
Structural and Functional Remodeling of Mitochondria in Cardiac Diseases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
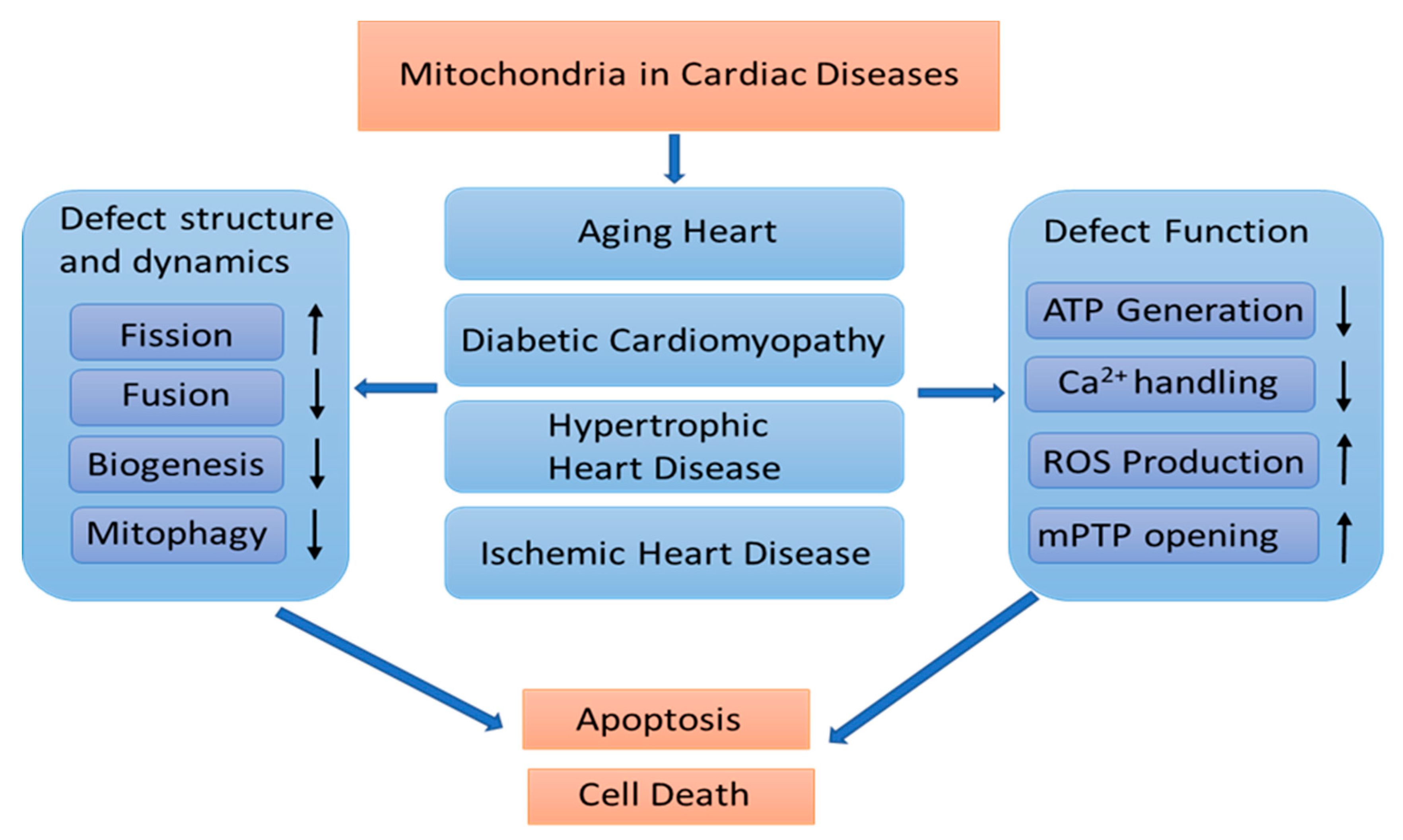
2. Mitochondrial Remodeling in Cardiac Diseases
2.1. The Overview of the Molecular Bases in Cardiac Mitochondrial Remodeling
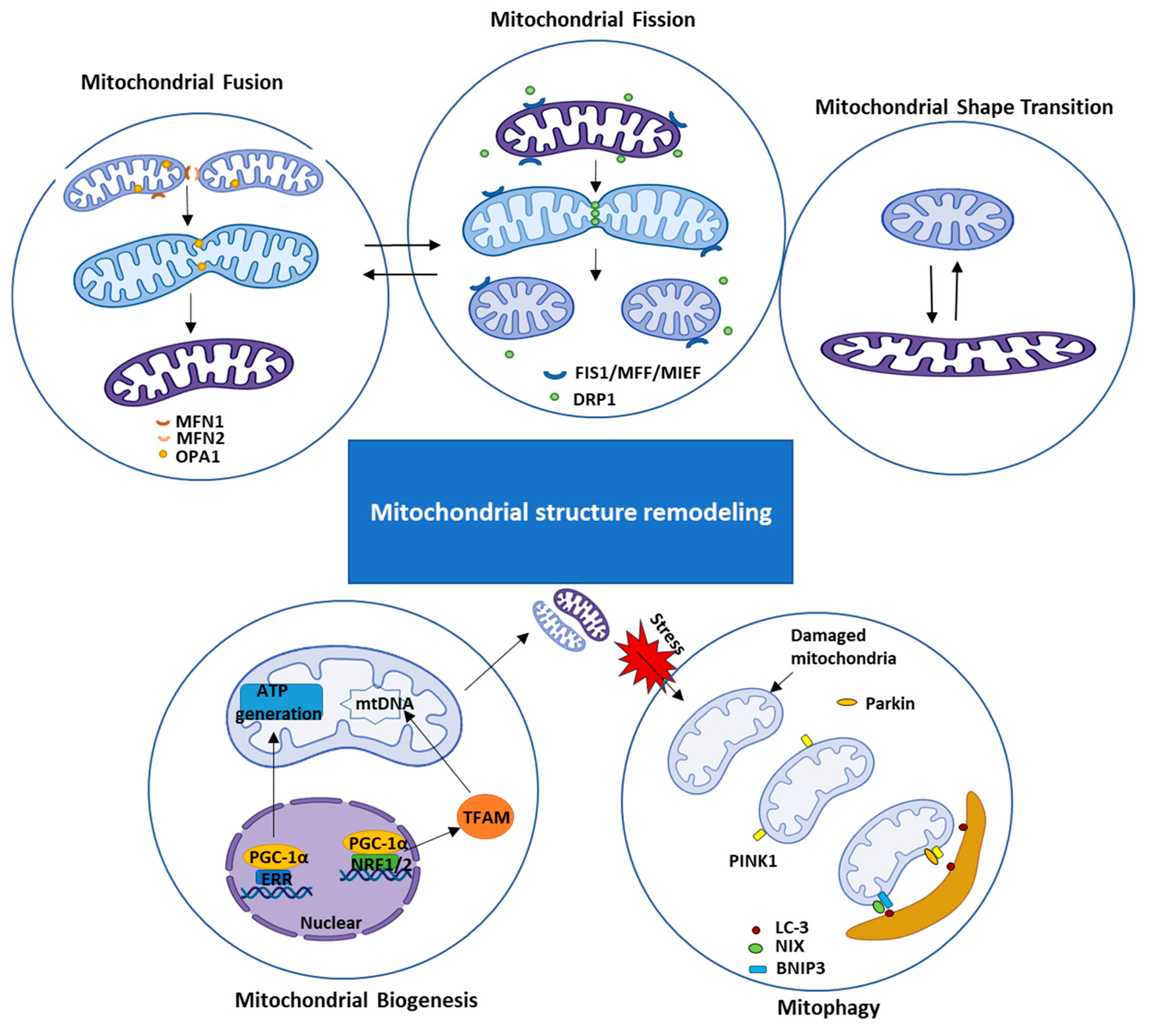
2.1.1. Mitochondrial Structural Remodeling
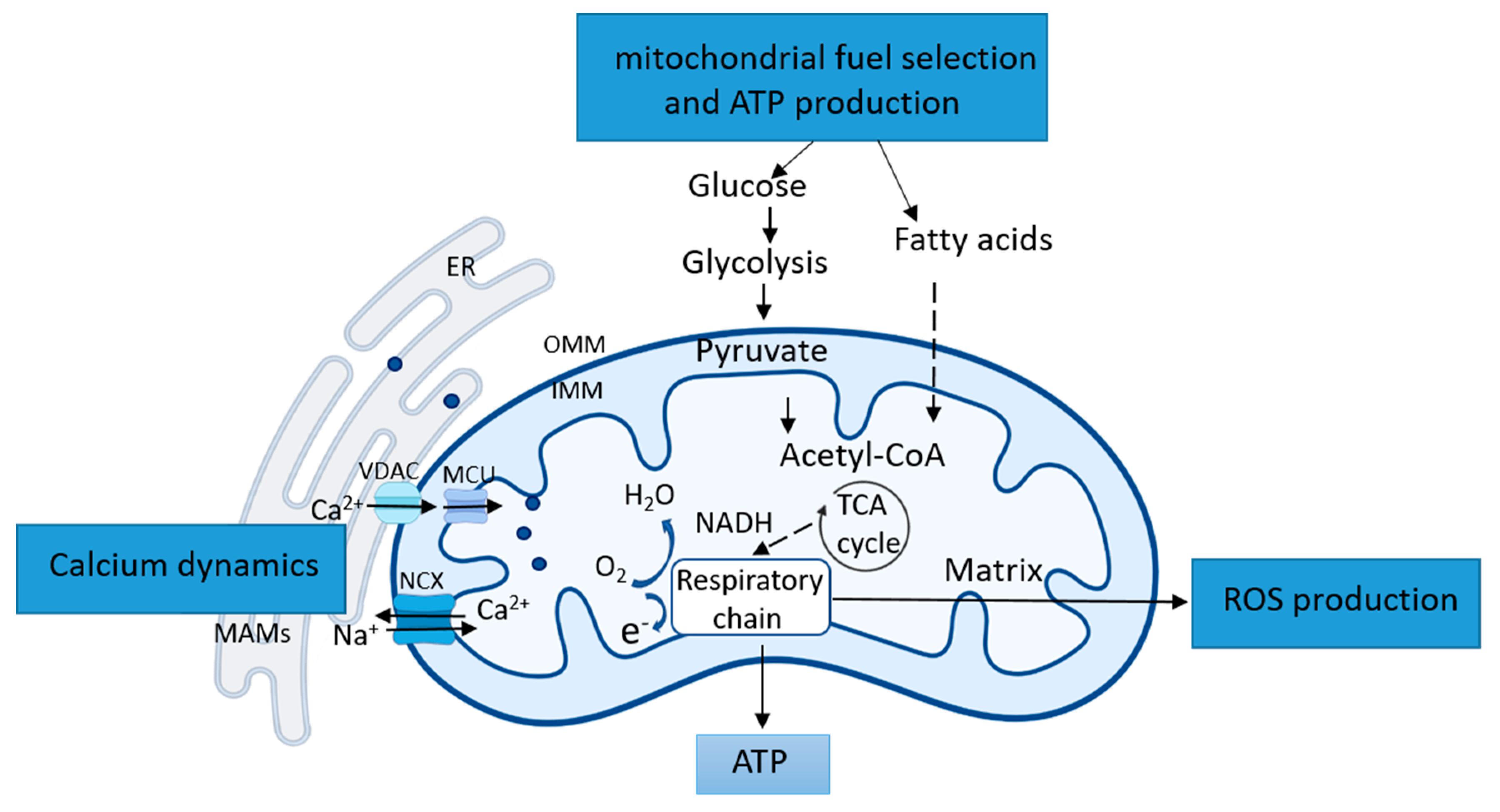
2.1.2. Mitochondrial Functional Remodeling
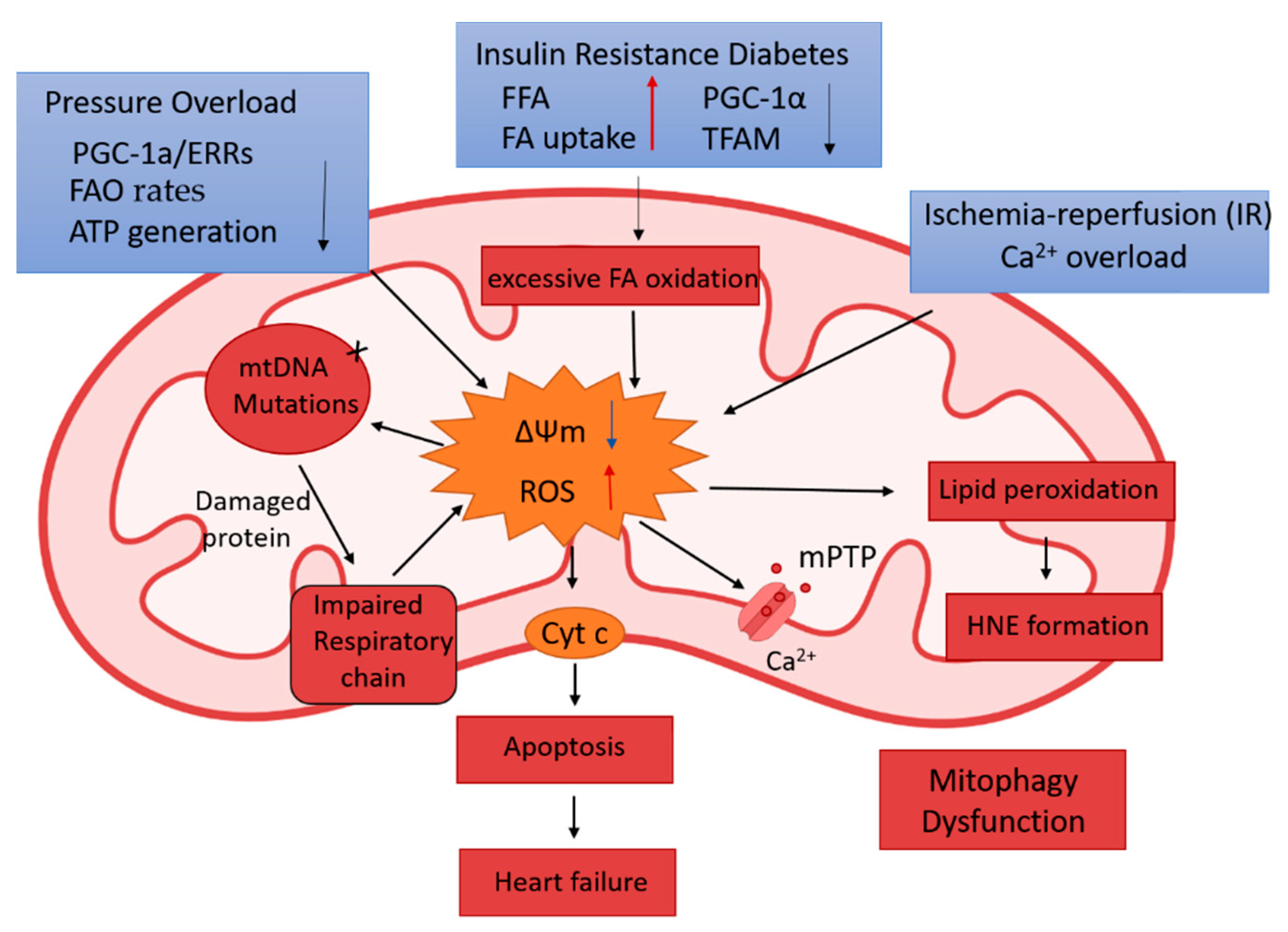
2.2. The Association of Mitochondrial Remodeling in Pathological Cardiac Conditions
2.2.1. Aging-Induced Cardiac Deterioration
2.2.2. Diabetic Cardiomyopathy (DCM)
2.2.3. Hypertrophic Heart Disease and Heart Failure
2.2.4. Acute Myocardial Infarction (AMI)
3. Conclusion, Clinical Potential, and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ii, G.W.D.; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Parra, V.; Verdejo, H.; Del Campo, A.; Pennanen, C.; Kuzmicic, J.; Iglewski, M.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. The complex interplay between mitochondrial dynamics and cardiac metabolism. J. Bioenerg. Biomembr. 2011, 43, 47–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Bernstein, D. Mitochondrial remodeling: Rearranging, recycling, and reprogramming. Cell Calcium 2016, 60, 88–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Kim, J.-A.; Wei, Y.; Sowers, J.R. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Donmez, G. Mitochondrial Biology and Neurological Diseases. Curr. Neuropharmacol. 2016, 14, 143–154. [Google Scholar] [CrossRef]
- Sabri, A.; Hughie, H.H.; Lucchesi, P.A. Regulation of Hypertrophic and Apoptotic Signaling Pathways by Reactive Oxygen Species in Cardiac Myocytes. Antioxidants Redox Signal. 2003, 5, 731–740. [Google Scholar] [CrossRef]
- Ni, R.; Zheng, N.; Xiong, S.; Hill, D.J.; Sun, T.; Gardiner, R.B.; Fan, G.-C.; Lu, Y.; Abel, E.D.; Greer, P.A.; et al. Mitochondrial Calpain-1 Disrupts ATP Synthase and Induces Superoxide Generation in Type 1 Diabetic Hearts: A Novel Mechanism Contributing to Diabetic Cardiomyopathy. Diabetes 2015, 65, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxidative Med. Cell. Longev. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Heath-Engel, H.M.; Shore, G.C. Mitochondrial membrane dynamics, cristae remodelling and apoptosis. Biochim. Biophys. Acta 2006, 1763, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 2010, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The Mitochondrial Protein hFis1 Regulates Mitochondrial Fission in Mammalian Cells through an Interaction with the Dynamin-Like Protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [Green Version]
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/Endoplasmic Reticulum Calcium ATPase Inhibitors: Beyond Anticancer Perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef]
- Jurášek, M.; Rimpelová, S.; Kmoníčková, E.; Drašar, P.; Ruml, T. Tailor-Made Fluorescent Trilobolide To Study Its Biological Relevance. J. Med. Chem. 2014, 57, 7947–7954. [Google Scholar] [CrossRef]
