes: Its dynamics and its relationship with DNA transcription. PLoS ONE 2020, 15, e0240844. [Google Scholar] [CrossRef] [PubMed]
- Chasovskikh, S.; Dimtchev, A.; Smulson, M.; Dritschilo, A. DNA transitions induced by binding of PARP-1 to cruciform structures in supercoiled plasmids. Cytom. Part. A 2005, 68, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.T.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.D.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Haussler, J.; Krone, W.; Bode, H.; Jenne, D.E.; Mehnert, K.U.; Tümmers, U.; Assum, G. The second case of a t(17;22) in a family with neurofibromatosis type 1: Sequence analysis of the breakpoint regions. Qual. Life Res. 1997, 99, 237–247. [Google Scholar] [CrossRef]
- Kurahashi, H.; Shaikh, T.; Takata, M.; Toda, T.; Emanuel, B.S. The Constitutional t(17;22): Another Translocation Mediated by Palindromic AT-Rich Repeats. Am. J. Hum. Genet. 2003, 72, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, M.B.; Kato, T.; Haldeman-Englert, C.; Jalali, G.R.; Milunsky, J.M.; Zou, Y.; Klaes, R.; Gimelli, G.; Gimelli, S.; Gemmill, R.M.; et al. A Palindrome-Mediated Recurrent Translocation with 3:1 Meiotic Nondisjunction: The t(8;22)(q24.13;q11.21). Am. J. Hum. Genet. 2010, 87, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Franconi, C.P.; Sheridan, M.B.; Hacker, A.M.; Inagakai, H.; Glover, T.W.; Arlt, M.F.; Drabkin, H.A.; Gemmill, R.M.; Kurahashi, H.; et al. Analysis of the t(3;8) of hereditary renal cell carcinoma: A palindrome-mediated translocation. Cancer Genet. 2014, 207, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotter, A.L.; Shaikh, T.H.; Budarf, M.L.; Rhodes, C.H.; Emanuel, B.S. A palindrome-mediated mechanism distinguishes translocations involving LCR-B of chromosome 22q11.2. Hum. Mol. Genet. 2004, 13, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmakayalu, M.A.; Gotter, A.L.; Shaikh, T.H.; Emanuel, B.S. A novel sequence-based approach to localize translocation breakpoints identifies the molecular basis of a t(4;22). Hum. Mol. Genet. 2003, 12, 2817–2825. [Google Scholar] [CrossRef]
- Kato, T.; Kurahashi, H.; Emanuel, B.S. Chromosomal translocations and palindromic AT-rich repeats. Curr. Opin. Genet. Dev. 2012, 22, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Anzick, S.L.; Khan, S.G.; Ueda, T.; Stone, G.; DiGiovanna, J.J.; Tamura, D.; Wattendorf, D.; Busch, D.; Brewer, C.C.; et al. Chimeric Negative Regulation ofp14ARFandTBX1by a t(9;22) Translocation Associated with Melanoma, Deafness, and DNA Repair Deficiency. Hum. Mutat. 2013, 34, 1250–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Shang, D.; Sun, M.; Choi, S.; Liu, Q.; Hao, J.; Figuera, L.E.; Zhang, F.; Choy, K.W.; Ao, Y.; et al. X-Linked Congenital Hypertrichosis Syndrome Is Associated with Interchromosomal Insertions Mediated by a Human-Specific Palindrome near SOX3. Am. J. Hum. Genet. 2011, 88, 819–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeStefano, G.M.; Fantauzzo, K.A.; Petukhova, L.; Kurban, M.; Tadin-Strapps, M.; Levy, B.; Warburton, D.; Cirulli, E.T.; Han, Y.; Sun, X.; et al. Position effect on FGF13 associated with X-linked congenital generalized hypertrichosis. Proc. Natl. Acad. Sci. USA 2013, 110, 7790–7795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimovski, A.J.; Baysal, E.; Efremov, D.G.; Prior, J.F.; Raven, J.L.; Efremov, G.D.; Huisman, T.H. A Large β-Thalassemia Deletion in A Fay of Indonesian-Malay Descent. Hemoglobin 1996, 20, 377–392. [Google Scholar] [CrossRef]
- Cardiero, G.; Prezioso, R.; Dembech, S.; Blanco, F.D.V.; Scarano, C.; Lacerra, G. Identification and molecular characterization of a novel 163 kb deletion: The Italian 0-thalassemia. Hematology 2016, 21, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svetec Miklenić, M.; Svetec, I.K. Palindromes in DNA—A Risk for Genome Stability and Implications in Cancer. Int. J. Mol. Sci. 2021, 22, 2840. https://doi.org/10.3390/ijms22062840
Svetec Miklenić M, Svetec IK. Palindromes in DNA—A Risk for Genome Stability and Implications in Cancer. International Journal of Molecular Sciences. 2021; 22(6):2840. https://doi.org/10.3390/ijms22062840
Chicago/Turabian StyleSvetec Miklenić, Marina, and Ivan Krešimir Svetec. 2021. "Palindromes in DNA—A Risk for Genome Stability and Implications in Cancer" International Journal of Molecular Sciences 22, no. 6: 2840. https://doi.org/10.3390/ijms22062840
APA StyleSvetec Miklenić, M., & Svetec, I. K. (2021). Palindromes in DNA—A Risk for Genome Stability and Implications in Cancer. International Journal of Molecular Sciences, 22(6), 2840. https://doi.org/10.3390/ijms22062840