- Gandre-Babbe, S.; Van Der Bliek, A.M. The Novel Tail-anchored Membrane Protein Mff Controls Mitochondrial and Peroxisomal Fission in Mammalian Cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, S.; DeVay, R.; Block, J.; Cassidy-Stone, A.; Wayson, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial Inner-Membrane Fusion and Crista Maintenance Requires the Dynamin-Related GTPase Mgm1. Cell 2006, 127, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lendahl, U.; Nistér, M. Regulation of mitochondrial dynamics: Convergences and divergences between yeast and vertebrates. Cell. Mol. Life Sci. 2013, 70, 951–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemani, N.; Carvalho, E.; Tomar, D.; Dong, Z.; Ketschek, A.; Breves, S.L.; Jaña, F.; Worth, A.M.; Heffler, J.; Palaniappan, P.; et al. MIRO-1 Determines Mitochondrial Shape Transition upon GPCR Activation and Ca2+ Stress. Cell Rep. 2018, 23, 1005–1019. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Ji, W.-K.; Higgs, H.N.; Chakrabarti, R. Two distinct actin filament populations have effects on mitochondria, with differences in stimuli and assembly factors. J. Cell Sci. 2019, 132, jcs234435. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xu, S.; Roelofs, B.A.; Boyman, L.; Lederer, W.J.; Sesaki, H.; Karbowski, M. Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J. Cell Biol. 2015, 208, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef] [Green Version]
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 2020, 219, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The estrogen-related receptor (ERR) functions in PPAR coactivator 1 (PGC-1)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Scarpulla, R.C. NRF-1: A trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990, 4, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handschin, C.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor γ Coactivator 1 Coactivators, Energy Homeostasis, and Metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in aging, disease and death: The true identity of a cell death impostor. Cell Death Differ. 2008, 16, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Kawajiri, S.; Saiki, S.; Sato, S.; Sato, F.; Hatano, T.; Eguchi, H.; Hattori, N. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 2010, 584, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Ii, G.W.D. Mitochondrial Pruning by Nix and BNip3: An Essential Function for Cardiac-Expressed Death Factors. J. Cardiovasc. Transl. Res. 2010, 3, 374–383. [Google Scholar] [CrossRef] [Green Version]
- Coronado, M.; Fajardo, G.; Nguyen, K.; Zhao, M.; Kooiker, K.; Jung, G.; Hu, D.-Q.; Reddy, S.; Sandoval, E.; Stotland, A.; et al. Physiological Mitochondrial Fragmentation Is a Normal Cardiac Adaptation to Increased Energy Demand. Circ. Res. 2018, 122, 282–295. [Google Scholar] [CrossRef]
- Fenton, A.R.; Jongens, T.A.; Holzbaur, E.L. Mitochondrial dynamics: Shaping and remodeling an organelle network. Curr. Opin. Cell Biol. 2021, 68, 28–36. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Chen, L.; Song, J.; Hu, S. Metabolic remodeling of substrate utilization during heart failure progression. Hear. Fail. Rev. 2018, 24, 143–154. [Google Scholar] [CrossRef]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.S.B.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyman, L.; Chikando, A.C.; Williams, G.S.; Khairallah, R.J.; Kettlewell, S.; Ward, C.W.; Smith, G.L.; Kao, J.P.; Lederer, W.J. Calcium Movement in Cardiac Mitochondria. Biophys. J. 2014, 107, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Qiu, H. Valosin-Containing Protein, a Calcium-Associated ATPase Protein, in Endoplasmic Reticulum and Mitochondrial Function and Its Implications for Diseases. Int. J. Mol. Sci. 2020, 21, 3842. [Google Scholar] [CrossRef]
- Aronsen, J.M.; Louch, W.E.; Sjaastad, I. Cardiomyocyte Ca2+dynamics: Clinical perspectives. Scand. Cardiovasc. J. 2016, 50, 65–77. [Google Scholar] [CrossRef]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Bravo, R.; Gutierrez, T.; Paredes, F.; Gatica, D.; Rodriguez, A.E.; Pedrozo, Z.; Chiong, M.; Parra, V.; Quest, A.F.; Rothermel, B.A.; et al. Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int. J. Biochem. Cell Biol. 2012, 44, 16–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Rosiers, C.D.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Circ. Physiol. 2011, 300, H144–H153. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Poláková, E.; Janíček, R.; Shirokova, N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta 2020, 1861, 148250. [Google Scholar] [CrossRef]
- Indo, H.P.; Davidson, M.; Yen, H.-C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Jian, C.; Hou, T.; Yin, R.; Cheng, H.; Wang, X. Regulation of superoxide flashes by transient and steady mitochondrial calcium elevations. Sci. China Life Sci. 2014, 57, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Ide, T.; Kinugawa, S. Mitochondrial Oxidative Stress, DNA Damage, and Heart Failure. Antioxid. Redox Signal. 2006, 8, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Catalá, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef]
- Uchida, K. 4-Hydroxy-2-nonenal: A product and mediator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Niimi, K.; Yasui, T.; Hirose, M.; Hamamoto, S.; Itoh, Y.; Okada, A.; Kubota, Y.; Kojima, Y.; Tozawa, K.; Sasaki, S.; et al. Mitochondrial permeability transition pore opening induces the initial process of renal calcium crystallization. Free. Radic. Biol. Med. 2012, 52, 1207–1217. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. BioMed Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bakula, D.; Scheibye-Knudsen, M. MitophAging: Mitophagy in Aging and Disease. Front. Cell Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef] [Green Version]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial Metabolism in Aging Heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [Green Version]
- Gems, D.; De La Guardia, Y. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxid. Redox Signal. 2013, 19, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Liu, Y.; Zhang, R.; Zhang, F.; Sui, W.; Chen, L.; Zheng, R.; Chen, X.; Wen, F.; Ouyang, H.-W.; et al. Apoptotic transition of senescent cells accompanied with mitochondrial hyper-function. Oncotarget 2016, 7, 28286–28300. [Google Scholar] [CrossRef]
- Fernández-Ortiz, M.; Sayed, R.K.A.; Fernández-Martínez, J.; Cionfrini, A.; Aranda-Martínez, P.; Escames, G.; De Haro, T.; Acuña-Castroviejo, D. Melatonin/Nrf2/NLRP3 Connection in Mouse Heart Mitochondria during Aging. Antioxidants 2020, 9, 1187. [Google Scholar] [CrossRef]
- Zhou, B.; Lu, Y.; Hajifathalian, K.; Bentham, J.; Di Cesare, M.; Danaei, G.; Bixby, H.; Cowan, M.; Ali, M.; Taddei, C.; et al. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4·4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2008, 32, S62–S67. [CrossRef] [Green Version]
- Donnelly, R.; Emslie-Smith, A.M.; Gardner, I.D.; Morris, A.D. ABC of arterial and venous disease: Vascular complications of diabetes. Br. Med. J. 2000, 320, 1062–1066. [Google Scholar] [CrossRef]
- Fischer, V.W.; Barner, H.B.; LaRose, L.S. Pathomorphologic aspects of muscular tissue in diabetes mellitus. Hum. Pathol. 1984, 15, 1127–1136. [Google Scholar] [CrossRef]
- Sharma, V.; Dhillon, P.; Wambolt, R.; Parsons, H.; Brownsey, R.; Allard, M.F.; McNeill, J.H. Metoprolol improves cardiac function and modulates cardiac metabolism in the streptozotocin-diabetic rat. Am. J. Physiol. Circ. Physiol. 2008, 294, H1609–H1620. [Google Scholar] [CrossRef] [Green Version]
- Rijzewijk, L.J.; van der Meer, R.W.; Lamb, H.J.; de Jong, H.W.; Lubberink, M.; Romijn, J.A.; Bax, J.J.; de Roos, A.; Twisk, J.W.; Heine, R.J.; et al. Altered Myocardial Substrate Metabolism and Decreased Diastolic Function in Nonischemic Human Diabetic Cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Xia, C.; Li, S.; Du, L.; Zhang, L.; Zhou, R. Defective mitophagy driven by dysregulation of rheb and KIF5B contributes to mitochondrial reactive oxygen species (ROS)-induced nod-like receptor 3 (NLRP3) dependent proinflammatory response and aggravates lipotoxicity. Redox Biol. 2014, 3, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.-I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Long, J. Phosphatase and tensin homolog-induced putative kinase 1 and Parkin in diabetic heart: Role of mitophagy. J. Diabetes Investig. 2015, 6, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Lu, Q.; Ding, Y.; Wu, Y.; Qiu, Y.; Wang, P.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.-H. Hyperglycemia-Driven Inhibition of AMP-Activated Protein Kinase α2 Induces Diabetic Cardiomyopathy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes In Vivo. Circulation 2019, 139, 1913–1936. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef] [Green Version]
- Duncan, J.G.; Fong, J.L.; Medeiros, D.M.; Finck, B.N.; Kelly, D.P. Insulin-Resistant Heart Exhibits a Mitochondrial Biogenic Response Driven by the Peroxisome Proliferator-Activated Receptor-α/PGC-1α Gene Regulatory Pathway. Circulation 2007, 115, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makrecka-Kuka, M.; Liepinsh, E.; Murray, A.J.; Lemieux, H.; Dambrova, M.; Tepp, K.; Puurand, M.; Käämbre, T.; Han, W.H.; De Goede, P.; et al. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. 2019, 228, e13430. [Google Scholar] [CrossRef] [Green Version]
- Mereweather, L.J.; Aparicio, C.N.M.; Heather, L.C. Positioning Metabolism as a Central Player in the Diabetic Heart. J. Lipid Atheroscler. 2020, 9, 92–109. [Google Scholar] [CrossRef]
- Belke, D.D.; Betuing, S.; Tuttle, M.J.; Graveleau, C.; Young, M.E.; Pham, M.; Zhang, D.; Cooksey, R.C.; McClain, D.A.; Litwin, S.E.; et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Investig. 2002, 109, 629–639. [Google Scholar] [CrossRef] [PubMed]
- How, O.-J.; Larsen, T.S.; Hafstad, A.D.; Khalid, A.; Myhre, E.S.P.; Murray, A.J.; Boardman, N.T.; Cole, M.; Clarke, K.; Severson, D.L.; et al. Rosiglitazone treatment improves cardiac efficiency in hearts from diabetic mice. Arch. Physiol. Biochem. 2007, 113, 211–220. [Google Scholar] [CrossRef]
- Onay-Besikci, A.; Guner, S.; Arioglu, E.; Ozakca, I.; Ozcelikay, A.T.; Altan, V.M. The effects of chronic trimetazidine treatment on mechanical function and fatty acid oxidation in diabetic rat hearts. Can. J. Physiol. Pharmacol. 2007, 85, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of fatty acid uptake and fatty acid β-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 2010, 1801, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Folmes, C.D.L.; Keung, W.; Fillmore, N.; Jaswal, J.S.; Cadete, V.J.; Beker, D.L.; Lam, V.H.; Zhang, L.; Lopaschuk, G.D. Inhibition of Serine Palmitoyl Transferase I Reduces Cardiac Ceramide Levels and Increases Glycolysis Rates following Diet-Induced Insulin Resistance. PLoS ONE 2012, 7, e37703. [Google Scholar] [CrossRef] [Green Version]
- Bagul, P.K.; Katare, P.B.; Bugga, P.; Dinda, A.K.; Banerjee, S.K. SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Nair, K.S.; Bergen, R.H.; Klaus, K.; Guenzel, A.J.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Integrity and Function in the Progression of Early Pressure Overload–Induced Left Ventricular Remodeling. J. Am. Hear. Assoc. 2017, 6, e005869. [Google Scholar] [CrossRef] [PubMed]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupte, A.A.; Hamilton, D.J.; Cordero-Reyes, A.M.; Youker, K.A.; Yin, Z.; Estep, J.D.; Stevens, R.D.; Wenner, B.; Ilkayeva, O.; Loebe, M.; et al. Mechanical Unloading Promotes Myocardial Energy Recovery in Human Heart Failure. Circ. Cardiovasc. Genet. 2014, 7, 266–276. [Google Scholar] [CrossRef]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR- coactivator 1. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [Green Version]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A Role for Peroxisome Proliferator-Activated Receptor γ Coactivator-1 in the Control of Mitochondrial Dynamics During Postnatal Cardiac Growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, L.D.L.; Soto, P.F.; Cupps, B.P.; Pasque, M.K.; Herrero, P.; Gropler, R.J.; Waggoner, A.D.; Davila-Roman, V.G. Hypertensive left ventricular hypertrophy is associated with abnormal myocardial fatty acid metabolism and myocardial efficiency. J. Nucl. Cardiol. 2006, 13, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Hear. Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R.; et al. Energy Metabolic Reprogramming in the Hypertrophied and Early Stage Failing Heart. Circ. Hear. Fail. 2014, 7, 1022–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Jaswal, J.S.; Ussher, J.R.; Sankaralingam, S.; Wagg, C.; Zaugg, M.; Lopaschuk, G.D. Cardiac Insulin-Resistance and Decreased Mitochondrial Energy Production Precede the Development of Systolic Heart Failure After Pressure-Overload Hypertrophy. Circ. Hear. Fail. 2013, 6, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Okamura, K.; Nakagama, Y.; Takeda, N.; Soma, K.; Sato, T.; Isagawa, T.; Kido, Y.; Sakamoto, M.; Manabe, I.; Hirata, Y.; et al. Therapeutic targeting of mitochondrial ROS ameliorates murine model of volume overload cardiomyopathy. J. Pharmacol. Sci. 2019, 141, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential Role of BNIP3 in Cardiac Remodeling, Myocardial Stiffness, and Endoplasmic Reticulum. Circ. Hear. Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Ma, Y.; You, J.; Li, Z.; Ding, Y.; He, P.; Lu, X.; Jiang, J.; Chen, S.; Liu, P. NMNAT3 is involved in the protective effect of SIRT3 in Ang II-induced cardiac hypertrophy. Exp. Cell Res. 2016, 347, 261–273. [Google Scholar] [CrossRef]
- Zhou, N.; Chen, X.; Xi, J.; Ma, B.; Leimena, C.; Stoll, S.; Qin, G.; Wang, C.; Qiu, H. Novel genomic targets of valosin-containing protein in protecting pathological cardiac hypertrophy. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Zhou, N.; Chen, X.; Xi, J.; Ma, B.; Leimena, C.; Stoll, S.; Qin, G.; Wang, C.; Qiu, H. Genomic characterization reveals novel mechanisms underlying the valosin-containing protein-mediated cardiac protection against heart failure. Redox Biol. 2020, 36, 101662. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Bagur, R.; Tanguy, S.; Foriel, S.; Grichine, A.; Sanchez, C.; Pernet-Gallay, K.; Kaambre, T.; Kuznetsov, A.V.; Usson, Y.; Boucher, F.; et al. The impact of cardiac ischemia/reperfusion on the mitochondria–cytoskeleton interactions. Biochim. Biophys. Acta 2016, 1862, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Sun, J.; Yoon, J.S.; Zhang, Y.; Zheng, L.; Murphy, E.; Mattson, M.P.; Lenardo, M.J. Mitochondrial Protein PGAM5 Regulates Mitophagic Protection against Cell Necroptosis. PLoS ONE 2016, 11, e0147792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddall, H.K.; Yellon, D.M.; Ong, S.-B.; Mukherjee, U.A.; Burke, N.; Hall, A.R.; Angelova, P.R.; Ludtmann, M.H.R.; Deas, E.; Davidson, S.M.; et al. Loss of PINK1 Increases the Heart’s Vulnerability to Ischemia-Reperfusion Injury. PLoS ONE 2013, 8, e62400. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Linbo, C.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, J.; Zhu, P.; Zhu, H.; Toan, S.; Hu, S.; Ren, J.; Chen, Y. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic Res. Cardiol. 2018, 113, 23. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Kurian, G.A. Preconditioning the rat heart with sodium thiosulfate preserved the mitochondria in response to ischemia-reperfusion injury. J. Bioenerg. Biomembr. 2019, 51, 189–201. [Google Scholar] [CrossRef]
- Shimizu, Y.; Lambert, J.P.; Nicholson, C.K.; Kim, J.J.; Wolfson, D.W.; Cho, H.C.; Husain, A.; Naqvi, N.; Chin, L.-S.; Li-Shen, C.; et al. DJ-1 protects the heart against ischemia–reperfusion injury by regulating mitochondrial fission. J. Mol. Cell. Cardiol. 2016, 97, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; Albertini, A.; Curello, S.; Ceconi, C.; Di Lisa, F.; Raddino, R.; Visioli, O. Myocardial recovery during post-ischaemic reperfusion: Effects of nifedipine, calcium and magnesium. J. Mol. Cell. Cardiol. 1986, 18, 487–498. [Google Scholar] [CrossRef]
- Ferrari, R.; Ceconi, C.; Curello, S.; Cargnoni, A.; Agnoletti, G.; Boffa, G.M.; Visioli, O. Intracellular effects of myocardial ischaemia and reperfusion: Role of calcium and oxygen. Eur. Hear. J. 1986, 7 (Suppl A), 3–12. [Google Scholar]
- Ferrari, R.; Ceconi, C.; Curello, S.; Cargnoni, A.; Pasini, E.; De Giuli, F.; Albertini, A. Role of oxygen free radicals in ischemic and reperfused myocardium. Am. J. Clin. Nutr. 1991, 53, 215S–222S. [Google Scholar] [CrossRef]
- Lizano, P.; Rashed, E.; Stoll, S.; Zhou, N.; Wen, H.; Hays, T.T.; Qin, G.; Xie, L.-H.; Depre, C.; Qiu, H. The valosin-containing protein is a novel mediator of mitochondrial respiration and cell survival in the heart in vivo. Sci. Rep. 2017, 7, srep46324. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Xi, J.; Ma, B.; Leimena, C.; Behringer, E.J.; Qin, G.; Qiu, H. The Valosin-Containing Protein Protects the Heart Against Pathological Ca2+ Overload by Modulating Ca2+ Uptake Proteins. Toxicol. Sci. 2019, 171, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Darley-Usmar, V.; Smith, D.; O’Leary, V. Hypoxia-reoxygenation induced increase in cellular Ca2+ in myocytes and perfused hearts: The role of mitochondria. J. Mol. Cell. Cardiol. 1989, 21, 963–973. [Google Scholar] [CrossRef]
- Ferrari, R.; Visioli, O. Protective Effects of Calcium Antagonists Against Ischaemia and Reperfusion Damage. Drugs 1991, 42, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Zong, W.-X. Necrotic death as a cell fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Alloush, J.; Beck, E.; Weisleder, N. A Murine Model of Myocardial Ischemia-reperfusion Injury through Ligation of the Left Anterior Descending Artery. J. Vis. Exp. 2014, e51329. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ding, W.; Ao, X.; Chu, X.; Wan, Q.; Wang, Y.; Xiao, D.; Yu, W.; Li, M.; Yu, F.; et al. ARC regulates programmed necrosis and myocardial ischemia/reperfusion injury through the inhibition of mPTP opening. Redox Biol. 2019, 20, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Weixler, V.; Lapusca, R.; Grangl, G.; Guariento, A.; Saeed, M.Y.; Cowan, D.B.; del Nido, P.J.; McCully, J.D.; Friehs, I. Autogenous mitochondria transplantation for treatment of right heart failure. J. Thorac. Cardiovasc. Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cowan, D.B.; Yao, R.; Akurathi, V.; Snay, E.R.; Thedsanamoorthy, J.K.; Zurakowski, D.; Ericsson, M.; Friehs, I.; Wu, Y.; Levitsky, S.; et al. Intracoronary Delivery of Mitochondria to the Ischemic Heart for Cardioprotection. PLoS ONE 2016, 11, e0160889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitzova, K.; Shin, B.; Liu, K.; Ramirez-Barbieri, G.; Guariento, A.; Blitzer, D.; Thedsanamoorthy, J.K.; Yao, R.; Snay, E.R.; Inkster, J.A.; et al. Mitochondrial transplantation prolongs cold ischemia time in murine heart transplantation. J. Hear. Lung Transplant. 2019, 38, 92–99. [Google Scholar] [CrossRef]
- Rimpelová, S.; Bříza, T.; Králová, J.; Záruba, K.; Kejík, Z.; Císařová, I.; Martásek, P.; Ruml, T.; Král, V. Rational Design of Chemical Ligands for Selective Mitochondrial Targeting. Bioconjugate Chem. 2013, 24, 1445–1454. [Google Scholar] [CrossRef]
- Krčová, L.; Rimpelová, S.; Havlík, M.; Dolenský, B.; Vellieux, F.; Ruml, T.; Martásek, P.; Král, V.; Bříza, T. Highly selective mitochondrial probes based on fluorinated pentamethinium salts: On two-photon properties and microscopic applications. Dye. Pigment. 2020, 172, 107802. [Google Scholar] [CrossRef]
- Garrido-Moreno, V.; Díaz-Vegas, A.; López-Crisosto, C.; Troncoso, M.F.; Navarro-Marquez, M.; García, L.; Estrada, M.; Cifuentes, M.; Lavandero, S. GDF-11 prevents cardiomyocyte hypertrophy by maintaining the sarcoplasmic reticulum-mitochondria communication. Pharmacol. Res. 2019, 146, 104273. [Google Scholar] [CrossRef]
- Junior, R.F.R.; Dabkowski, E.R.; Shekar, K.C.; O´Connell, K.A.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free. Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Lu, H.-I.; Huang, T.-H.; Sung, P.-H.; Chen, Y.-L.; Chua, S.; Chai, H.-Y.; Chung, S.-Y.; Liu, C.-F.; Sun, C.-K.; Chang, H.-W.; et al. Administration of antioxidant peptide SS-31 attenuates transverse aortic constriction-induced pulmonary arterial hypertension in mice. Acta Pharmacol. Sin. 2016, 37, 589–603. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.-Y.; Shao, P.-L.; Wallace, C.G.; Chua, S.; Sung, P.-H.; Ko, S.-F.; Chai, H.-T.; Chung, S.-Y.; Chen, K.-H.; Lu, H.-I.; et al. Combined Therapy with SS31 and Mitochondria Mitigates Myocardial Ischemia-Reperfusion Injury in Rats. Int. J. Mol. Sci. 2018, 19, 2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-X.; Cheng, Y.; Liu, D.-Z.; Liu, M.; Cui, H.; Zhang, B.-L.; Mei, Q.-B.; Zhou, S.-Y. Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. J. Nanobiotechnol. 2019, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Alford, J.; Qiu, H. Structural and Functional Remodeling of Mitochondria in Cardiac Diseases. Int. J. Mol. Sci. 2021, 22, 4167. https://doi.org/10.3390/ijms22084167
Sun X, Alford J, Qiu H. Structural and Functional Remodeling of Mitochondria in Cardiac Diseases. International Journal of Molecular Sciences. 2021; 22(8):4167. https://doi.org/10.3390/ijms22084167
Chicago/Turabian StyleSun, Xiaonan, Jalen Alford, and Hongyu Qiu. 2021. "Structural and Functional Remodeling of Mitochondria in Cardiac Diseases" International Journal of Molecular Sciences 22, no. 8: 4167. https://doi.org/10.3390/ijms22084167
APA StyleSun, X., Alford, J., & Qiu, H. (2021). Structural and Functional Remodeling of Mitochondria in Cardiac Diseases. International Journal of Molecular Sciences, 22(8), 4167. https://doi.org/10.3390/ijms22084167